
Mitochondrial Permeability Transition Causes Mitochondrial Reactive Oxygen Species- and Caspase 3-Dependent Atrophy of Single Adult Mouse Skeletal Muscle Fibers

 , , and
, , and 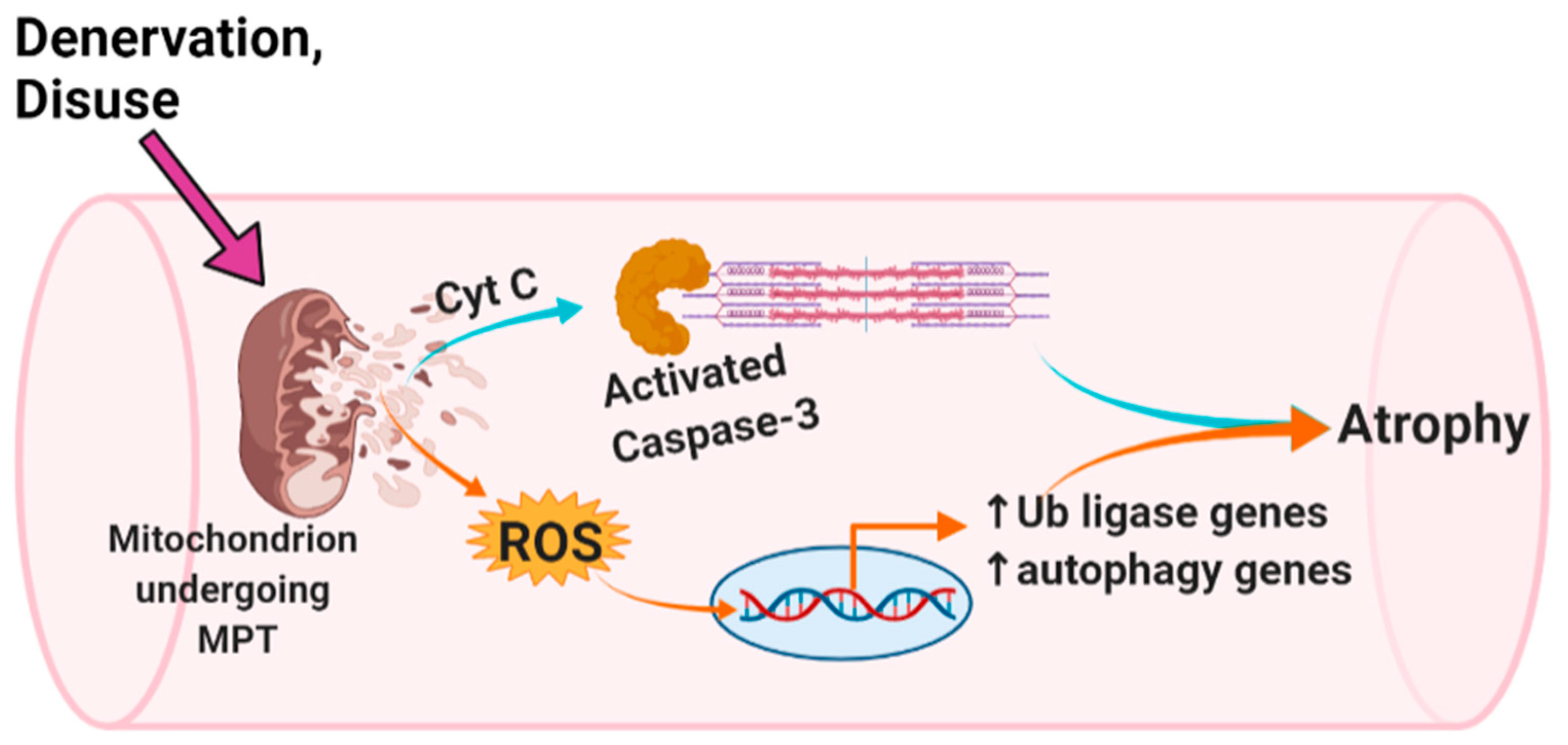{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Surgical Methods
2.2. Single Fiber Isolation and Culture
2.3. mROS Emission and Caspase-3 Activity
2.4. Acute Inhibition of MPT, mROS and Caspase-3 Activity
2.5. ‘Disuse’ Atrophy Experiments
2.6. Statistics
3. Results
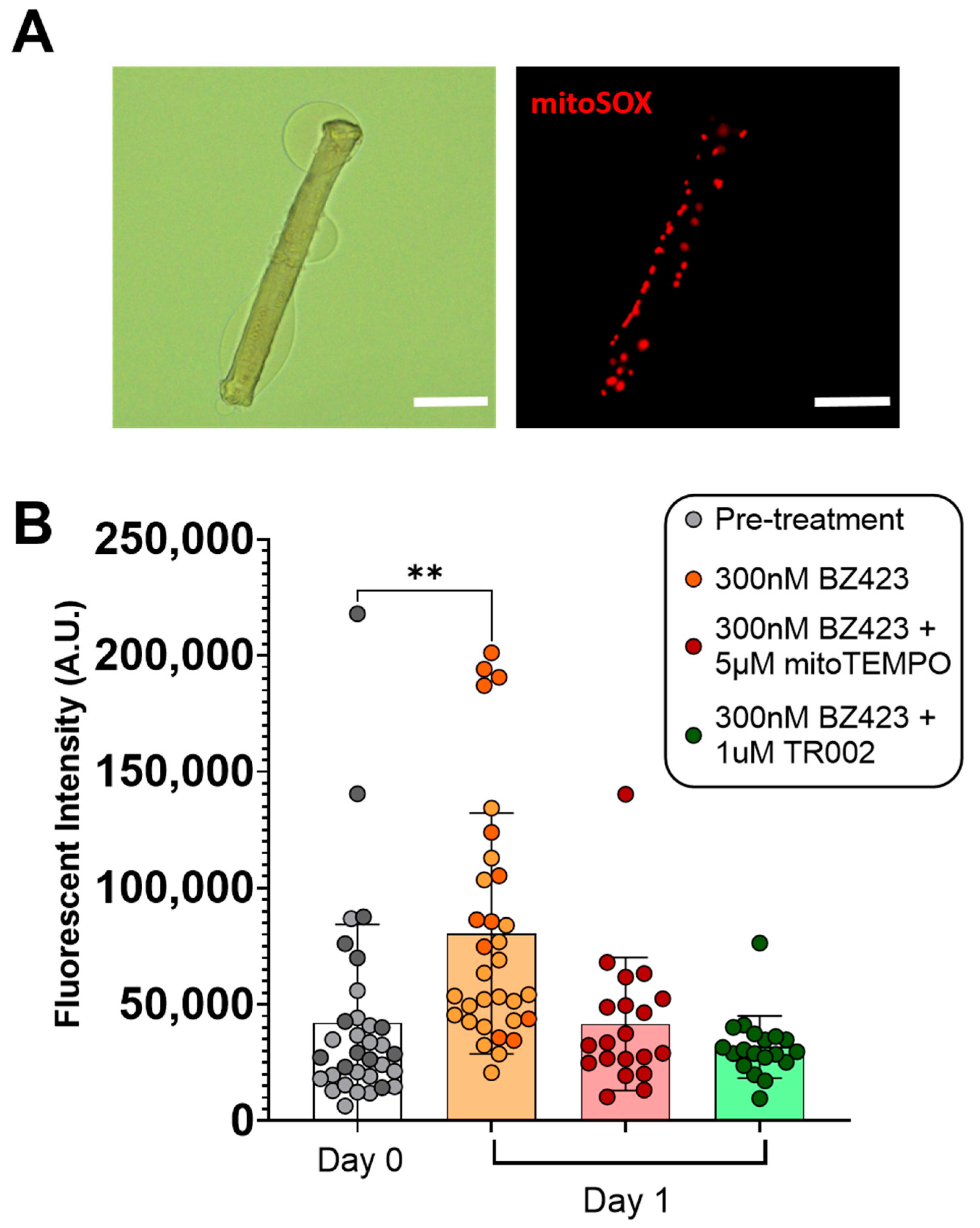
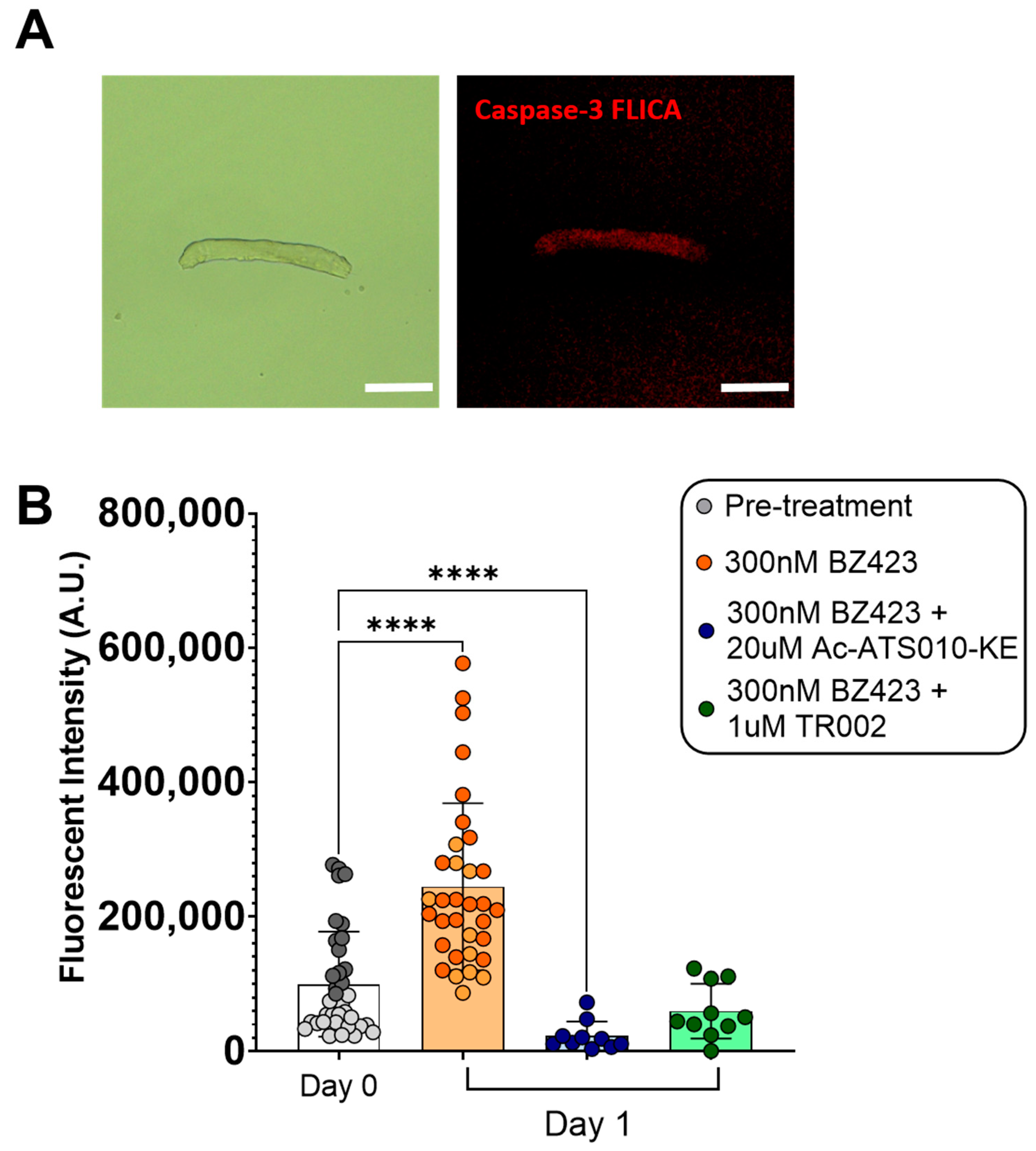
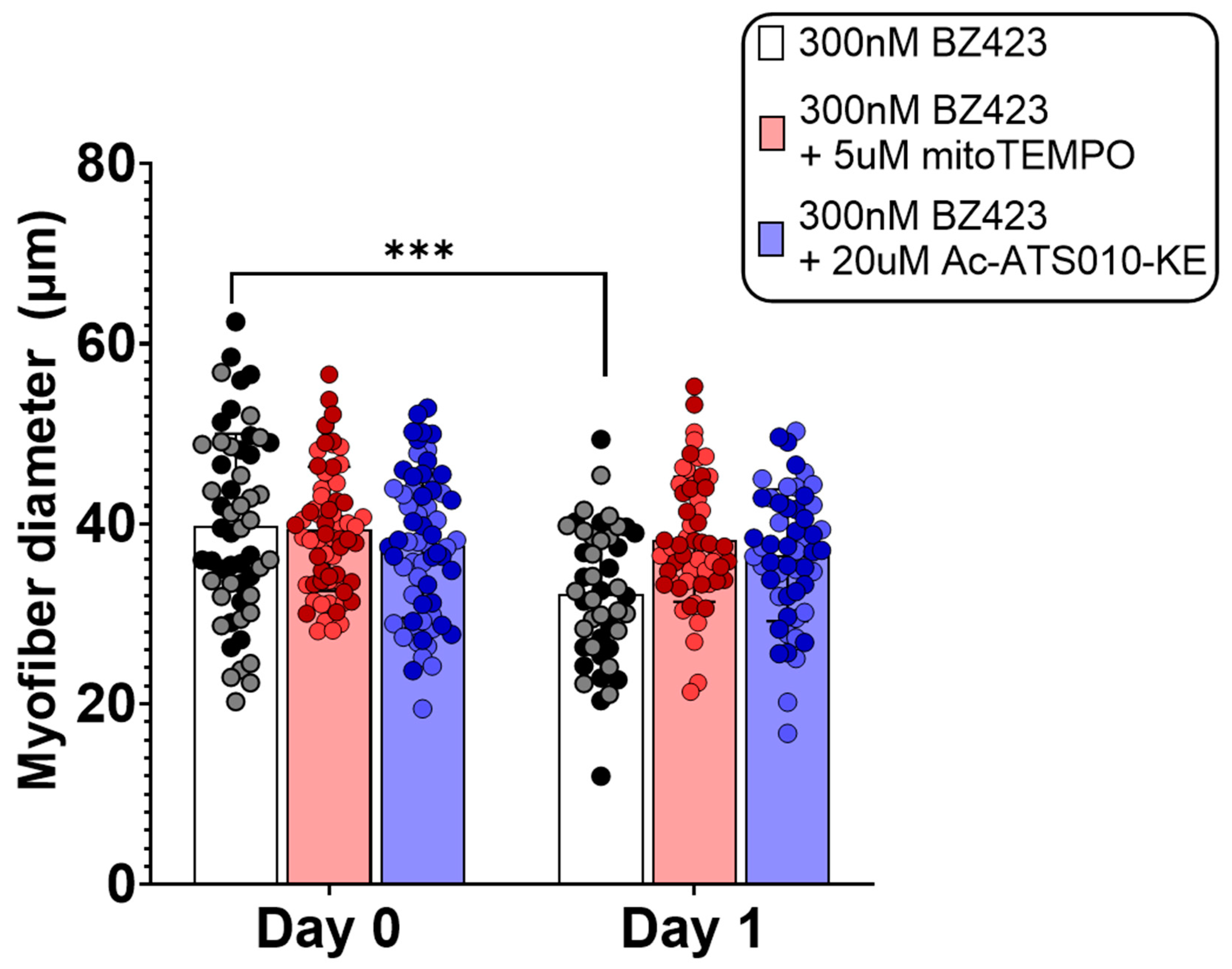
3.1. Acute Myofiber Culture Experiments
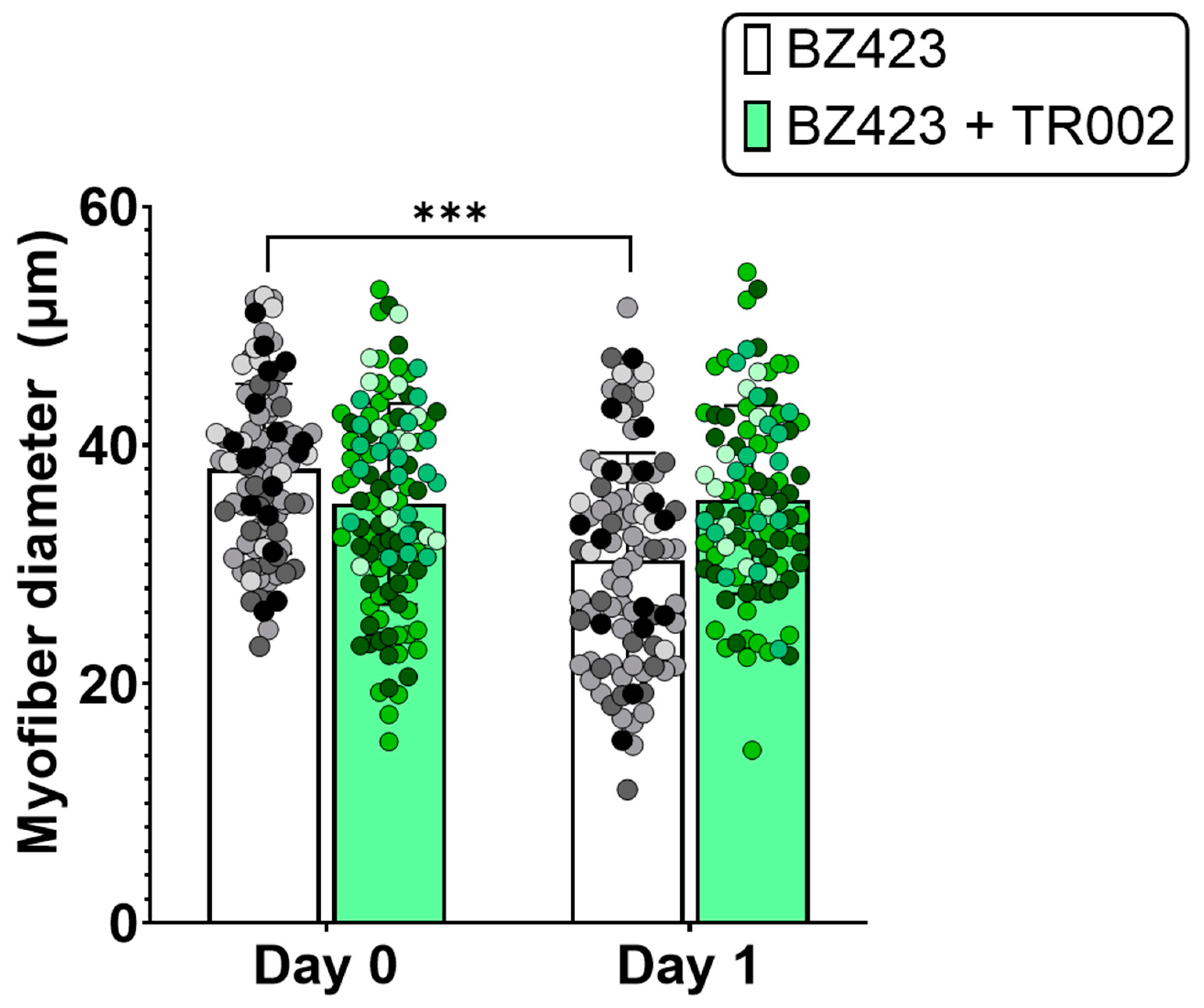
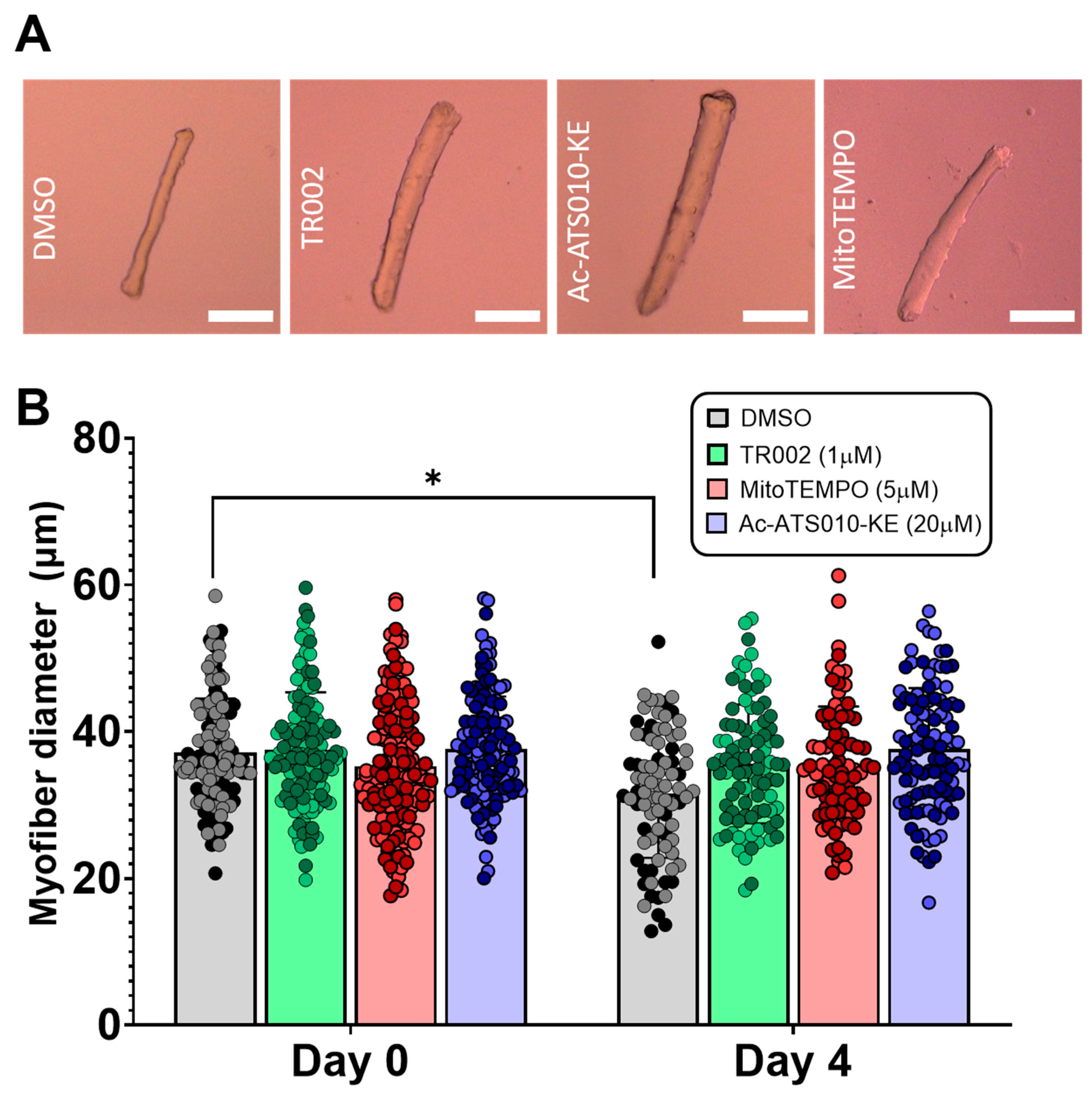
3.2. ‘Disuse’ Muscle Atrophy Model Experiments
4. Discussion
4.1. mROS Emission and Caspase-3 Activation Increase Following MPT in Single Isolated Mouse Muscle Fibers, and Are Accompanied by a Decrease in Myofiber Diameter
4.2. MPT, mROS, and Caspase-3 in a Single Muscle Fiber Model of ‘Disuse’
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef]
- Kapchinsky, S.; Vuda, M.; Miguez, K.; Elkrief, D.; De Souza, A.R.; Baglole, C.J.; Aare, S.; Macmillan, N.J.; Baril, J.; Rozakis, P.; et al. Smoke-induced neuromuscular junction degeneration precedes the fibre type shift and atrophy in chronic obstructive pulmonary disease. J. Physiol. 2018, 596, 2865–2881. [Google Scholar] [CrossRef]
- Baloh, R.H.; Rakowicz, W.; Gardner, R.; Pestronk, A. Frequent atrophic groups with mixed-type myofibers is distinctive to motor neuron syndromes. Muscle Nerve 2007, 36, 107–110. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Ebner, N.; dos Santos, M.R.; Ishida, J.; Hasenfuss, G.; Haehling, S. Cachexia, muscle wasting, and frailty in cardiovascular disease. Eur. J. Hear. Fail. 2020, 22, 2314–2326. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Mitch, W.E. Muscle wasting from kidney failure—A model for catabolic conditions. Int. J. Biochem. Cell Biol. 2013, 45, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Smuder, A.J.; Kwon, O.-S.; Kavazis, A.N.; Szeto, H.H.; Powers, S.K. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 2011, 111, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Plant, P.J.; Bain, J.R.; Correa, J.E.; Woo, M.; Batt, J. Absence of caspase-3 protects against denervation-induced skeletal muscle atrophy. J. Appl. Physiol. 2009, 107, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin–proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Hughes, D.C.; Baehr, L.M.; Driscoll, J.R.; Lynch, S.A.; Waddell, D.S.; Bodine, S.C. Identification and characterization of Fbxl22, a novel skeletal muscle atrophy-promoting E3 ubiquitin ligase. Am. J. Physiol. Physiol. 2020, 319, C700–C719. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Zhang, L.; Mitch, W.E.; LeDoux, J.M.; Hu, J.; Du, J. Caspase-3 Cleaves Specific 19 S Proteasome Subunits in Skeletal Muscle Stimulating Proteasome Activity. J. Biol. Chem. 2010, 285, 21249–21257. [Google Scholar] [CrossRef]
- Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef]
- Biasutto, L.; Azzolini, M.; Szabo, I.; Zoratti, M. The mitochondrial permeability transition pore in AD 2016: An update. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2016, 1863, 2515–2530. [Google Scholar] [CrossRef]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef]
- Powers, S.K.; Wiggs, M.P.; Duarte, J.A.; Zergeroglu, A.M.; Demirel, H.A. Mitochondrial signaling contributes to disuse muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E31–E39. [Google Scholar] [CrossRef]
- Adhihetty, P.J.; O’Leary, M.F.N.; Chabi, B.; Wicks, K.L.; Hood, D.A. Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J. Appl. Physiol. 2007, 102, 1143–1151. [Google Scholar] [CrossRef]
- Sesso, A.; Marques, M.; Monteiro, M.M.; Schumacher, R.I.; Colquhoun, A.; Belizário, J.; Konno, S.N.; Felix, T.B.; Botelho, L.A.; Santos, V.Z.; et al. Morphology of mitochondrial permeability transition: Morphometric volumetry in apoptotic cells. Anat. Rec. Part A: Discov. Mol. Cell. Evol. Biol. 2004, 281A, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Strubbe-Rivera, J.O.; Schrad, J.; Pavlov, E.; Conway, J.; Parent, K.; Bazil, J.N. The mitochondrial permeability transition phenomenon elucidated by cryo-EM reveals the genuine impact of calcium overload on mitochondrial structure and function. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H.; et al. Absence of physiological Ca2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Anderson, J.E.; Akahoshi, M.; Nakamura, M.; Tatsumi, R.; Ikeuchi, Y.; Mizunoya, W. Protocol for rat single muscle fiber isolation and culture. Anal. Biochem. 2015, 482, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Chen, X.; Boitano, A.; Swenson, L.; Opipari, A.W.; Glick, G.D. Identification and Validation of the Mitochondrial F1F0-ATPase as the Molecular Target of the Immunomodulatory Benzodiazepine Bz-423. Chem. Biol. 2005, 12, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Sambri, I.; Massa, F.; Gullo, F.; Meneghini, S.; Cassina, L.; Carraro, M.; Dina, G.; Quattrini, A.; Patanella, L.; Carissimo, A.; et al. Impaired flickering of the permeability transition pore causes SPG7 spastic paraplegia. EBioMedicine 2020, 61, 103050. [Google Scholar] [CrossRef]
- Solania, A.; Gonzalez-Paez, G.; Wolan, D.W. Selective and Rapid Cell-Permeable Inhibitor of Human Caspase-3. ACS Chem. Biol. 2019, 14, 2463–2470. [Google Scholar] [CrossRef]
- Antonucci, S.; Di Sante, M.; Sileikyte, J.; Deveraux, J.; Bauer, T.; Bround, M.J.; Menabò, R.; Paillard, M.; Alanova, P.; Carraro, M.; et al. A novel class of cardioprotective small-molecule PTP inhibitors. Pharmacol. Res. 2020, 151, 104548. [Google Scholar] [CrossRef]
- Bodine, S.C. Disuse-induced muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2200–2208. [Google Scholar] [CrossRef]
- Duddy, W.J.; Cohen, T.; Duguez, S.; Partridge, T.A. The isolated muscle fibre as a model of disuse atrophy: Characterization using PhAct, a method to quantify f-actin. Exp. Cell Res. 2011, 317, 1979–1993. [Google Scholar] [CrossRef]
- Halestrap, A.; Connern, C.; Griffiths, E.; Kerr, P. Cyclosporin A binding in mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 1997, 174, 167–172. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Carraro, M.; Checchetto, V.; Szabo, I.; Bernardi, P. F-ATPsynthase and the permeability transition pore: Fewer doubts, more certainties. FEBS Lett. 2019, 593, 1542–1553. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.M.; Bround, M.J.; Khalil, H.; Sargent, M.A.; Latchman, N.; Terada, N.; Peixoto, P.M.; Molkentin, J.D. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 2019, 5, eaaw4597. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Daugas, E.; Nochy, D.; Ravagnan, L.; Loeffler, M.; Susin, S.A.; Zamzami, N.; Kroemer, G. Apoptosis-inducing factor (AIF): A ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000, 476, 118–123. [Google Scholar] [CrossRef]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nat. Cell Biol. 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Powers, S.K.; Morton, A.; Ahn, B.; Smuder, A.J. Redox control of skeletal muscle atrophy. Free. Radic. Biol. Med. 2016, 98, 208–217. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Brocca, L.; Toniolo, L.; Reggiani, C.; Bottinelli, R.; Sandri, M.; Pellegrino, M.A. FoxO-dependent atrogenes vary among catabolic conditions and play a key role in muscle atrophy induced by hindlimb suspension. J. Physiol. 2016, 595, 1143–1158. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Nowak, K.J.; Jackaman, C.; Clément, S.; Lyons, M.A.; Gallagher, S.; Bakker, A.J.; Laing, N.G. Dissociated flexor digitorum brevis myofiber culture system—A more mature muscle culture system. Cell Motil. Cytoskelet. 2007, 64, 727–738. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skinner, S.K.; Solania, A.; Wolan, D.W.; Cohen, M.S.; Ryan, T.E.; Hepple, R.T. Mitochondrial Permeability Transition Causes Mitochondrial Reactive Oxygen Species- and Caspase 3-Dependent Atrophy of Single Adult Mouse Skeletal Muscle Fibers. Cells 2021, 10, 2586. https://doi.org/10.3390/cells10102586
Skinner SK, Solania A, Wolan DW, Cohen MS, Ryan TE, Hepple RT. Mitochondrial Permeability Transition Causes Mitochondrial Reactive Oxygen Species- and Caspase 3-Dependent Atrophy of Single Adult Mouse Skeletal Muscle Fibers. Cells. 2021; 10(10):2586. https://doi.org/10.3390/cells10102586
Chicago/Turabian StyleSkinner, Sarah K., Angelo Solania, Dennis W. Wolan, Michael S. Cohen, Terence E. Ryan, and Russell T. Hepple. 2021. "Mitochondrial Permeability Transition Causes Mitochondrial Reactive Oxygen Species- and Caspase 3-Dependent Atrophy of Single Adult Mouse Skeletal Muscle Fibers" Cells 10, no. 10: 2586. https://doi.org/10.3390/cells10102586