
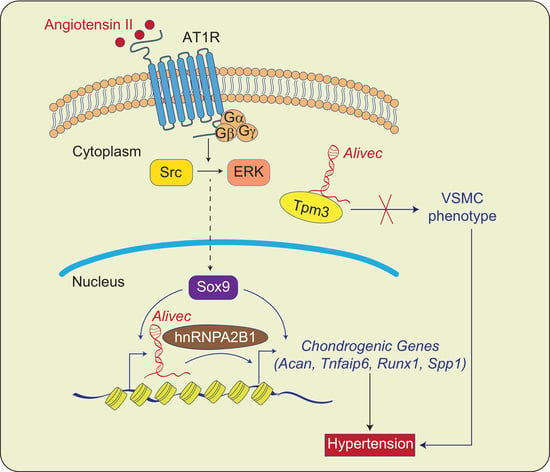
Angiotensin II-Induced Long Non-Coding RNA Alivec Regulates Chondrogenesis in Vascular Smooth Muscle Cells

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Studies
2.2. Cell Culture and Treatment
2.3. Treatment with Inhibitors of the AT1R and Signal Transduction Pathways
2.4. RNA Isolation and RT-qPCR
2.5. Cellular Fractionation
2.6. RNA Fluorescence In Situ Hybridization (RNA-FISH)
2.7. 5′ and 3′ Rapid Amplification of cDNA Ends (RACE)
2.8. In Vitro Transcription and Translation
2.9. Transient Transfection of RVSMCs with Plasmids, GapmeRs and siRNAs
2.10. Affymetrix Gene Array Analyses
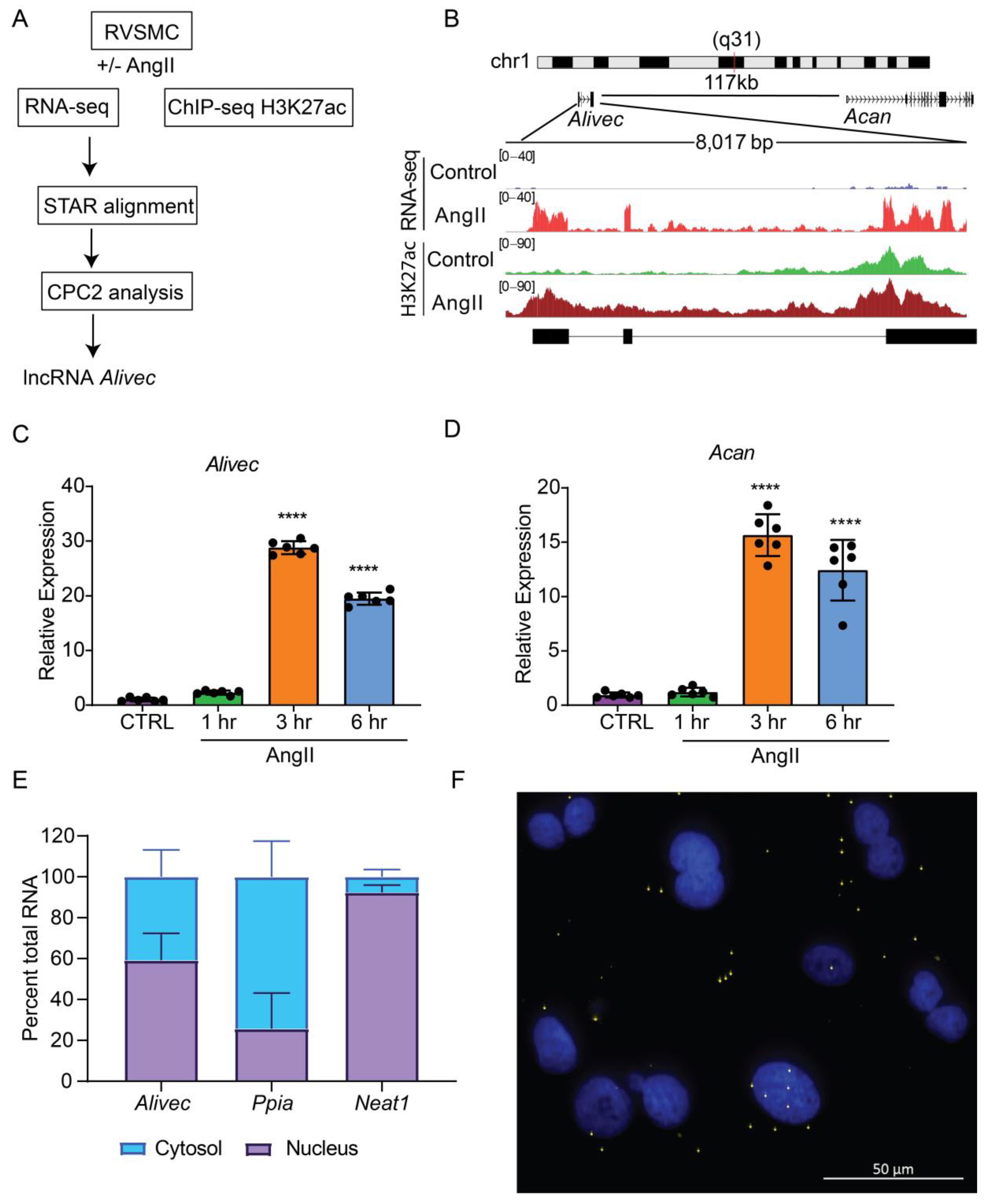
2.11. Identification of Rat lncRNA Alivec
2.12. Alcian Blue Staining to Determine Chondrogenic Phenotype
2.13. AngII-Infused Rat Model of Hypertension and Vasculopathy
2.14. Tissue Staining and Immunohistochemistry
2.15. Alivec RNA-Pulldown and Mass Spectrometry
2.16. UV-RNA Immunoprecipitation (RIP) Assay
2.17. Data Deposition
2.18. Statistical Analysis
3. Results
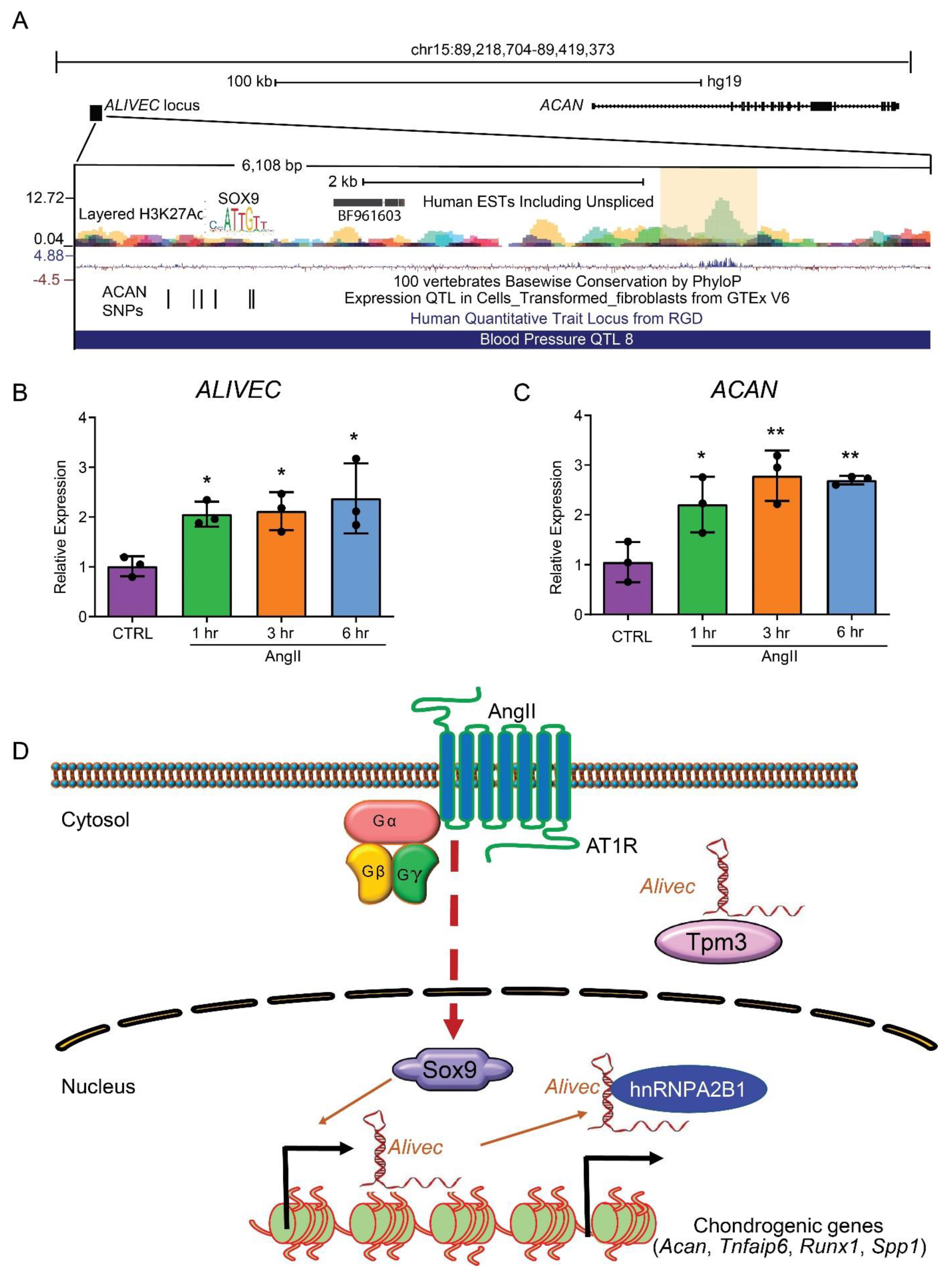
3.1. Alivec Is an AngII-Induced lncRNA Adjacent to Chondrogenic Gene Acan in RVSMCs
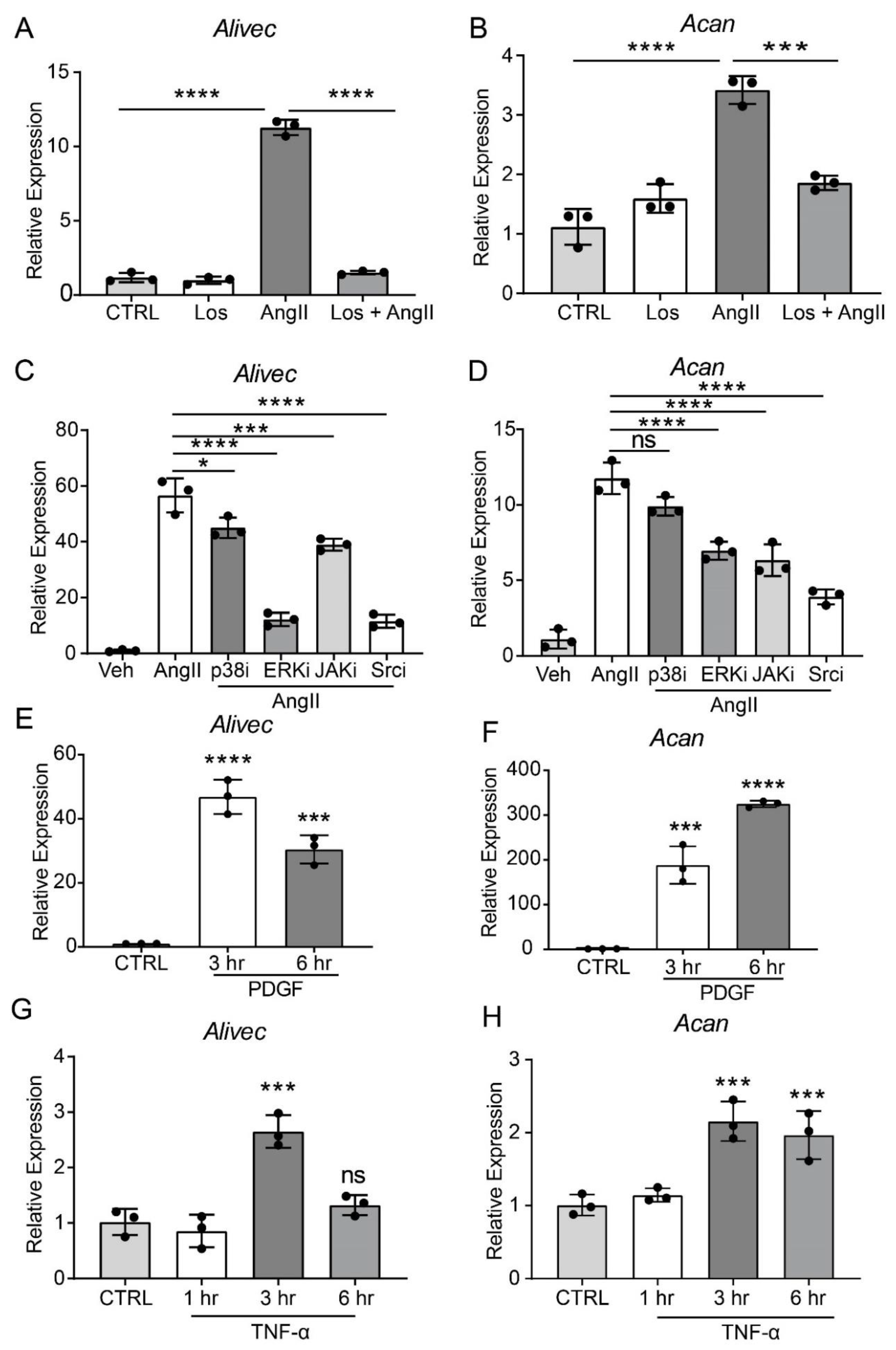
3.2. AngII-induces Alivec and Acan Expression via Activation of AT1R and Src Kinase
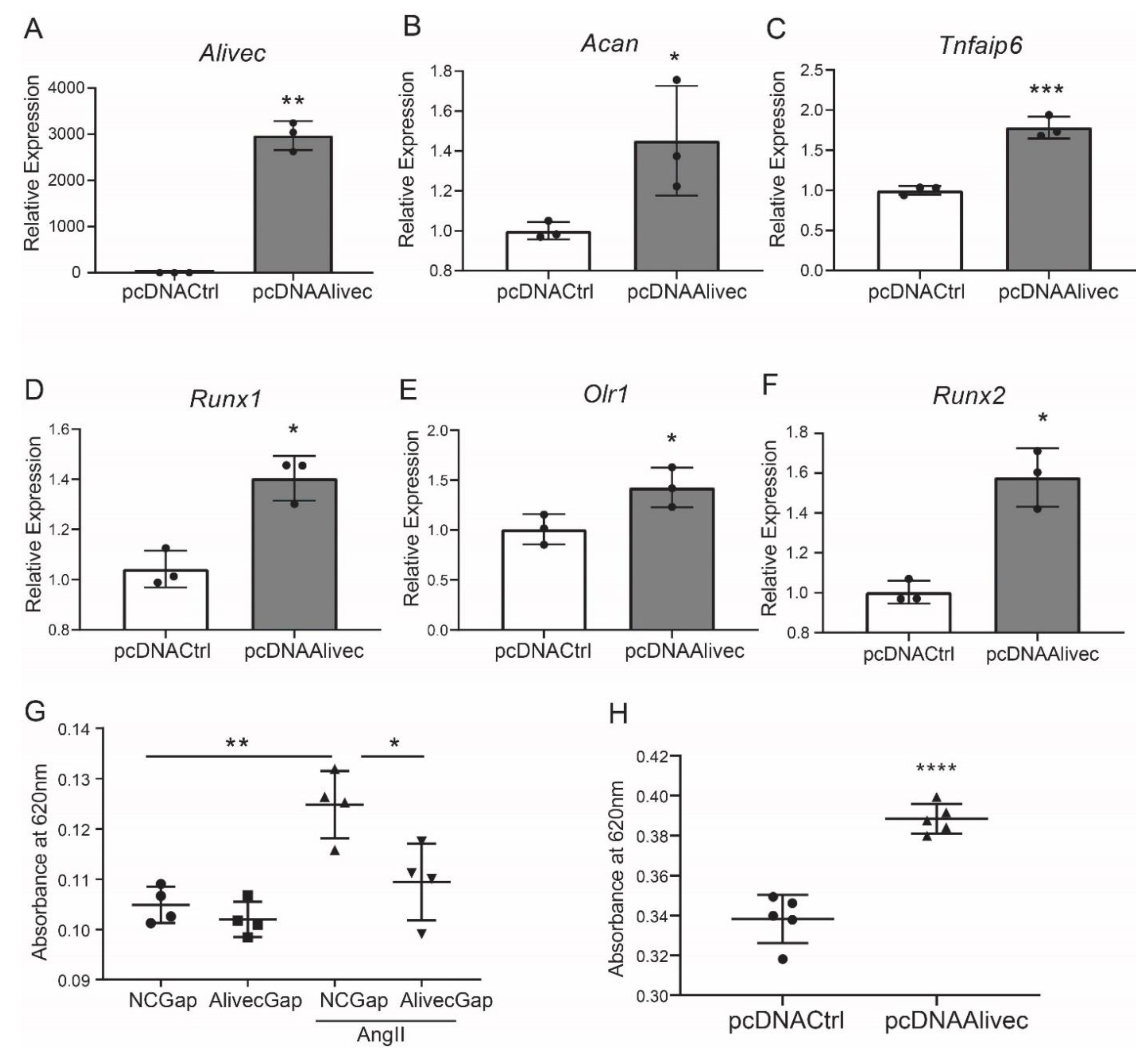
3.3. Alivec Knockdown Attenuates AngII-Induced Upregulation of Genes Associated with Chondrogenesis
3.4. Alivec Mediates a Chondrogenic/Osteogenic Phenotype in RVSMCs
3.5. Transcription Factor Sox9 Controls Alivec Expression in RVSMCs
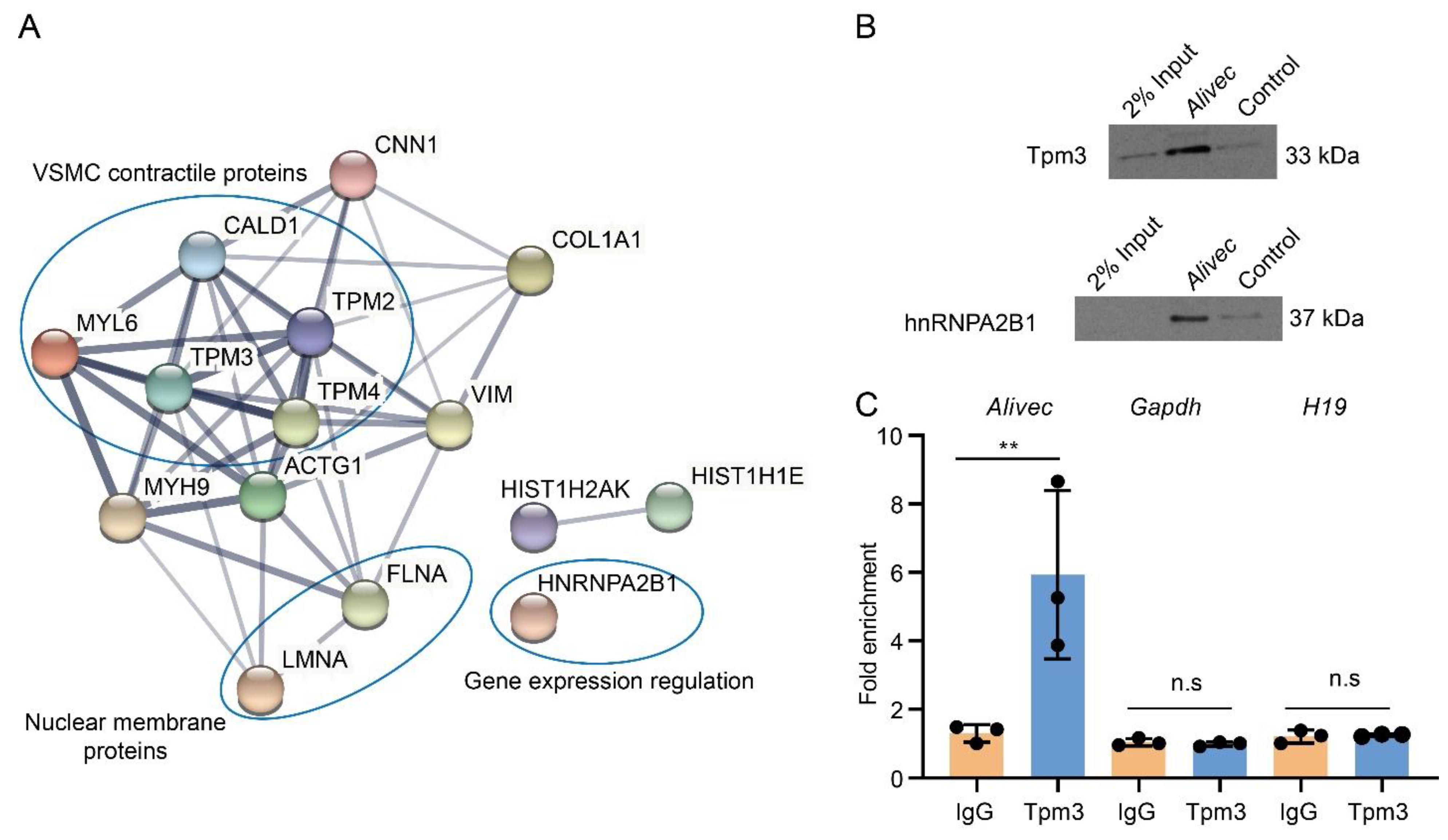
3.6. Alivec RNA Interacts with hnRNPA2B1 as well as with Tropomyosin alpha-3 Chain, a Protein with Putative Association with the Contractile Phenotype of RVSMCs
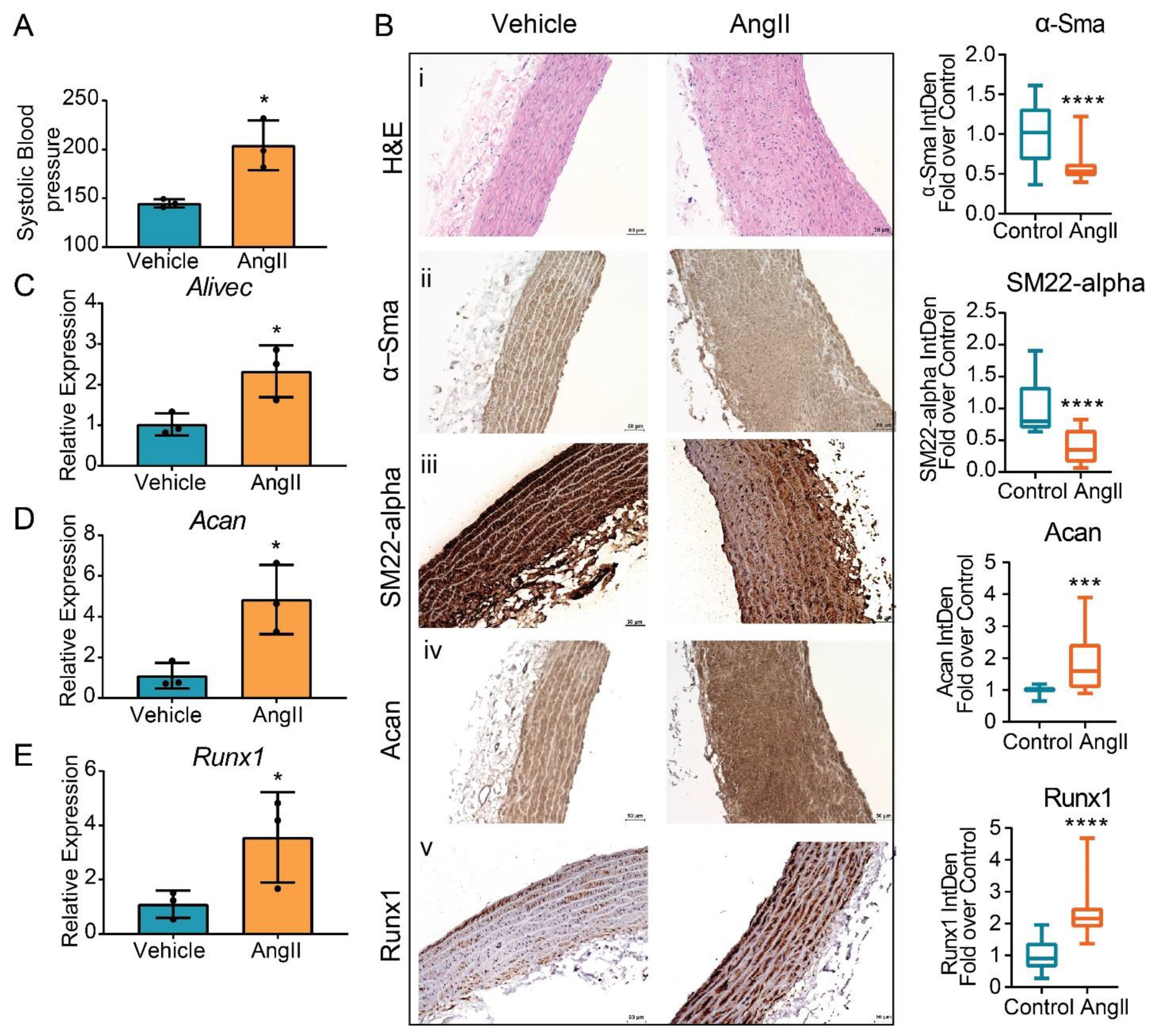
3.7. AngII Treatment Increases Aortic Expression of Alivec in Rats
3.8. The Human ALIVEC Locus Contains ACAN Regulatory Elements and a Blood Pressure Quantitative Trait Locus (QTL)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, E.R.; Jaeger, J.; Sengupta, R.; Ruzzo, W.L.; Reimer, C.; Lukito, S.; Schwartz, S.M. Atherosclerotic Plaque Smooth Muscle Cells Have a Distinct Phenotype. Arter. Thromb. Vasc. Biol. 2004, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahar, S.; Dwarakanath, R.S.; Reddy, M.A.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II enhances interleukin-18 me-diated inflammatory gene expression in vascular smooth muscle cells: A novel cross-talk in the pathogenesis of atherosclerosis. Circ. Res. 2005, 96, 1064–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montezano, A.C.; Cat, A.N.D.; Rios, F.; Touyz, R.M. Angiotensin II and Vascular Injury. Curr. Hypertens. Rep. 2014, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116. [Google Scholar] [CrossRef]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef] [Green Version]
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of Advanced Atherosclerotic Lesions in the Innominate Arteries of ApoE-Deficient Mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1420–1425. [Google Scholar] [CrossRef] [Green Version]
- Fakhry, M.; Roszkowska, M.; Briolay, A.; Bougault, C.; Guignandon, A.; Diaz-Hernandez, J.I.; Diaz-Hernandez, M.; Pikuła, S.; Buchet, R.; Hamade, E.; et al. TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 643–653. [Google Scholar] [CrossRef]
- Augstein, A.; Mierke, J.B.; Poitz, D.M.; Strasser, R.H. Sox9 is increased in arterial plaque and stenosis, associated with synthetic phenotype of vascular smooth muscle cells and causes alterations in extracellular matrix and calcification. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2526–2537. [Google Scholar] [CrossRef]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Ghanbari, M.; Peters, M.J.; de Vries, P.; Boer, C.; Van Rooij, J.G.J.; Lee, Y.-C.; Kumar, V.; Uitterlinden, A.G.; Ikram, M.A.; Wijmenga, C.; et al. A systematic analysis highlights multiple long non-coding RNAs associated with cardiometabolic disorders. J. Hum. Genet. 2018, 63, 431–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2013, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Haemmig, S.; Feinberg, M.W. LncRNAs in vascular biology and disease. Vasc. Pharmacol. 2018, 114, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Amaram, V.; Natarajan, R. Linking diabetic vascular complications with LncRNAs. Vasc. Pharmacol. 2019, 114, 139–144. [Google Scholar] [CrossRef]
- Leung, A.; Trac, C.; Jin, W.; Lanting, L.; Akbany, A.; Sætrom, P.; Schones, D.E.; Natarajan, R. Novel Long Noncoding RNAs Are Regulated by Angiotensin II in Vascular Smooth Muscle Cells. Circ. Res. 2013, 113, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Tanwar, V.S.; Reddy, M.A.; Natarajan, R. Emerging Role of Long Non-Coding RNAs in Diabetic Vascular Complications. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, M.D.; Pinel, K.; Dakin, R.S.; Vesey, A.; A Diver, L.; MacKenzie, R.M.; Garcia, R.; Welsh, P.; Sattar, N.A.; Hamilton, G.; et al. Smooth Muscle Enriched Long Noncoding RNA (SMILR) Regulates Cell Proliferation. Circulation 2016, 133, 2050–2065. [Google Scholar] [CrossRef]
- Bell, R.D.; Long, X.; Lin, M.; Bergmann, J.H.; Nanda, V.; Cowan, S.L.; Zhou, Q.; Han, Y.; Spector, D.L.; Zheng, D.; et al. Identification and Initial Functional Characterization of a Human Vascular Cell–Enriched Long Noncoding RNA. Arter. Thromb. Vasc. Biol. 2014, 34, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.S.I.; Dong, K.; Liu, J.; Wen, T.; Yu, L.; Xu, F.; Kang, X.; Osman, I.; Hu, G.; Bunting, K.M.; et al. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 2018, 115, E8660–E8667. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Zhang, E.; Senapati, P.; Amaram, V.; Reddy, M.A.; Stapleton, K.; Leung, A.; Lanting, L.; Wang, M.; Chen, Z.; et al. A Novel Angiotensin II–Induced Long Noncoding RNA Giver Regulates Oxidative Stress, Inflammation, and Proliferation in Vascular Smooth Muscle Cells. Circ. Res. 2018, 123, 1298–1312. [Google Scholar] [CrossRef]
- Das, S.; Senapati, P.; Chen, Z.; Reddy, M.A.; Ganguly, R.; Lanting, L.; Mandi, V.; Bansal, A.; Leung, A.; Zhang, S.; et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat. Commun. 2017, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Reddy, M.A.; Chen, Z.; Putta, S.; Lanting, L.; Kato, M.; Park, J.T.; Chandra, M.; Wang, C.; Tangirala, R.K.; et al. Small RNA Sequencing Reveals MicroRNAs That Modulate Angiotensin II Effects in Vascular Smooth Muscle Cells. J. Biol. Chem. 2012, 287, 15672–15683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessueille, L.; Fakhry, M.; Hamade, E.; Badran, B.; Magne, D. Glucose stimulates chondrocyte differentiation of vascular smooth muscle cells and calcification: A possible role for IL-1β. FEBS Lett. 2015, 589, 2797–2804. [Google Scholar] [CrossRef]
- Sullivan, J.C.; Bhatia, K.; Yamamoto, T.; Elmarakby, A.A. Angiotensin (1-7) Receptor Antagonism Equalizes Angiotensin II–Induced Hypertension in Male and Female Spontaneously Hypertensive Rats. Hypertension 2010, 56, 658–666. [Google Scholar] [CrossRef] [Green Version]
- Man, H.S.J.; Sukumar, A.N.; Lam, G.C.; Turgeon, P.J.; Yan, M.S.; Ku, K.H.; Dubinsky, M.K.; Ho, J.J.D.; Wang, J.J.; Das, S.; et al. Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc. Natl. Acad. Sci. USA 2018, 115, 2401–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Feng, Y.; Hu, Z.; Hu, X.; Yuan, C.-X.; Fan, Y.; Zhang, L. Characterization of Long Noncoding RNA-Associated Proteins by RNA-Immunoprecipitation. Methods Mol. Biol. 2016, 1402, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Yang, M.; Jiang, H.; Ju, D.; Zheng, J.-P.; Xu, Z.; Liao, T.-D.; Li, L. Arterial injury promotes medial chondrogenesis in Sm22 knockout mice. Cardiovasc. Res. 2010, 90, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zheng, B.; Meszaros, L.B.; Vella, J.B.; Usas, A.; Matsumoto, T.; Huard, J. Identification and characterization of chondrogenic progenitor cells in the fascia of postnatal skeletal muscle. J. Mol. Cell Biol. 2011, 3, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Kim, J.-E.; Nakashima, K.; Balmes, G.; Iwai, N.; Deng, J.M.; Zhang, Z.; Martin, J.F.; Behringer, R.R.; Nakamura, T.; et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 2005, 102, 14665–14670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef] [Green Version]
- Ferrè, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein–lncRNA interaction. Briefings Bioinform. 2015, 17, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-S.; Yim, S.-H.; Xu, H.-D.; Jung, S.-H.; Shin, S.-H.; Hu, H.-J.; Jung, C.-K.; Choi, J.Y.; Chung, Y.-J. Tropomyosin3 overexpression and a potential link to epithelial-mesenchymal transition in human hepatocellular carcinoma. BMC Cancer 2010, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Kashiwada, K.; Nishida, W.; Hayashi, K.; Ozawa, K.; Yamanaka, Y.; Saga, H.; Yamashita, T.; Tohyama, M.; Shimada, S.; Sato, K.; et al. Coordinate Expression of α-Tropomyosin and Caldesmon Isoforms in Association with Phenotypic Modulation of Smooth Muscle Cells. J. Biol. Chem. 1997, 272, 15396–15404. [Google Scholar] [CrossRef] [Green Version]
- Sobue, K.; Hayashi, K.; Nishida, W. Expressional regulation of smooth muscle cell-specific genes in association with phe-notypic modulation. Mol. Cell Biochem. 1999, 190, 105–118. [Google Scholar] [CrossRef]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Briefings Bioinform. 2012, 14, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Rapp, J.P. Genetic Analysis of Inherited Hypertension in the Rat. Physiol. Rev. 2000, 80, 135–172. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Burger, D.; Nishigaki, N.; Montezano, A.; Touyz, R. Angiotensin II and the vascular phenotype in hypertension. Expert Rev. Mol. Med. 2011, 13, e11. [Google Scholar] [CrossRef] [PubMed]
- Doege, K.J.; Sasaki, M.; Kimura, T.; Yamada, Y. Complete coding sequence and deduced primary structure of the human cartilage large aggregating proteoglycan, aggrecan. Human-specific repeats, and additional alternatively spliced forms. J. Biol. Chem. 1991, 266, 894–902. [Google Scholar] [CrossRef]
- Lee, G.-L.; Yeh, C.-C.; Wu, J.-Y.; Lin, H.-C.; Wang, Y.-F.; Kuo, Y.-Y.; Hsieh, Y.-T.; Hsu, Y.-J.; Kuo, C.-C. TLR2 Promotes Vascular Smooth Muscle Cell Chondrogenic Differentiation and Consequent Calcification via the Concerted Actions of Osteoprotegerin Suppression and IL-6–Mediated RANKL Induction. Arter. Thromb. Vasc. Biol. 2019, 39, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Barallobre-Barreiro, J.; Oklu, R.; Lynch, M.; Fava, M.; Baig, F.; Yin, X.; Barwari, T.; Potier, D.N.; Albadawi, H.; Jahangiri, M.; et al. Extracellular matrix remodelling in response to venous hypertension: Proteomics of human varicose veins. Cardiovasc. Res. 2016, 110, 419–430. [Google Scholar] [CrossRef]
- Suna, G.; Wojakowski, W.; Lynch, M.; Barallobre-Barreiro, J.; Yin, X.; Mayr, U.; Baig, F.; Lu, R.; Fava, M.; Hayward, R.; et al. Extracellular Matrix Proteomics Reveals Interplay of Aggrecan and Aggrecanases in Vascular Remodeling of Stented Coronary Arteries. Circulation 2018, 137, 166–183. [Google Scholar] [CrossRef]
- Parkar, A.A.; Kahmann, J.D.; Howat, S.L.; Bayliss, M.T.; Day, A.J. TSG-6 interacts with hyaluronan and aggrecan in a pH-dependent manner via a common functional element: Implications for its regulation in inflamed cartilage. FEBS Lett. 1998, 428, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Limor, R.; Kaplan, M.; Sawamura, T.; Sharon, O.; Keidar, S.; Weisinger, G.; Knoll, E.; Naidich, M.; Stern, N. Angiotensin II increases the expression of lectin-like oxidized low-density lipoprotein receptor-1 in human vascular smooth muscle cells via a lipoxygenase-dependent pathway. Am. J. Hypertens. 2005, 18, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.H.; Choi, M.; Chang, S.; Lee, M.Y.; Lee, D.J.; Choi, K.; Park, J.; Han, E.C.; Hwang, D.; Kwon, K.; et al. Vascular Proteomics Reveal Novel Proteins Involved in SMC Phenotypic Change: OLR1 as a SMC Receptor Regulating Proliferation and Inflammatory Response. PLoS ONE 2015, 10, e0133845. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.J.; Bourne, L.E.; Davies, B.K.; Arnett, T.R.; MacRae, V.E.; Wheeler-Jones, C.P.; Orriss, I.R. Differing calcification processes in cultured vascular smooth muscle cells and osteoblasts. Exp. Cell Res. 2019, 380, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Schinzari, F.; Tesauro, M.; Bertoli, A.; Valentini, A.; Veneziani, A.; Campia, U.; Cardillo, C. Calcification biomarkers and vascular dysfunction in obesity and type 2 diabetes: Influence of oral hypoglycemic agents. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E658–E666. [Google Scholar] [CrossRef] [PubMed]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arterioscler Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadson, P.F.; Rossignol, C.; McCoy, J.; Rosenquist, T.H. Expression of elastin, smooth muscle alpha-actin, and c-Jun as a function of the embryonic lineage of vascular smooth muscle cells. In Vitro Cell. Dev. Biol. Anim. 1993, 29, 773–781. [Google Scholar] [CrossRef]
- Gunning, P.W.; Hardeman, E.C.; Lappalainen, P.; Mulvihill, D. Tropomyosin—Master regulator of actin filament function in the cytoskeleton. J. Cell Sci. 2015, 128, 2965–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Ma, W.-D.; Zhang, C.-Y.; Zhang, Y.; Yao, Z.-H.; Quan, X.-H.; Guo, X.; Wang, C.-X. Tanshinone IIA attenuates high glucose induced human VSMC proliferation and migration through miR-21-5p-mediated tropomyosin 1 downregulation. Arch. Biochem. Biophys. 2019, 677, 108154. [Google Scholar] [CrossRef]
- Gao, C.; Guo, H.; Mi, Z.; Wai, P.Y.; Kuo, P.C. Transcriptional Regulatory Functions of Heterogeneous Nuclear Ribonucleoprotein-U and -A/B in Endotoxin-Mediated Macrophage Expression of Osteopontin. J. Immunol. 2005, 175, 523–530. [Google Scholar] [CrossRef]
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A Long Noncoding RNA Mediates Both Activation and Repression of Immune Response Genes. Science 2013, 341, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.L.; Pjanic, M.; Wang, T.; Nguyen, T.; Cohain, A.; Lee, J.; Perisic, L.; Hedin, U.; Kundu, R.K.; Majmudar, D.; et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 2016, 7, 12092. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samara, V.A.; Das, S.; Reddy, M.A.; Tanwar, V.S.; Stapleton, K.; Leung, A.; Abdollahi, M.; Ganguly, R.; Lanting, L.; Natarajan, R. Angiotensin II-Induced Long Non-Coding RNA Alivec Regulates Chondrogenesis in Vascular Smooth Muscle Cells. Cells 2021, 10, 2696. https://doi.org/10.3390/cells10102696
Samara VA, Das S, Reddy MA, Tanwar VS, Stapleton K, Leung A, Abdollahi M, Ganguly R, Lanting L, Natarajan R. Angiotensin II-Induced Long Non-Coding RNA Alivec Regulates Chondrogenesis in Vascular Smooth Muscle Cells. Cells. 2021; 10(10):2696. https://doi.org/10.3390/cells10102696
Chicago/Turabian StyleSamara, Vishnu Amaram, Sadhan Das, Marpadga A. Reddy, Vinay Singh Tanwar, Kenneth Stapleton, Amy Leung, Maryam Abdollahi, Rituparna Ganguly, Linda Lanting, and Rama Natarajan. 2021. "Angiotensin II-Induced Long Non-Coding RNA Alivec Regulates Chondrogenesis in Vascular Smooth Muscle Cells" Cells 10, no. 10: 2696. https://doi.org/10.3390/cells10102696