
YAP-Dependent BiP Induction Is Involved in Nicotine-Mediated Oral Cancer Malignancy

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Line and Cell Culture
2.2. Drugs and Antibodies
2.3. RNA Interference
2.4. RNA Extraction and Quantitative Real-Time PCR (RT-PCR)
2.5. Scratch Wound Healing Assay
2.6. Transwell Invasion Assay
2.7. Co-Immunoprecipitation
2.8. Chromatin Immunoprecipitation
2.9. Luciferase Dual Assay
2.10. Immunohistochemistry
2.11. Western Blot Analysis
2.12. Animal Studies
2.13. Statistical Analysis
3. Results
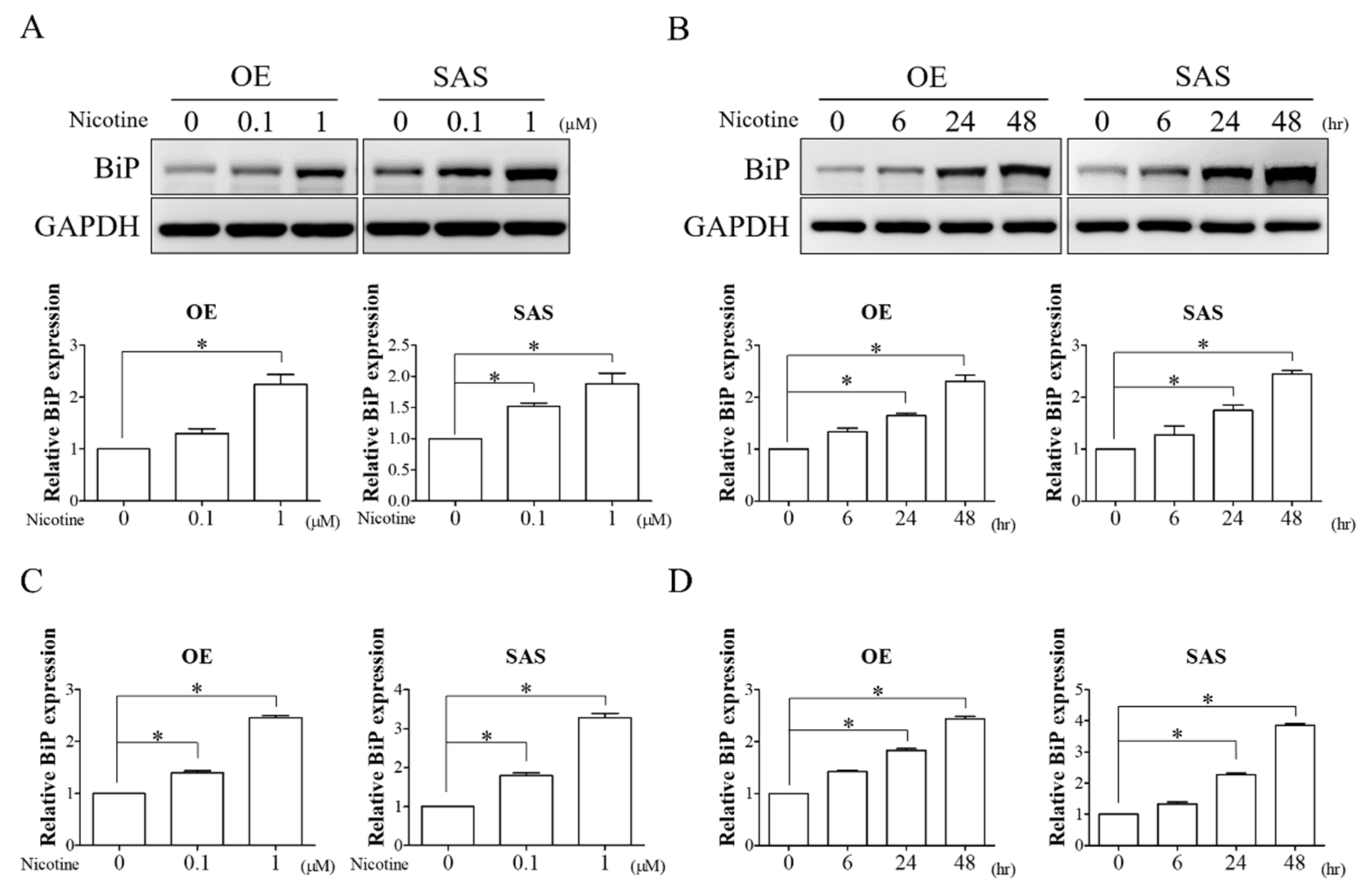
3.1. Nicotine Remarkably Induced BiP Expression in OSCC Cells
3.2. BiP Was Involved in Nicotine-Stimulated Malignant Behaviors in OSCC Cells
3.3. Nicotine Induced BiP Expression and Tumor Progression via α7-nAChR-Akt Signaling in OSCC Cells
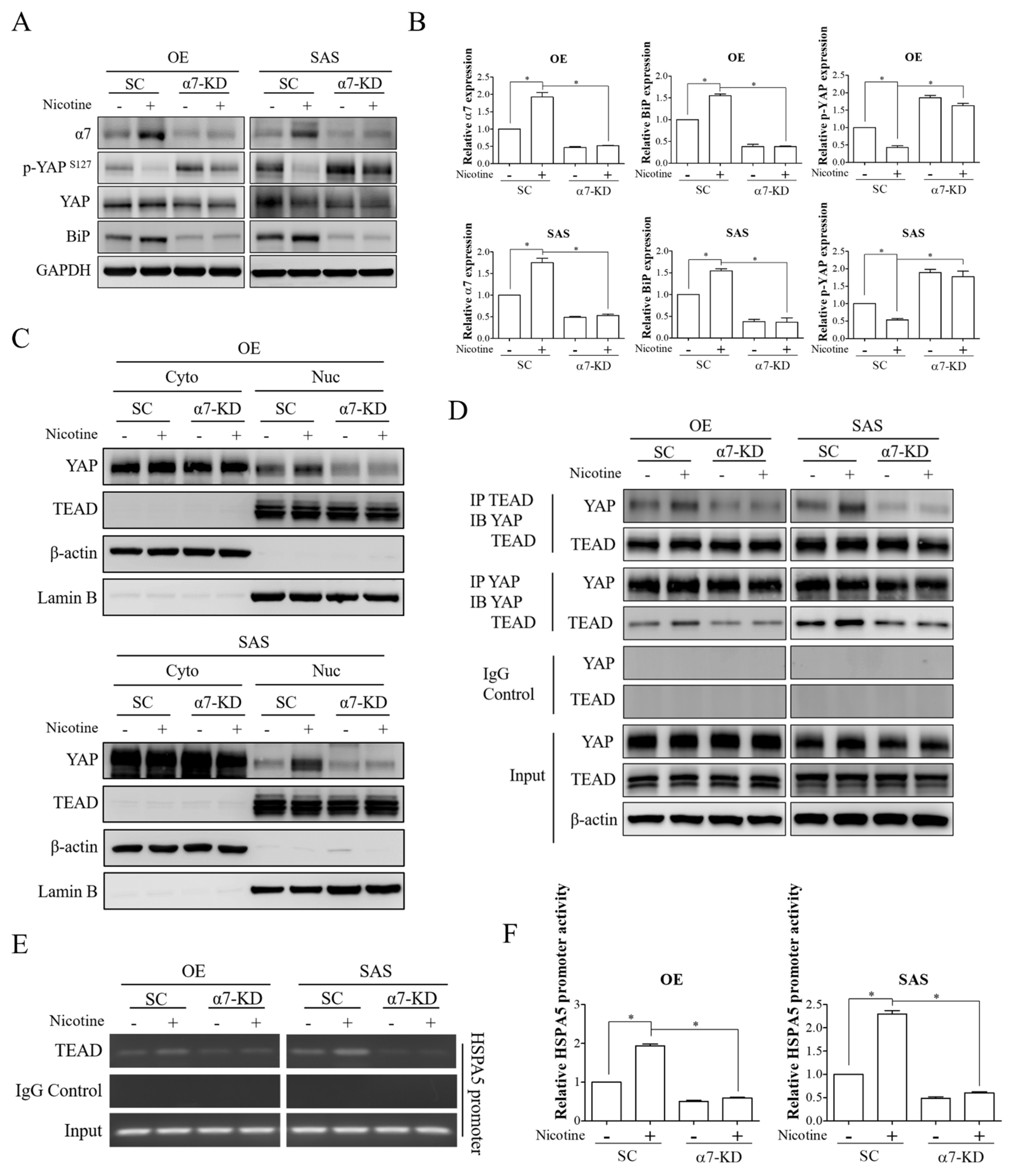
3.4. The YAP-TEAD Transcriptional Complex Was the Downstream Effector of α7-nAChR-Akt Signaling in Nicotine-Induced BiP Expression and Malignant Behaviors
3.5. BiP Silencing Decreased Nicotine-Induced Cell Growth and EMT in Tumor-Bearing Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.; Wu, J.; Wang, J.; Huang, R. Tobacco and oral squamous cell carcinoma: A review of carcinogenic pathways. Tob. Induc. Dis. 2019, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Nanavati, R.; Modi, T.G.; Dobariya, C. Oral cancer: Etiology and risk factors: A review. J. Cancer Res. Ther. 2016, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.C. Tobacco smoking, E-cigarettes, and nicotine harm. Proc. Natl. Acad. Sci. USA 2018, 115, 1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Huang, H.; Pan, C.; Zhang, B.; Liu, X.; Zhang, L. Nicotine inhibits apoptosis induced by cisplatin in human oral cancer cells. Int. J. Oral Maxillofac. Surg. 2007, 36, 739. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Tada, H.; Ibaragi, S.; Chen, C.; Sasano, T. Nicotine exposure induces the proliferation of oral cancer cells through the alpha subunit of the nicotinic acetylcholine receptor. Biochem. Biophys. Res. Commun. 2019, 509, 514. [Google Scholar] [CrossRef]
- Wang, M.; Niu, W.; Qi, M.; Chen, H.; Zhang, M.; Wang, C.; Ge, L.; Yang, J.; Miao, C.; Shi, N.; et al. Nicotine promotes cervical metastasis of oral cancer by regulating peroxiredoxin 1 and epithelial-mesenchymal transition in mice. Onco Targets Ther. 2019, 12, 3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, E.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.K.; Holloway, A.C.; Hardy, D.B. Nicotine Directly Induces Endoplasmic Reticulum Stress Response in Rat Placental Trophoblast Giant Cells. Toxicol. Sci. 2016, 151, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonthal, A.H. Endoplasmic reticulum stress: Its role in disease and novel prospects for therapy. Scientifica 2012, 2012, 857516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252. [Google Scholar] [CrossRef]
- Li, Z.; Li, Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim. Biophys. Acta 2012, 1826, 13. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chen, W.H.; Liao, C.T.; Chen, I.H.; Chiu, C.C.; Wang, H.M.; Yen, T.C.; Lee, L.Y.; Chang, J.T.; Cheng, A.J. Positive association of glucose-regulated protein 78 during oral cancer progression and the prognostic value in oral precancerous lesions. Head Neck 2010, 32, 1028. [Google Scholar] [CrossRef]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.C.; Lin, C.Y.; Lee, L.Y.; Chen, Y.J.; Kuo, T.F.; Chang, J.T.; Liao, C.T.; Wang, H.M.; Yen, T.C.; Shen, C.R.; et al. Glucose-regulated protein 78 regulates multiple malignant phenotypes in head and neck cancer and may serve as a molecular target of therapeutic intervention. Mol. Cancer Ther. 2008, 7, 2788. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.; Xu, J.C.; Zhang, P.; Zhang, Y.Y.; Zhang, Q.W.; Chao, Z.H.; Wang, F. Glucose-regulated protein 78 and heparanase expression in oral squamous cell carcinoma: Correlations and prognostic significance. World J. Surg. Oncol. 2014, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Crowley-Weber, C.L.; Dvorakova, K.; Crowley, C.; Bernstein, H.; Bernstein, C.; Garewal, H.; Payne, C.M. Nicotine increases oxidative stress, activates NF-kappaB and GRP78, induces apoptosis and sensitizes cells to genotoxic/xenobiotic stresses by a multiple stress inducer, deoxycholate: Relevance to colon carcinogenesis. Chem. Biol. Interact. 2003, 145, 53. [Google Scholar] [CrossRef]
- Repo, J.K.; Pesonen, M.; Mannelli, C.; Vahakangas, K.; Loikkanen, J. Exposure to ethanol and nicotine induces stress responses in human placental BeWo cells. Toxicol. Lett. 2014, 224, 264. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. The Oncogenic Functions of Nicotinic Acetylcholine Receptors. J. Oncol. 2016, 2016, 9650481. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Pillai, S.; Chellappan, S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J. Oncol. 2011, 2011, 456743. [Google Scholar] [CrossRef]
- Wang, S.; Hu, Y. α7 nicotinic acetylcholine receptors in lung cancer. Oncol. Lett. 2018, 16, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhao, Y.H.; Tang, X.F.; Hou, M. Effect of nicotine on the proliferation and cell apoptosis of oral squamous cell carcinoma SCC15 cells. Chin. J. Stomatol. 2012, 47, 233. (In Chinese) [Google Scholar] [CrossRef]
- Wang, C.; Niu, W.; Chen, H.; Shi, N.; He, D.; Zhang, M.; Ge, L.; Tian, Z.; Qi, M.; Chen, T.; et al. Nicotine suppresses apoptosis by regulating α7nAChR/Prx1 axis in oral precancerous lesions. Oncotarget 2017, 8, 75065. [Google Scholar] [CrossRef]
- Ishibashi, T.; Morita, S.; Kishimoto, S.; Uraki, S.; Takeshima, K.; Furukawa, Y.; Inaba, H.; Ariyasu, H.; Iwakura, H.; Furuta, H.; et al. Nicotinic acetylcholine receptor signaling regulates inositol-requiring enzyme 1α activation to protect β-cells against terminal unfolded protein response under irremediable endoplasmic reticulum stress. J. Diabetes Investig. 2020, 11, 801. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.; Rizwani, W.; Banerjee, S.; Kovacs, M.; Haura, E.; Coppola, D.; Chellappan, S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 2009, 4, e7524. [Google Scholar] [CrossRef]
- Underwood, P.W.; Zhang, D.Y.; Cameron, M.E.; Gerber, M.H.; Delitto, D.; Maduka, M.U.; Cooper, K.J.; Han, S.; Hughes, S.J.; Judge, S.M.; et al. Nicotine Induces IL-8 Secretion from Pancreatic Cancer Stroma and Worsens Cancer-Induced Cachexia. Cancers 2020, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.-L.; Chen, K.-Y.; Lee, K.-Y.; Feng, P.-H.; Wu, S.-M. Nicotinic-nAChR signaling mediates drug resistance in lung cancer. J. Cancer 2020, 11, 1125. [Google Scholar] [CrossRef] [Green Version]
- Omori, H.; Nishio, M.; Masuda, M.; Miyachi, Y.; Ueda, F.; Nakano, T.; Sato, K.; Mimori, K.; Taguchi, K.; Hikasa, H.; et al. YAP1 is a potent driver of the onset and progression of oral squamous cell carcinoma. Sci. Adv. 2020, 6, eaay3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhou, W.; Xue, L.; Zhang, W.; Zhan, Q. Nicotine activates YAP1 through nAChRs mediated signaling in esophageal squamous cell cancer (ESCC). PLoS ONE 2014, 9, e90836. [Google Scholar] [CrossRef]
- Lin, K.C.; Park, H.W.; Guan, K.L. Regulation of the Hippo Pathway Transcription Factor TEAD. Trends Biochem. Sci. 2017, 42, 862. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Singh, A.; Chien, C.-Y.; Warnakulasuriya, S. Tobacco related oral cancer. BMJ 2019, 365, l2142. [Google Scholar] [CrossRef]
- Sanner, T.; Grimsrud, T.K. Nicotine: Carcinogenicity and Effects on Response to Cancer Treatment—A Review. Front. Oncol. 2015, 5, 196. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.C.; Girard, L.; Ramirez, R.; Chau, W.S.; Suen, W.S.; Sheridan, S.; Tin, V.P.; Chung, L.P.; Wong, M.P.; Shay, J.W.; et al. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007, 67, 4638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaal, C.; Chellappan, S. Nicotine-Mediated Regulation of Nicotinic Acetylcholine Receptors in Non-Small Cell Lung Adenocarcinoma by E2F1 and STAT1 Transcription Factors. PLoS ONE 2016, 11, e0156451. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; Morini, V.; Coluccia, P.; Proietti, S.; D’Anselmi, F.; Pasqualato, A.; Masiello, M.G.; Palombo, A.; De Toma, G.; Bizzarri, M.; et al. Nicotine increases survival in human colon cancer cells treated with chemotherapeutic drugs. Toxicol. In Vitro 2013, 27, 2256. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, P.; Zhu, L.; Zhao, Q.; Lu, X.; Bo, S. Blockade of alpha7 nicotinic acetylcholine receptors inhibit nicotine-induced tumor growth and vimentin expression in non-small cell lung cancer through MEK/ERK signaling way. Oncol. Rep. 2017, 38, 3309. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Richards, C.I.; Xiao, C.; Rhee, D.; Pantoja, R.; Dougherty, D.A.; Miwa, J.M.; Lester, H.A. Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the endoplasmic reticulum stress response. Mol. Pharmacol. 2012, 81, 759. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. BioEssays News Rev. Mol. Cell. Dev. Biol. 2020, 42, e1900162. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gu, W.; Wang, Q.; Fu, X.; Wang, Y.; Xu, X.; Wen, Y. C-MYC and BCL-2 mediate YAP-regulated tumorigenesis in OSCC. Oncotarget 2017, 9, 668. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, G.; Yang, Y.; Mei, Z.; Liang, Z.; Cui, A.; Wu, T.; Liu, C.Y.; Cui, L. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene 2016, 35, 2789. [Google Scholar] [CrossRef]
- Huang, T.T.; Chen, J.Y.; Tseng, C.E.; Su, Y.C.; Ho, H.C.; Lee, M.S.; Chang, C.T.; Wong, Y.K.; Chen, H.R. Decreased GRP78 protein expression is a potential prognostic marker of oral squamous cell carcinoma in Taiwan. J. Formos. Med. Assoc. 2010, 109, 326. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Rocchi, S.; Benhida, R. Expression level of GRP78/BiP as a predictor of favorable or unfavorable outcomes in cancer patients. Mediastinum 2018, 2, 26. [Google Scholar] [CrossRef]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Vig, S.; Buitinga, M.; Rondas, D.; Crèvecoeur, I.; van Zandvoort, M.; Waelkens, E.; Eizirik, D.L.; Gysemans, C.; Baatsen, P.; Mathieu, C.; et al. Cytokine-induced translocation of GRP78 to the plasma membrane triggers a pro-apoptotic feedback loop in pancreatic beta cells. Cell Death Dis. 2019, 10, 309. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, V.; Gomez, M.; Prasad, V.; Kimlinger, T.; Painuly, U.; Mukhopadhyay, B.; Haug, J.; Bi, L.; Rajkumar, S.V.; Kumar, S. Smac mimetic LCL161 overcomes protective ER stress induced by obatoclax, synergistically causing cell death in multiple myeloma. Oncotarget 2016, 7, 56253. [Google Scholar] [CrossRef] [Green Version]
- Ogata, S.; Kameda, K.; Kono, T.; Ozeki, Y.; Hashimoto, H.; Tominaga, S.; Nakanishi, K. Expressions of ATF6, XBP1, and GRP78 in normal tissue, atypical adenomatous hyperplasia, and adenocarcinoma of the lung. Hum. Pathol. 2019, 83, 22. [Google Scholar] [CrossRef]
- Aksoy, M.O.; Kim, V.; Cornwell, W.D.; Rogers, T.J.; Kosmider, B.; Bahmed, K.; Barrero, C.; Merali, S.; Shetty, N.; Kelsen, S.G. Secretion of the endoplasmic reticulum stress protein, GRP78, into the BALF is increased in cigarette smokers. Respir. Res. 2017, 18, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-J.; Jan, C.-I.; Tsay, Y.-G.; Yu, Y.-H.; Huang, C.-Y.; Lin, S.-C.; Liu, C.-J.; Chen, Y.-S.; Lo, J.-F.; Yu, C.-C. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol. Cancer 2010, 9, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, J.; Untergasser, G.; Zenzmaier, C.; Sarg, B.; Gastl, G.; Gunsilius, E.; Steurer, M. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 2009, 114, 3960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Yang, P.; Wu, H.L.; Li, Z.W.; Li, Z.Y. GRP78 secreted by colon cancer cells facilitates cell proliferation via PI3K/Akt signaling. Asian Pac. J. Cancer Prev. 2014, 15, 7245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, R.; Feith, M.; Siewert, J.R.; Wester, H.J.; Hoefler, H. Expression and clinical significance of glucose regulated proteins GRP78 (BiP) and GRP94 (GP96) in human adenocarcinomas of the esophagus. BMC Cancer 2008, 8, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 5′-3′ | Sequences |
|---|---|---|
| BiP | Forward | TGA CAT TGA AGA CTT CAA AGC T |
| Reverse | CTG CTG TAT CCT CTT CAC CAG T | |
| α7-nAchR | Forward | GCT GGT CAA GAA CTA CAA TCC C |
| Reverse | CTC ATC CAC GTC CAT GAT CTG | |
| YAP | Forward | TGA ACA AAC GTC CAG CAA GAT AC |
| Reverse | CAG CCC CCA AAA TGA ACA GTA G | |
| GAPDH | Forward | CCA CAT CGC TCA GAC ACC AT |
| Reverse | TGA CCA GGC GCC CAA TA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chien, C.-Y.; Chen, Y.-C.; Hsu, C.-C.; Chou, Y.-T.; Shiah, S.-G.; Liu, S.-Y.; Hsieh, A.C.-T.; Yen, C.-Y.; Lee, C.-H.; Shieh, Y.-S. YAP-Dependent BiP Induction Is Involved in Nicotine-Mediated Oral Cancer Malignancy. Cells 2021, 10, 2080. https://doi.org/10.3390/cells10082080
Chien C-Y, Chen Y-C, Hsu C-C, Chou Y-T, Shiah S-G, Liu S-Y, Hsieh AC-T, Yen C-Y, Lee C-H, Shieh Y-S. YAP-Dependent BiP Induction Is Involved in Nicotine-Mediated Oral Cancer Malignancy. Cells. 2021; 10(8):2080. https://doi.org/10.3390/cells10082080
Chicago/Turabian StyleChien, Chu-Yen, Ying-Chen Chen, Chia-Chen Hsu, Yu-Ting Chou, Shine-Gwo Shiah, Shyun-Yeu Liu, Alexander Cheng-Ting Hsieh, Ching-Yu Yen, Chien-Hsing Lee, and Yi-Shing Shieh. 2021. "YAP-Dependent BiP Induction Is Involved in Nicotine-Mediated Oral Cancer Malignancy" Cells 10, no. 8: 2080. https://doi.org/10.3390/cells10082080