
Applying Bioinformatic Platforms, In Vitro, and In Vivo Functional Assays in the Characterization of Genetic Variants in the GH/IGF Pathway Affecting Growth and Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
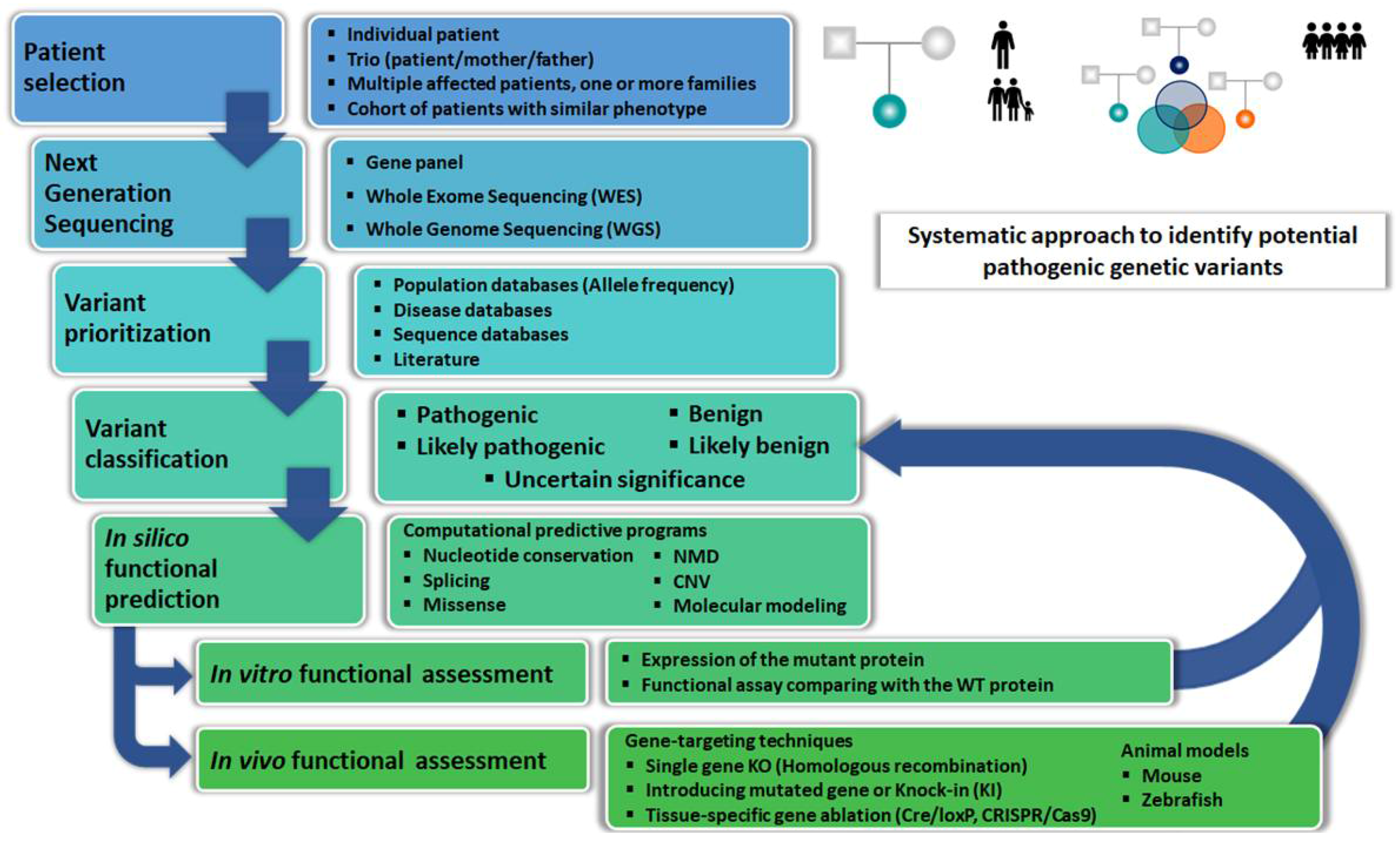
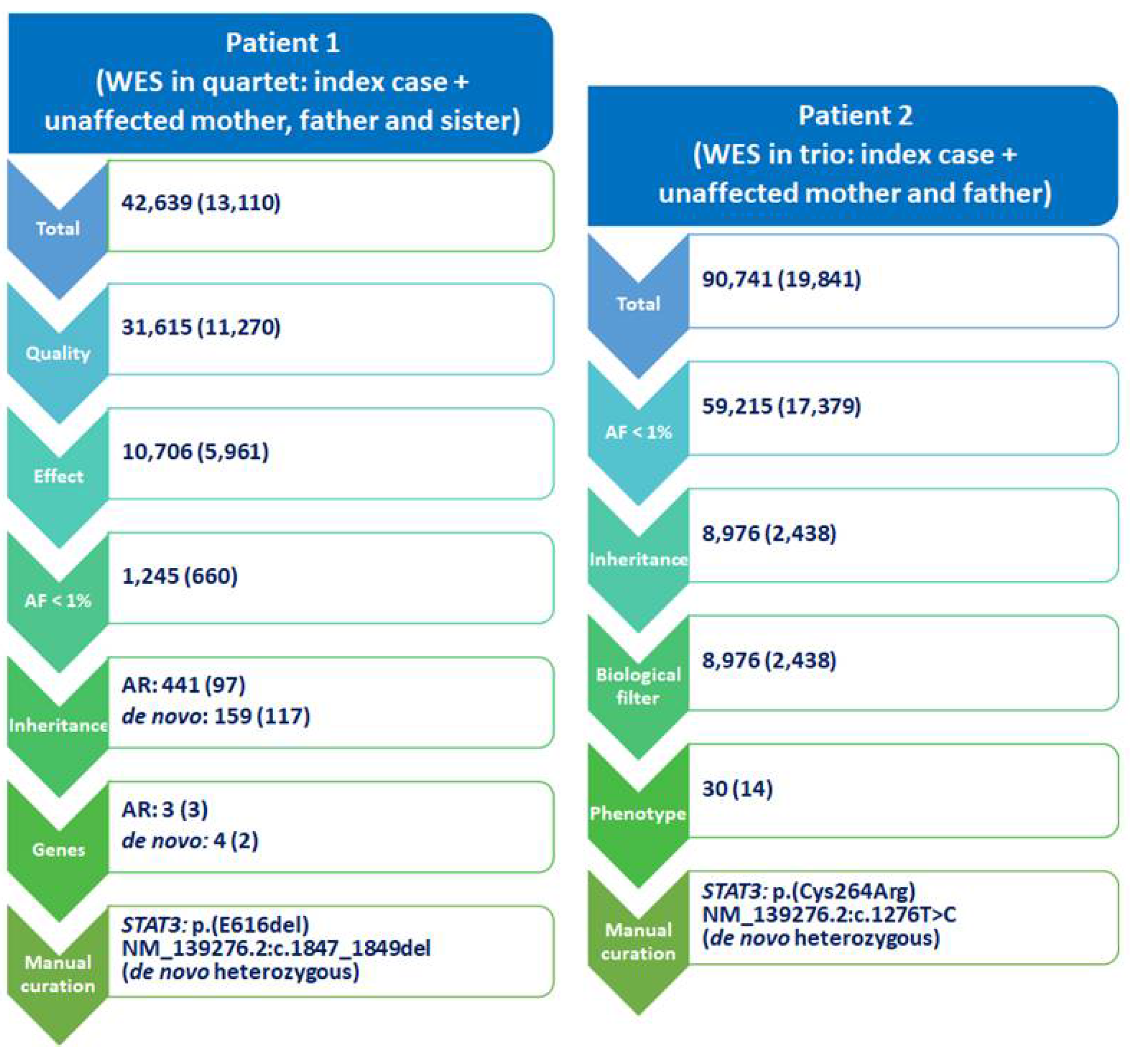
2. Bioinformatic Tools
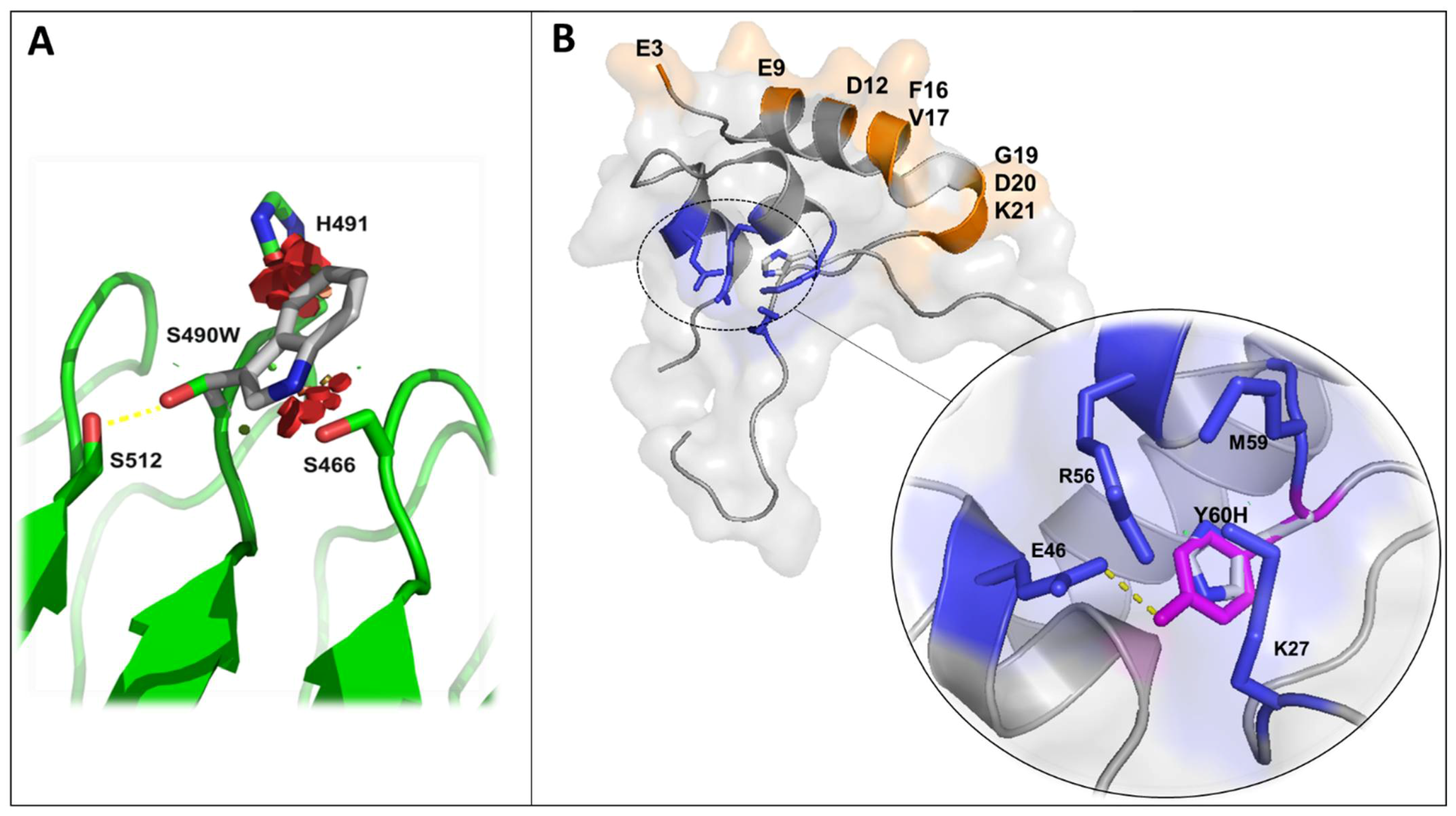
3. In Silico Modeling
4. In Vitro Functional Studies
4.1. Membrane-Bound Receptors
4.2. Intracellular Transcription Factors
4.3. Soluble Secreted Proteins
4.4. Soluble Secreted Enzymes
5. In Vivo Functional Studies
5.1. Membrane-Bound Receptors
5.2. Intracellular Transcription Factors
5.3. Soluble Secreted Proteins
5.4. Soluble Secreted Enzymes
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silventoinen, K.; Sammalisto, S.; Perola, M.; Boomsma, D.I.; Cornes, B.K.; Davis, C.; Dunkel, L.; De Lange, M.; Harris, J.R.; Hjelmborg, J.V.; et al. Heritability of adult body height: A comparative study of twin cohorts in eight countries. J. Twin Res. 2003, 6, 399–408. [Google Scholar] [CrossRef]
- Wood, A.R.; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; Kutalik, Z.; et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.A., III; Hjelle, B.L.; Seeburg, P.H.; Zachman, M. Molecular basis for familial isolated growth hormone deficiency. Proc. Natl. Acad. Sci. USA 1981, 78, 6372–6375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wit, J.M.; Oostdijk, W.; Losekoot, M.; van Duyvenvoorde, H.A.; Ruivenkamp, C.A.; Kant, S.G. Mechanisms in Endocrinology: Novel genetic causes of short stature. Eur. J. Endocrinol. 2016, 174, R145–R173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domené, S.; Domené, H.M. Genetic mutations in the GH/IGF Axis. Pediatr. Endocrinol. Rev. 2018, 16, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Plachy, L.; Strakova, V.; Elblova, L.; Obermannova, B.; Kolouskova, S.; Snajderova, M.; Zemkova, D.; Dusatkova, P.; Sumnik, Z.; Lebl, J.; et al. High prevalence of growth plate gene variants in children with familial short stature treated with GH. J. Clin. Endocrinol. Metab. 2019, 104, 4273–4281. [Google Scholar] [CrossRef]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Coban-Akdemir, Z.; Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Muller, H.J. Further studies on the nature and causes of gene mutations. Proc. Sixth Int. Cong. Genet. 1932, 1, 213–255. [Google Scholar]
- Argente, J. Challenge in the management of short stature. Horm. Res. Pediatr. 2016, 85, 2–10. [Google Scholar] [CrossRef]
- Dauber, A.; Rosenfeld, R.G.; Hirschhorn, J.N. Genetic evaluation of short stature. J. Clin. Endocrinol. Metab. 2014, 99, 3080–3092. [Google Scholar] [CrossRef]
- Sun, Y.; Ruivenkamp, C.A.L.; Hoffer, M.J.V.; Vrijenhoek, T.; Kriek, M.; van Asperen, C.J.; den Dunnen, J.T.; Santen, G.W.E. Next-generation diagnostics: Gene panel, exome, or whole genome? Hum. Mutat. 2015, 36, 648–655. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [Green Version]
- Dauber, A.; Yu, Y.; Turchin, M.C.; Chiang, C.W.; Meng, Y.A.; Demerath, E.W.; Patel, S.R.; Rich, S.S.; Rotter, J.I.; Schreiner, P.J.; et al. Genome-wide association of copy-number variation reveals an association between short stature and the presence of low-frequency genomic deletions. Am. J. Hum. Genet. 2011, 89, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Canton, A.P.; Costa, S.S.; Rodrigues, T.C.; Bertola, D.R.; Malaquias, A.C.; Correa, F.A.; Arnhold, I.J.; Rosenberg, C.; Jorge, A.A. Genome-wide screening of copy number variants in children born small for gestational age reveals several candidate genes involved in growth pathways. Eur. J. Endocrinol. 2014, 171, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Wit, J.M.; van Duyvenvoorde, H.A.; van Klinken, J.B.; Caliebe, J.; Bosch, C.A.; Lui, J.C.; Gijsbers, A.C.; Bakker, E.; Breuning, M.H.; Oostdijk, W.; et al. Copy number variants in short children born small for gestational age. Horm. Res. Paediatr. 2014, 82, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Zahnleiter, D.; Uebe, S.; Ekici, A.B.; Hoyer, J.; Wiesener, A.; Wieczorek, D.; Kunstmann, E.; Reis, A.; Doerr, H.G.; Rauch, A.; et al. Rare copy number variants are a common cause of short stature. PLoS Genet. 2013, 9, e1003365. [Google Scholar] [CrossRef] [Green Version]
- Van Duyvenvoorde, H.A.; Lui, J.C.; Kant, S.G.; Oostdijk, W.; Gijsbers, A.C.; Hoffer, M.J.; Karperien, M.; Walenkamp, M.J.; Noordam, C.; Voorhoeve, P.G.; et al. Copy number variants in patients with short stature. Eur. J. Hum. Genet. 2014, 22, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, T.K.; Krepischi, A.; Furuya, T.K.; Honjo, R.S.; Malaquias, A.C.; Bertola, D.R.; Costa, S.S.; Canton, A.P.; Roela, R.A.; Freire, B.L.; et al. Recurrent Copy Number Variants Associated with Syndromic Short Stature of Unknown Cause. Horm. Res. Paediatr. 2018, 89, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Cabrera, J.M.; Del Valle, J.; Castellanos, E.; Feliubadaló, L.; Pineda, M.; Brunet, J.; Serra, E.; Capellà, G.; Lázaro, C.; Gel, B. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur. J. Hum. Genet. 2020, 28, 1645–1655. [Google Scholar] [CrossRef]
- Guo, M.H.; Shen, Y.; Walvoord, E.C.; Miller, T.C.; Moon, J.E.; Hirschhorn, J.N.; Dauber, A. Whole exome sequencing to identify genetic causes of short stature. Horm. Res. Paediatr. 2014, 82, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.C.; Jee, Y.H.; Nilsson, O. New genetic diagnoses of short stature provide insights into local regulation of childhood growth. Horm. Res. Paediatr. 2017, 88, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Wit, J.M.; Kamp, G.A.; Oostdijk, W.; On Behalf of the Dutch Working Group on Triage and Diagnosis of Growth Disorders in Children. Towards a Rational and Efficient Diagnostic Approach in Children Referred for Growth Failure to the General Paediatrician. Horm. Res. Paediatr. 2019, 91, 223–240. [Google Scholar] [CrossRef]
- Argente, J.; Tatton-Brown, K.; Lehwalder, D.; Pfäffle, R. Genetics of growth disorders-which patients require genetic testing? Front. Endocrinol. 2019, 10, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.; Holmen, O.L.; Dauber, A.; Vatten, L.; Havulinna, A.S.; Kvaløy, K.; Silander, K.; Nguyen, T.T.; Wiler, C.; Boehnke, M.; et al. Common Variants Show Predicted Polygenic Effects on Height in the Tails of the Distribution, Except in Extremely Short Individuals. PLoS Genet. 2011, 7, e1002439. [Google Scholar] [CrossRef] [Green Version]
- Prokop, J.W.; May, T.; Strong, K.; Bilinovich, S.M.; Bupp, C.; Rajasekaran, S.; Worthey, E.A.; Lazar, J. Genome sequencing in the clinic: The past, present, and future of genomic medicine. Physiol. Genom. 2018, 50, 563–579. [Google Scholar] [CrossRef]
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Brunner, H.G.; Buske, O.J.; Carey, K.; Doll, C.; et al. The Matchmaker Exchange: A platform for rare disease gene discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Bromberg, Y. Predicting functional effects of synonymous variants: A systematic review and perspectives. Front. Genet. 2019, 10, 914. [Google Scholar] [CrossRef]
- Vihinen, M. Problems in variation interpretation guidelines and in their implementation in computational tools. Mol. Genet. Genom. Med. 2020, 8, e1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef]
- Abel, H.J.; Duncavage, E.J. Detection of structural DNA variation from next generation sequencing data: A review of informatic approaches. Cancer Genet. 2013, 206, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Mason-Suares, H.; Landry, L.S.; Lebo, M. Detecting copy number variation via next generation technology. Curr. Genet. Med. Rep. 2016, 4, 74–85. [Google Scholar] [CrossRef]
- Teo, S.M.; Pawitan, Y.; Ku, C.S.; Chia, K.S.; Salim, A. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics 2012, 28, 2711–2718. [Google Scholar] [CrossRef] [Green Version]
- Hoskinson, D.C.; Dubuc, A.M.; Mason-Suares, H. The current state of clinical interpretation of sequence variants. Curr. Opin. Genet. Dev. 2017, 42, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazazian, J.; Boehm, C.D.; Seltzer, W.K. ACMG recommendations for standards for interpretation of sequence variations. Genet. Med. 2000, 2, 302–303. [Google Scholar]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E.; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 2, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 7, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, M.; Scaglia, P.; Keselman, A.; Martucci, L.; Karabatas, L.; Domené, S.; Martin, A.; Pennisi, P.; Blanco, M.; Sanguineti, N.; et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol. Cell. Endocrinol. 2018, 473, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, P.; Li, Z.; Moult, J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Saunders, A.J. An emerging role for Ubiquilin 1 in regulating protein quality control system and in disease pathogenesis. Discov. Med. 2009, 8, 18–22. [Google Scholar]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.R.; Parker, A.L.; Kalkman, E.R.; White, K.; Kovalskyy, D.; Kelly, S.M.; Baker, A.H. Identification of novel small molecule inhibitors of adenovirus gene transfer using a high throughput screening approach. J. Control. Release 2013, 170, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Rogozin, I.B.; Panchenko, A.R. Oncogenic potential is related to activating effect of cancer single and double somatic mutations in receptor tyrosine kinases. Hum. Mutat. 2012, 33, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Schlebach, J.P.; Narayan, M.; Alford, C.; Mittendorf, K.F.; Carter, B.D.; Li, J.; Sanders, C.R. Conformational stability and pathogenic mis-folding of the integral membrane protein PMP22. J. Am. Chem. Soc. 2015, 137, 8758–8768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein-protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar] [CrossRef] [Green Version]
- Amberg, A. In silico methods. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays; Vogel, H.G., Maas, J., Hock, F.J., Mayer, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1273–1296. [Google Scholar]
- Oliver, G.R.; Zimmermann, M.T.; Klee, E.W.; Urrutia, R.A. “The molecule’s the thing:” the promise of molecular modeling and dynamic simulations in aiding the prioritization and interpretation of genomic testing results. F1000Research 2016, 5, 766. [Google Scholar] [CrossRef]
- Bonneau, R.; Baker, D. Ab initio protein structure prediction: Progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.; Ngan, S.; Samudrala, R. De novo protein structure prediction. In Computational Methods for Protein Structure Prediction and Modeling; Xu, Y., Xu, D., Liang, J., Eds.; Springer: New York, NY, USA, 2007; pp. 43–64. [Google Scholar]
- Lee, J.; Freddolino, P.L.; Zhang, Y. Ab initio protein structure prediction. In From Protein Structure to Function with Bioinformatics; Rigden, D.J., Ed.; Springer: Dordrecht, The Netherlands, 2017; pp. 3–35. [Google Scholar]
- Dunbrack, R.L., Jr.; Karplus, M. Conformational analysis of the backbone-dependent rotamer preferences of protein sidechains. Nat. Struct. Biol. 1994, 1, 334–340. [Google Scholar] [CrossRef]
- Vasquez, M. Modeling side-chain conformation. Curr. Opin. Struct. Biol. 1996, 6, 217–221. [Google Scholar] [CrossRef]
- Levitt, M.; Gerstein, M.; Huang, E.; Subbiah, S.; Tsai, J. Protein folding: The endgame. Annu. Rev. Biochem. 1997, 66, 549–579. [Google Scholar] [CrossRef]
- Xiang, Z.; Honig, B. Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 2001, 311, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fiser, A.; ter Kuile, B.; Sali, A.; Muller, M. Convergent evolution of Trichomonas vaginalis lactate dehydrogenase from malate dehydrogenase. Proc. Natl. Acad. Sci. USA 1999, 96, 6285–6290. [Google Scholar] [CrossRef] [Green Version]
- Martucci, L.C.; Gutiérrez, M.L.; Karabatas, L.M.; Scaglia, P.A.; Rey, R.A.; Domené, H.M.; Jasper, H.G.; Domené, S. Assessment of pathogenicity of natural IGFALS gene variants by in silico bioinformatics tools and in vitro functional studies. Mol. Cell. Endocrinol. 2016, 429, 19–28. [Google Scholar] [CrossRef]
- Keselman, A.C.; Martin, A.; Scaglia, P.A.; Sanguineti, N.M.; Armando, R.; Gutiérrez, M.; Braslavsky, D.; Ballerini, M.G.; Ropelato, M.G.; Ramirez, L.; et al. A homozygous mutation in the highly conserved Tyr60 of the mature IGF1 peptide broadens the spectrum of IGF1 deficiency. Eur. J. Endocrinol. 2019, 181, K43–K53. [Google Scholar] [CrossRef]
- Ramírez, L.; Sanguineti, N.; Scaglia, P.; Keselman, A.; Ballerini, M.G.; Karabatas, L.; Landi, E.; Castro, J.; Domené, S.; Pennisi, P.; et al. A novel heterozygous STAT5B variant in a patient with short stature and partial growth hormone insensitivity (GHI). Growth Horm. IGF Res. 2020, 50, 61–70. [Google Scholar] [CrossRef]
- Laron, Z.; Pertzelan, A.; Mannheimer, S. Genetic pituitary dwarfism with high serum concentration of growth hormone—A new inborn error of metabolism? Israel J. Med. Sci. 1966, 2, 152–155. [Google Scholar] [PubMed]
- Godowski, P.J.; Leung, D.W.; Meacham, L.R.; Galgani, J.P.; Hellmiss, R.; Keret, R.; Rotwein, P.S.; Parks, J.S.; Laron, Z.; Wood, W.I. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc. Nat. Acad. Sci. USA 1989, 86, 8083–8087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbloom, A.L.; Aguirre, J.G.; Rosenfeld, R.G.; Fielder, P.J. The little women of Loja—Growth hormone-receptor deficiency in an inbred population of southern Ecuador. N. Engl. J. Med. 1990, 323, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.A.; Guevara-Aguirre, J.; Rosenbloom, A.L.; Rosenfeld, R.G.; Francke, U. Mutation creating a new splice site in the growth hormone receptor genes of 37 Ecuadorean patients with Laron syndrome. Hum. Mutat. 1992, 1, 24–32. [Google Scholar] [CrossRef]
- Gonçalves, F.T.; Fridman, C.; Pinto, E.M.; Guevara-Aguirre, J.; Shevah, O.; Rosembloom, A.L.; Hwa, V.; Cassorla, F.; Rosenfeld, R.G.; Lins, T.S.; et al. The E180splice mutation in the GHR gene causing Laron syndrome: Witness of a Sephardic Jewish exodus from the Iberian Peninsula to the New World? Am. J. Med. Genet. A 2014, 164, 1204–1208. [Google Scholar] [CrossRef]
- Ayling, R.M.; Ross, R.; Towner, P.; Von Laue, S.; Finidori, J.; Moutoussamy, S.; Buchanan, C.R.; Clayton, P.E.; Norman, M.R. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nat. Genet. 1997, 16, 13–14. [Google Scholar] [CrossRef]
- Iida, K.; Takahashi, Y.; Kaji, H.; Nose, O.; Okimura, Y.; Abe, H.; Chihara, K. Growth hormone (GH) insensitivity syndrome with high serum GH-binding protein levels caused by a heterozygous splice site mutation of the GH receptor gene producing a lack of intracellular domain. J. Clin. Endocrinol. Metab. 1998, 83, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vairamani, K.; Merjaneh, L.; Casano-Sancho, P.; Sanli, M.E.; David, A.; Metherell, L.A.; Savage, M.O.; Del Pozo, J.S.; Backeljauw, P.F.; Rosenfeld, R.G.; et al. Novel dominant-negative GH receptor mutations expands the spectrum of GHI and IGF-I deficiency. J. Endocr. Soc. 2017, 1, 345–358. [Google Scholar] [CrossRef]
- Wojcik, J.; Berg, M.A.; Esposito, N.; Geffner, M.E.; Sakati, N.; Reiter, E.O.; Dower, S.; Francke, U.; Postel-Vinay, M.C.; Finidori, J. Four contiguous amino acid substitutions, identified in patients with Laron syndrome, differently affect the binding affinity and intracellular trafficking of the growth hormone receptor. J. Clin. Endocrinol. Metab. 1998, 83, 4481–4489. [Google Scholar] [CrossRef] [Green Version]
- Fang, P.; Riedl, S.; Amselem, S.; Pratt, K.L.; Little, B.M.; Haeusler, G.; Hwa, V.; Frisch, H.; Rosenfeld, R.G. Primary growth hormone (GH) insensitivity and insulin-like growth factor deficiency caused by novel compound heterozygous mutations of the GH receptor gene: Genetic and functional studies of simple and compound heterozygous states. J. Clin. Endocrinol. Metab. 2007, 92, 2223–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derr, M.A.; Aisenberg, J.; Fang, P.; Tenenbaum-Rakover, Y.; Rosenfeld, R.G.; Hwa, V. The growth hormone receptor (GHR) c.899dupC mutation functions as a dominant negative: Insights into the pathophysiology of intracellular GHR defects. J. Clin. Endocrinol. Metab. 2011, 96, E1896–E1904. [Google Scholar] [CrossRef] [Green Version]
- Aisenberg, J.; Auyeung, V.; Pedro, H.F.; Sugalski, R.; Chartoff, A.; Rothenberg, R.; Derr, M.A.; Hwa, V.; Rosenfeld, R.G. Atypical GH insensitivity syndrome and severe insulin-like growth factor-I deficiency resulting from compound heterozygous mutations of the GH receptor, including a novel frameshift mutation affecting the intracellular domain. Horm. Res. Paediatr. 2010, 74, 406–411. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth hormone insensitivity associated with a STAT5b mutation. N. Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, P.A.; Martínez, A.S.; Feigerlová, E.; Bezrodnik, L.; Gaillard, M.; Di Giovanni, D.; Ballerini, M.G.; Jasper, H.G.; Heinrich, J.J.; Fang, P.; et al. A novel missense mutation in the SH2 domain of the STAT5B gene results in a transcriptionally inactive STAT5b associated with severe IGF-I deficiency, immune dysfunction, and lack of pulmonary disease. J. Clin. Endocrinol. Metab. 2012, 97, E830–E839. [Google Scholar] [CrossRef] [PubMed]
- Scalco, R.C.; Hwa, V.; Domené, H.M.; Jasper, H.G.; Belgorosky, A.; Marino, R.; Pereira, A.M.; Tonelli, C.A.; Wit, J.M.; Rosenfeld, R.G.; et al. STAT5B mutations in heterozygous state have negative impact on height: Another clue in human stature heritability. Eur. J. Endocrinol. 2015, 173, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Klammt, J.; Neumann, D.; Gevers, E.F.; Andrew, S.F.; Schwartz, I.D.; Rockstroh, D.; Colombo, R.; Sanchez, M.A.; Vokurkova, D.; Kowalczyk, J.; et al. Dominant-negative STAT5B mutations cause growth hormone insensitivity with short stature and mild immune dysregulation. Nat. Commun. 2018, 9, 2105. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Ramelius, A.; Barton, J.; et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef]
- Haapaniemi, E.M.; Kaustio, M.; Rajala, H.L.; van Adrichem, A.J.; Kainulainen, L.; Glumoff, V.; Doffinger, R.; Kuusanmäki, H.; Heiskanen-Kosma, T.; Trotta, L.; et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 2015, 125, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Milner, J.D.; Vogel, T.P.; Forbes, L.; Ma, C.A.; Stray-Pedersen, A.; Niemela, J.E.; Lyons, J.J.; Engelhardt, K.R.; Zhang, Y.; Topcagic, N.; et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 2015, 125, 591–599. [Google Scholar] [CrossRef]
- Boisclair, Y.R.; Rhoads, R.P.; Ueki, I.; Wang, J.; Ooi, G.T. The acid-labile subunit (ALS) of the 150 kDa IGF-binding protein complex: An important but forgotten component of the circulating IGF system. J. Endocrinol. 2001, 170, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Domené, H.M.; Bengolea, S.V.; Martinez, A.S.; Ropelato, M.G.; Pennisi, P.; Scaglia, P.; Heinrich, J.J.; Jasper, H.G. Deficiency of the circulating IGF system associated with inactivation of the acid-labile subunit gene. N. Engl. J. Med. 2004, 350, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Heath, K.E.; Argente, J.; Barrios, V.; Pozo, J.; Díaz-González, F.; Martos-Moreno, G.A.; Caimari, M.; Gracia, R.; Campos-Barros, A. Primary acid-labile subunit deficiency due to recessive IGFALS mutations results in postnatal growth deficit associated with low circulating insulin growth factor (IGF)-I, IGF binding protein-3 levels, and hyperinsulinemia. J. Clin. Endocrinol. Metab. 2008, 93, 1616–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domené, S.; Domené, H.M. The role of acid-labile subunit (ALS) in the modulation of GH-IGF-I action. Mol. Cell. Endocrinol. 2020, 518, 111006. [Google Scholar] [CrossRef] [PubMed]
- Domené, H.M.; Scaglia, P.A.; Martínez, A.S.; Keselman, A.C.; Karabatas, L.M.; Pipman, V.R.; Bengolea, S.V.; Guida, M.C.; Ropelato, M.G.; Ballerini, M.G.; et al. Heterozygous IGFALS gene variants in idiopathic short stature and normal children: Impact on height and the IGF system. Horm. Res. Paediatr. 2013, 80, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Firth, S.M.; Yan, X.; Baxter, R.C. D440N mutation in the acid-labile subunit of insulin-like growth factor complexes inhibits secretion and complex formation. Mol. Endocrinol. 2011, 25, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Woods, K.A.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J.L. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef]
- Walenkamp, M.J.; Karperien, M.; Pereira, A.M.; Hilhorst-Hofstee, Y.; van Doorn, J.; Chen, J.W.; Mohan, S.; Denley, A.; Forbes, B.; van Duyvenvoorde, H.A.; et al. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J. Clin. Endocrinol. Metab. 2005, 90, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Netchine, I.; Azzi, S.; Houang, M.; Seurin, D.; Perin, L.; Ricort, J.M.; Daubas, C.; Legay, C.; Mester, J.; Herich, R.; et al. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J. Clin. Endocrinol. Metab. 2009, 94, 3913–3921. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, M.T.; Boldt, H.B.; Laursen, L.S.; Sottrup-Jensen, L.; Conover, C.A.; Oxvig, C. Pregnancy-associated plasma protein-A2 (PAPP-A2), a novel insulin-like growth factor-binding protein-5 proteinase. J. Biol. Chem. 2001, 276, 21849–21853. [Google Scholar] [CrossRef] [Green Version]
- Dauber, A.; Muñoz-Calvo, M.T.; Barrios, V.; Domené, H.M.; Kloverpris, S.; Serra-Juhé, C.; Desikan, V.; Pozo, J.; Muzumdar, R.; Martos-Moreno, G.Á.; et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol. Med. 2016, 8, 363–374. [Google Scholar] [CrossRef]
- Smithies, O.; Gregg, R.G.; Boggs, S.S.; Koralewski, M.A.; Kucherlapati, R.S. Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 1985, 317, 230–234. [Google Scholar] [CrossRef]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Muller, U. Ten years of gene targeting: Targeted mouse mutants, from vector design to phenotype analysis. Mech. Deve. 1999, 82, 3–21. [Google Scholar] [CrossRef]
- Koller, B.H.; Smithies, O. Inactivating the beta 2-microglobulin locus in mouse embryonic stem cells by homologous recombination. Proc. Natl. Acad. Sci. USA 1989, 86, 8932–8935. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, K.L.; Harvey, S. Growth hormone resistance: Clinical states and animal models. J. Endocrinol. 1999, 163, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef] [Green Version]
- Yakar, S.; Liu, J.L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; LeRoith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329. [Google Scholar] [CrossRef] [Green Version]
- Niederriter, A.R.; Davis, E.E.; Golzio, C.; Oh, E.C.; Tsai, I.C.; Katsanis, N. In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp. 2013, 24, e50338. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Hu, P.; Marino, J.; Hufnagel, S.B.; Hopkin, R.J.; Toromanović, A.; Richieri-Costa, A.; Ribeiro-Bicudo, L.A.; Kruszka, P.; Roessler, E.; et al. Dominant-negative kinase domain mutations in FGFR1 can explain the clinical severity of Hartsfield syndrome. Hum. Mol. Genet. 2016, 25, 1912–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, E.O.; Berryman, D.E.; Jensen, E.A.; Kulkarni, P.; McKenna, S.; Kopchick, J.J. New insights of growth hormone (GH) actions from tissue-specific GH receptor knockouts in mice. Arch. Endocrinol. Metab. 2019, 63, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, E.O.; Duran-Ortiz, S.; Kopchick, J.J. Effects of tissue-specific GH receptor knockouts in mice. Mol. Cell. Endocrinol. 2020, 15, 515:110919. [Google Scholar] [CrossRef]
- Ahmed, A.S.I.; Xiong, F.; Pang, S.-C.; He, M.-D.; Waters, M.J.; Zhu, Z.-Y.; Sun, Y.-H. Activation of GH signaling and GH-independent stimulation of growth in zebrafish by introduction of a constitutively activated GHR construct. Transgenic Res. 2011, 20, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.A.; Mareco, E.A.; Silva, M.D.; Marins, L.F. Muscle-specific growth hormone receptor (GHR) overexpression induces hyperplasia but not hypertrophy in transgenic zebrafish. Transgenic Res. 2012, 21, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.G.; Almeida, D.V.; Nornberg, B.F.; Figueiredo, M.A.; Romano, L.A.; Marins, L.F. Effects of Double Transgenesis of Somatotrophic Axis (GH/GHR) on Skeletal Muscle Growth of Zebrafish (Danio rerio). Zebrafish 2015, 12, 408–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMenamin, S.K.; Minchin, J.E.; Gordon, T.N.; Rawls, J.F.; Parichy, D.M. Dwarfism and increased adiposity in the gh1 mutant zebrafish vizzini. Endocrinology 2013, 154, 1476–1487. [Google Scholar] [CrossRef] [Green Version]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [Green Version]
- Xiong, S.; Mei, J.; Huang, P.; Jing, J.; Li, Z.; Kang, J.; Gui, J.F. Essential roles of stat5.1/stat5b in controlling fish somatic growth. J. Genet. Genom. 2017, 44, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, T.; Zhang, S.S.; Wang, T.; Zhang, Z.; Hesslein, D.G.T.; Yin, Z.; Kano, A.; Iwamoto, Y.; Li, E.; Craft, J.E.; et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: A critical role of STAT3 in innate immunity. Proc. Natl. Acad. Sci. USA 2003, 100, 1879–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Schlessinger, K.; Zhu, X.; Meffre, E.; Quimby, F.; Levy, D.E.; Darnell, J.E., Jr. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell. Biol. 2004, 24, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Miyagi, C.; Carmany-Rampey, A.; Shimizu, T.; Fujii, R.; Schier, A.F.; Hirano, T. Stat3 controls cell movements during zebrafish gastrulation. Dev. Cell. 2002, 2, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, C.; Yamashita, S.; Ohba, Y.; Yoshizaki, H.; Matsuda, M.; Hirano, T. STAT3 noncell-autonomously controls planar cell polarity during zebrafish convergence and extension. J. Cell Biol. 2004, 166, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sepich, D.S.; Solnica-Krezel, L. Stat3/Cdc25a-dependent cell proliferation promotes embryonic axis extension during zebrafish gastrulation. PLoS Genet. 2017, 13, e1006564. [Google Scholar] [CrossRef]
- Ueki, I.; Ooi, G.T.; Tremblay, M.L.; Hurst, K.R.; Bach, L.A.; Boisclair, Y.R. Inactivation of the acid labile subunit gene in mice results in mild retardation of postnatal growth despite profound disruptions in the circulating insulin-like growth factor system. Proc. Natl. Acad. Sci. USA 2000, 97, 6868–6873. [Google Scholar] [CrossRef] [Green Version]
- Silha, J.V.; Gui, Y.; Modric, T.; Suwanichkul, A.; Durham, S.K.; Powell, D.R.; Murphy, L.J. Overexpression of the acid-labile subunit of the IGF ternary complex in transgenic mice. Endocrinology 2001, 142, 4305–4313. [Google Scholar] [CrossRef]
- Landi, E.; Karabatas, L.; Scaglia, P.; Pisciottano, F.; Gutiérrez, M.; Ramírez, L.; Bergadá, I.; Rey, R.A.; Jasper, H.G.; Domené, H.M.; et al. Expression of acid-labile subunit (ALS) in developing and adult zebrafish and its role in dorso-ventral patterning during development. Gen. Comp. Endocrinol. 2020, 299, 113591. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Powell-Braxton, L.; Hollingshead, P.; Warburton, C.; Dowd, M.; Pitts-Meek, S.; Dalton, D.; Gillett, N.; Stewart, T.A. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993, 7, 2609–2617. [Google Scholar] [CrossRef]
- Yakar, S.; Rosen, C.J.; Beamer, W.G.; Ackert-Bicknell, C.L.; Wu, Y.; Liu, J.-L.; Ooi, G.T.; Setser, J.; Frystyk, J.; Boisclair, Y.R.; et al. Circulating levels of IGF-1 directly regulate bone growth and density. J. Clin. Investig. 2002, 110, 771–781. [Google Scholar] [CrossRef]
- Elis, S.; Wu, Y.; Courtland, H.W.; Cannata, D.; Sun, H.; Beth-On, M.; Liu, C.; Jasper, H.; Domené, H.; Karabatas, L.; et al. Unbound (bioavailable) IGF1 enhances somatic growth. Dis. Model. Mech. 2011, 4, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serbanovic-Canic, J.; de Luca, A.; Warboys, C.; Ferreira, P.F.; Luong, L.A.; Hsiao, S.; Gauci, I.; Mahmoud, M.; Feng, S.; Souilhol, C.; et al. Zebrafish model for functional screening of flow-responsive genes. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 130–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conover, C.A.; Boldt, H.B.; Bale, L.K.; Clifton, K.B.; Grell, J.A.; Mader, J.R.; Mason, E.J.; David R Powell, D.R. Pregnancy associated plasma protein-A2 (PAPP-A2): Tissue expression and biological consequences of gene knockout in mice. Endocrinology 2011, 152, 2837–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Andrew, M.; Liao, L.; Zhang, D.; Yildirim, G.; Sluss, P.; Kalra, B.; Kumar, A.; Yakar, S.; Hwa, V.; et al. Low IGF-I bioavailability impairs growth and glucose metabolism in a mouse model of human PAPPA2 p.Ala1033Val mutation. Endocrinology 2019, 160, 1363–1376. [Google Scholar] [CrossRef]
- Kjaer-Sorensen, K.; Engholm, D.H.; Jepsen, M.R.; Morch, M.G.; Weyer, K.; Hefting, L.L.; Skov, L.L.; Laursen, L.S.; Oxvig, C. Papp-a2 modulates development of cranial cartilage and angiogenesis in zebrafish embryos. J. Cell Sci. 2014, 127, 5027–5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domené, S.; Scaglia, P.A.; Gutiérrez, M.L.; Domené, H.M. Applying Bioinformatic Platforms, In Vitro, and In Vivo Functional Assays in the Characterization of Genetic Variants in the GH/IGF Pathway Affecting Growth and Development. Cells 2021, 10, 2063. https://doi.org/10.3390/cells10082063
Domené S, Scaglia PA, Gutiérrez ML, Domené HM. Applying Bioinformatic Platforms, In Vitro, and In Vivo Functional Assays in the Characterization of Genetic Variants in the GH/IGF Pathway Affecting Growth and Development. Cells. 2021; 10(8):2063. https://doi.org/10.3390/cells10082063
Chicago/Turabian StyleDomené, Sabina, Paula A. Scaglia, Mariana L. Gutiérrez, and Horacio M. Domené. 2021. "Applying Bioinformatic Platforms, In Vitro, and In Vivo Functional Assays in the Characterization of Genetic Variants in the GH/IGF Pathway Affecting Growth and Development" Cells 10, no. 8: 2063. https://doi.org/10.3390/cells10082063