
A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression?

Department of Anesthesiology, Intensive Care and Pain Medicine, University Hospital Muenster, 48149 Muester, Germany
*
Author to whom correspondence should be addressed.
Cells 2021, 10(8), 1932; https://doi.org/10.3390/cells10081932
Submission received: 30 June 2021
/
Revised: 22 July 2021
/
Accepted: 28 July 2021
/
Published: 29 July 2021
(This article belongs to the Special Issue Neutrophil Extracellular Traps: From Host Defence to the Pathophysiology of Disease)
Abstract
:Neutrophils act as the first line of defense during infection and inflammation. Once activated, they are able to fulfil numerous tasks to fight inflammatory insults while keeping a balanced immune response. Besides well-known functions, such as phagocytosis and degranulation, neutrophils are also able to release “neutrophil extracellular traps” (NETs). In response to most stimuli, the neutrophils release decondensed chromatin in a NADPH oxidase-dependent manner decorated with histones and granule proteins, such as neutrophil elastase, myeloperoxidase, and cathelicidins. Although primarily supposed to prevent microbial dissemination and fight infections, there is increasing evidence that an overwhelming NET response correlates with poor outcome in many diseases. Lung-related diseases especially, such as bacterial pneumonia, cystic fibrosis, chronic obstructive pulmonary disease, aspergillosis, influenza, and COVID-19, are often affected by massive NET formation. Highly vascularized areas as in the lung are susceptible to immunothrombotic events promoted by chromatin fibers. Keeping this fragile equilibrium seems to be the key for an appropriate immune response. Therapies targeting dysregulated NET formation might positively influence many disease progressions. This review highlights recent findings on the pathophysiological influence of NET formation in different bacterial, viral, and non-infectious lung diseases and summarizes medical treatment strategies.

1. Introduction
The lung comprises different mucosal and alveolar compartments harboring resident immune cells maintaining a well-balanced milieu of protection versus potentially infectious inhaled pathogens. Lung infections may aggravate and turn into life-threatening diseases. Excessive neutrophil recruitment is a major risk factor, and the well-balanced activation of neutrophils is a prerequisite for an adequate immune response. Once pathogens have infiltrated the lung, epithelial cells, lung resident macrophages and dendritic cells (DCs) produce inflammatory mediators leading to immune cell recruitment, which occurs in a tightly regulated cascade. Activated neutrophils can fulfil numerous tasks to fight infection, such as degranulation, the release of reactive oxygen species (ROS), phagocytosis and the release of neutrophil extracellular traps (NETs). NETs are composed of extracellular decondensed chromatin in the majority of the nuclear but also with mitochondrial origin. The chromatin fibers are decorated with a variety of proteins, e.g., neutrophil elastase (NE), myeloperoxidase (MPO), histones, calprotectin, α-defensins, cathelicidins, and cytoskeletal proteins [1,2]. Initially, 24 proteins were identified following a PMA stimulation of neutrophils, but some studies extended this list to up to 50 different proteins and suggest a stimulus-dependent protein composition [3,4].
The underlying mechanisms leading to NET formation show variable characteristics, and studies demonstrated that the signaling pathways vary depending on the respective stimulus. NET formation can be stimulated via G protein-coupled receptors (GPCRs), chemokine and cytokine receptors, Toll-like receptors (TLR), and Fc receptors (FcR). The subsequent downstream signaling comprises mostly the activation of the NADPH oxidase (NOX) complex, but exceptions were also described [5]. Upstream of oxidant production, the molecules Raf-MEK-ERK have been shown to be involved [6]. Cytoskeletal rearrangement [7] and glycolytic ATP production [8] are both required for NET formation and are dependent on ROS produced in the context of mitochondrial dysfunction and NOX activation [9]. ROS initiates the dissociation of NE from a membrane-associated complex into the cytosol and activates its proteolytic activity in an MPO-dependent manner. Subsequently, NE degrades F-actin to arrest actin dynamics followed by translocation into the nucleus, where NE and MPO drive chromatin decondensation and histone cleavage [10,11], which can be supported by PAD4-dependent histone citrullination [12] (Figure 1). Nevertheless, NE- and PAD4-independent pathways have been described, too [13,14]. Van Avondt and colleagues demonstrated that the inhibition of the signal inhibitory receptor on leukocytes-1 (SIRL-1) could prevent NET production without affecting oxidant production [15]. Cell cycle proteins [16] support nuclear envelope breakdown followed by the release of chromatin into the cytosol, where nuclear and cytosolic proteins are mixed [17]. The final cell lysis and NET release involves Gasdermin D (GSDMD), which is able to form pores in granule and plasma membranes [18,19]. This kind of NET formation ends up with cell death and is often described as lytic NET release or NETosis and occurs within a rather long time frame of three to eight hours. In contrast, the non-lytic NET release can be observed rapidly after 5–60 min of stimulation. Here, neutrophils do not undergo cell death, which was observed for neutrophils in close contact with activated platelets [20,21] or in response to Staphylococcus aureus infections [22]. Similar to NETosis, the non-lytic NET formation also involves the translocation of NE to the nucleus, histone citrullination, and chromatin decondensation [23]. Conversely, the membrane does not disintegrate, and the protein-decorated chromatin is released via vesicles [24] (Figure 1). Even the remnants of non-lytic NET formation, cytoplasts, are able to keep their mobility and fulfill important functions, such as phagocytosis, the activation of DCs, and the release of cytotoxic molecules [24,25,26].
The direct capture and clearance of pathogens by neutrophils can occur via phagocytosis or NET formation. Branzk and colleagues published a study suggesting that the pathogen size determines the neutrophil response. Large pathogens such as hyphae initiate NET formation, whereas small single-cell bacteria are eliminated via phagocytosis. Both events do not occur at the same time, since phagocytosis can prevent NET release by inhibiting NE translocation to the nucleus [27]. It is reasonable to extend this finding to cocci- or biofilm-forming bacteria, such as S. aureus, which also induce NET formation rather than phagocytosis.
Taken together, exteriorized chromatin fibers are able to entrap and alleviate the elimination of bacteria, fungi, protozoa, and even viruses [1,2,28,29,30,31]. Nevertheless, these properties are not only deleterious for invading pathogens but can also harm the host as well. NETs are able to kill epithelial and endothelial cells [32], and histones especially have a cytotoxic capability disturbing membrane integrity [33,34]. Additionally, NETs are able to promote vaso-occlusion, initiated by the hypoxia-induced release of von Willebrand factor (VWF) and endothelial P-selectin, resulting in neutrophil recruitment and activation [35,36]. Another proposed mechanism is the P-selectin-dependent neutrophil and platelet recruitment. Here, neutrophils initiate thromboxane A2 production in platelets, which induces the upregulation of intercellular adhesion molecule-1 (ICAM-1), further strengthening neutrophil–endothelium interactions [37]. This process induces NET formation implicating platelet-derived high mobility group protein B1 (HMGB1), ROS, and integrins [36,38]. Platelet-dependent NET formation requires the pro-inflammatory heterodimerized CXCL4 and CCL5, as well as the simultaneous stimulation of GPCRs and integrins [39]. NETs can further contribute to vessel occlusion through recruitment of factor XIIa, which mobilizes Weibel-Palade bodies containing VWF, P-Selectin, and factor XIIa [38,40]. Extracellular NET histones bind VWF and fibrin to recruit red blood cells and platelets [41,42], whereas NE cleaves the coagulation-inhibiting tissue factor pathway inhibitor (TFPI) and, in parallel, activates platelet receptors to increase platelet accumulation [43,44]. Thrombotic events can disturb the microcirculation particularly in the lungs, resulting in small pulmonary vessel occlusion [42,45], and, therefore, contribute to the pathogenesis of numerous diseases [46] (Figure 2). Although accumulated NETs especially seem to worsen disease outcome, the mechanisms of NET resolution and how NETs influence the resolution of inflammation are poorly understood.
Beside the degradation of NETs through DNases, some studies also suggest a contribution of macrophages to NET resolution and degradation. In vitro experiments with human monocyte-derived macrophages and PMA-stimulated human neutrophils demonstrated that macrophages are able to internalize NETs in a cathelicidin LL37-dependent manner and degrade DNA via TREX1/DNAseIII. In this setting, DCs contribute to extracellular NET degradation by DNase1L3. Here, cytokine profiling indicated that NETs alone are non-inflammatory but could also be immune modulatory in presence of LPS [47]. In contrast, Apel and colleagues describe a mechanism where phagocytosed NETs are able to activate the innate immune sensor cyclic GMP-AMP synthase (cGAS), thereby inducing the production of pro-inflammatory type I interferons [48]. Another study suggested a dual, phenotype-dependent role of macrophages: in the early phase, M2 macrophages induced a pro-inflammatory response and sustained the inflammatory state. In the second phase, M1 macrophages underwent cell death with nuclear decondensation, which took place in a PAD4-dependent manner and resulted in a local release of extracellular DNA. In the late phase, M1 macrophages degraded this DNA in a caspase-activated DNase-dependent manner resulting in the clearance of extracellular DNA within 24 h [49]. The release of nuclear DNA by macrophages or monocytes has been described by different groups and is referred to as macrophage extracellular traps (METs). They are also attributed to offer anti-microbial functions and contribute to pathology. Nevertheless, the respective studies show conflicting results, which has been discussed in more detail before [50].
NET aggregates (aggNETs), first described in a murine gout model [51], are formed at sites of high neutrophil density and contain enzymes that cleave, bind or modify autologous and foreign macromolecules. AggNETs are able to sequester and degrade histones and, thus, attenuate their cytotoxic effect on epithelial cells [52]. This process was executed by at least two aggNET-borne serine proteases, NE and PR3. Further, they are capable of resolving inflammation by the proteolytical degradation of inflammatory cytokines and chemokines [51,53]. Nevertheless, the physiological relevance of these proposed mechanisms remains elusive, and further work is required to shed light on the mechanisms of NET resolution and degradation.
This review focuses on the role of NET formation during virus-induced lung infections, as well as primary and secondary bacterial infections, and summarizes possible therapeutic interventions.
2. The Role of NETs in Virus-Induced Lung Diseases
Up to the beginning of 2020, NET formation in virus-induced lung diseases was of minor interest compared to bacterial lung infections. The current COVID-19 pandemic spotlighted NET formation within this disease, since several studies demonstrated a strong contribution of NETs to thrombosis and pulmonary vessel occlusion in COVID-19 patients. Here, we summarize recent research data and put them in context with other virus-induced diseases, such as respiratory syncytial virus (RSV) and influenza, with a special focus on NETs in the lung.
2.1. COVID-19
Similar to other SARS viruses, SARS-CoV-2 enters cells through angiotensin-converting enzyme 2 receptor (ACE2), expressed on renal and pulmonary endothelial cells [54,55]. Infected cells release paracrine factors influencing epithelial cells, neutrophils, and pneumocytes [56] and recruit further immune cells. The damaged lung during COVID-19 displays deformed capillaries, alveolar capillary damage, fluid-filled alveoli, hemorrhage, fibrin deposition, signs of compensatory neovascularization, and immune cell infiltration [57,58,59,60], which are altogether responsible for respiratory symptoms and shortness of breath. Disease severity was correlated to neutrophilia, indicating a direct contribution [61]. NET components, such as MPO-DNA, and citrullinated histone H3 are increased in patients with lung distress and higher severity of disease [62] and might serve as prognostic factors. In fact, viable SARS-CoV-2 can induce NET formation in human neutrophils in a dose-dependent manner [63] and requires virus replication, serine protease activity, and ACE2. NE, which is associated with NETs, is supposed to increase the susceptibility to SARS-CoV-2 infection by facilitating cell entry and virulence [64].
Histopathological lung analysis of SARS-CoV-2-related acute respiratory distress syndrome (ARDS) patients revealed small pulmonary vessel occlusion by NET aggregates, which was less often observed in histological analysis of influenza-induced pneumonia [45,65]. COVID-19-associated NETs are decorated with TF, which has been attributed to complement activation [66]. Complement activation may trigger coagulopathy and cytokine storm, both described as critical symptoms during COVID-19 [67]. Complement deficiencies in COVID-19 patients seem to be protective [67], whereas macular degeneration, which is associated with complement activation or a history of coagulation disorders, are rather poor prognostic factors [68].
Additionally, neutrophil activation markers as well as neutrophil–platelet aggregates are strongly elevated in patients with severe disease, whereas cases of intermediate severity displayed a hypo-reactive neutrophil phenotype and exhausted platelets [69]. The severe course has been linked to dysregulated immunothrombosis, resulting in ARDS and systemic hypercoagulability.
Formed NETs can further activate and damage endothelial cells, weakening the endothelial barrier integrity [70,71,72]. Circulating NET components can therefore reach other organs and, once accumulated, trigger microvascular thrombosis [45,66,73]. NET components with a high density of cationic residues, such as histone H4, can bind to negatively charged plasma membranes, which result in cell lysis through pore formation, finally leading to inflammation and tissue damage [74].
2.2. Respiratory Syncitial Virus
RSV is one of the most common causes for bronchiolitis in young children worldwide. At the age of 3, nearly all children have acquired at least one infection with the virus [75,76], and estimations suggest it is responsible for more than 3 million hospitalizations and almost 200,000 deaths per year for children <5 years. Characteristic symptoms are massive neutrophil accumulation in the lungs and occlusion of small airways by DNA-rich mucus plugs, resulting in coughing, wheezing, and labored breathing [77]. RSV activates neutrophils, induces IL-8 secretion, degranulation [78], and inhibits neutrophil apoptosis in a phosphoinositide 3-kinase (PI3K) and nuclear factor κb (NF-κB)-dependent manner [79].
The DNA content fosters the viscosity of the mucus, and it derives from necrotic inflammatory and epithelial cells, but also from RSV-induced NETosis, as demonstrated independently from two different groups [80,81]. Muraro and colleagues revealed that RSV induces lytic NET formation with 3 h of incubation dose dependently in human neutrophils in vitro. They suggest a mechanism where RSV fusion protein induces NET formation via TLR4 activation, further signaling via PI3K/AKT, ERK, p38-MAPK, NADPH-oxidase, and PAD4 [81,82]. Further evidence that NET components in the mucus contribute to worsening inflammation was provided by demonstrating that the bronchoalveolar lavage fluid (BALF) of severe RSV-induced lower respiratory tract disease (LRTD) in children contains NETs, involving NE, and citrullinated histone H3 [80]. In vitro assays revealed that NET formation and RSV trapping resulted in a reduced infection of epithelial cells. Nevertheless, only a minor fraction of NET-containing small airway occlusions in lung tissue sections of bRSV-LRTD in calves contained RSV antigens, suggesting further NET-inducing stimuli. This further raises the question of which events can turn the formerly positive NET function, the trapping of virus and diminishing of further infection, into something detrimental. The detailed investigation of the underlying mechanisms is a prerequisite to identify potential targets for effective therapies.
2.3. Influenza
Influenza A is a recurring infection with varying severity. Clinical symptoms include fever and upper respiratory tract complications, such as runny nose, cough, and sore throat [83]. Patients at high risk for complications are of older age or have pre-existing medical conditions. Complications include pneumonia, bronchiolitis, toxic shock syndrome, seizures, and bacterial pneumonia. Infection occurs via the carbohydrate-binding protein haemagglutinin (HA) and the enzyme neuraminidase (NA) to cleave the glycosidic bonds of the sialic acid residues of plasma membranes [84].
Similar to RSV, excessive neutrophil infiltration of the lung is a characteristic feature of influenza A [85,86,87]. Influenza A infection of mice revealed that neutrophil recruitment is promoted by the epithelial and endothelial expression of G-CSF and CXCL4 [88,89], and it depends on CXCR2 [90]. Beside the positive disease-limiting effects of neutrophils [86,87,91] and the ability to predispose neutrophils for possible secondary bacterial infections [92], the abundance of neutrophils can also exert harmful functions. Transcriptional analysis revealed a chemokine-driven feedforward circuit, potentially leading to lethal influenza infection [93]. Additionally, it was demonstrated that influenza infection leads to C3 release of platelets, triggering NET formation in a TLR7-dependent manner [94]. In influenza H1N1-infected mice, lung-recruited neutrophils showed an upregulation of several chemokine receptors (CCR1, CCR2, CCR3, CCR5, CXCR1, CXCR3, and CXCR4) compared to circulating neutrophils. In vitro experiments suggest their possible role in modulating chemotaxis, phagocytosis, and NET formation [95]. Recent data suggest subtype-specific differences, e.g., that subtype H1N1 induces NET formation with rather less chemokine and cytokine transcription, whereas H5N1 does not trigger NET formation but induces a higher expression of inflammatory cytokines [96].
Excessive NET release was correlated with poor outcome following influenza A infection [97]. Similar to RSV or COVID-19, NETs decorated with histones and MMP-9 were found in mice infected with influenza A-H1N1 inducing alveolar capillary damage and an obstruction of small airways [98], but they were also attributed to protective features in the liver following poxvirus infection [31]. Nevertheless, in mice deficient in PAD4, influenza A infection severity is not altered compared to WT mice. In contrast, in vitro studies suggest an anti-viral role of α-defensin, a cationic antimicrobial molecule. Here, cell treatment with human α-defensin-1 results in a significant inhibition of influenza virus replication and viral protein synthesis, probably through a protein kinase C-dependent mechanism [99]. Taken together, the well-balanced formation of NETs is essential for a mild course of disease and for an early defense against secondary bacterial infections. Inappropriate NET formation seems to be one of the main factors for a poor prognosis during influenza infection.
3. The Role of NETs in Bacteria-Associated Lung Diseases
Community-acquired pneumonia (CAP) is one of the most common infectious diseases and remains a burden worldwide. It is responsible for hospitalization and represents a cause of considerable morbidity and mortality. The development of CAP and secondary bacterial infections of the lung occurs likely by translocation or aspiration of nasal colonizing bacteria. These bacteria usually act as commensals but can infect the lung upon the expression of a wide array of species-specific virulence factors. Preceding viral infections can potentiate lung infection by damage and alterations in pulmonary antibacterial immunity. The most frequent causes of CAP admitted to intensive care units are infections with Streptococcus pneumoniae and Staphylococcus aureus [100]. In this section, we focus on the role of NETs during these bacterial infections. The infection with Pseudomonas aeruginosa is also of interest and is discussed later in the context of cystic fibrosis.
3.1. Streptococcus Pneumoniae
S. pneumoniae, also known as pneumococcus, is a Gram-positive diplococcus and is one of the main causes for bacterial pneumonia. It colonizes the mucosa of the human nasopharynx. Aspiration of nasopharyngeal secretions enables the invasion of the lung parenchyma, leading to pulmonary infection [101]. Following the secretion of inflammatory chemokines, neutrophils are recruited and fight infections by phagocytosis with subsequent degradation by proteases such as NE and cathepsin G stored in azurophilic granules [102] and by forming NETs with significant antibacterial activity against S. pneumoniae [103]. Invasive serotypes often express polysaccharide capsules, which confer resistance against phagocytic killing [104] and also reduce entrapment within NETs. NET formation was directly correlated with the thickness of the pneumococcal capsule, further augmenting disease severity in mice [105]. However, a higher incidence of NET components in CAP patients was associated with increased mortality [106]. Virulent pneumococci release EndA, a membrane-localized endonuclease capable of degrading NETs in vitro, contributing to host response evasion [107,108]. Additionally, EndA can foster bacteria spreading from the upper airways to the lungs and further to the bloodstream of infected mice [107]. Furthermore, the extracellular vesicle-associated endodeoxyribonuclease TatD is also capable of degrading NETs, and TatD-deficient pneumococci displayed compromised virulence with improved lung pathology during murine sepsis compared to the wild-type strain [109].
However, the release of NE by neutrophils either through the pneumolysin-induced leakage of neutrophils or the degradation of NETs is not only detrimental to invading microorganisms but also to the host. It degrades extracellular matrix components, such as elastin, fibronectin, collagen, and proteoglycan [110,111]. Additionally, it reduces the phagocytic activity of macrophages [112], impairs the pulmonary endothelial barrier, and injures alveolar epithelial cells [113], altogether contributing to host tissue damage [114]. NE is further capable of influencing the immune response by cleaving cell surface receptors, such as TLR2, TLR4, CD14, TNFR, and C5a [115,116,117], and degrading multiple inflammatory cytokines and chemokines, such as interleukin (IL)-1β, IL-2, IL-6, IL-8, IL-12p40, IL-12p70, and TNFα [115,117,118]. Recent studies demonstrate that NE cleaves human leukocyte antigen class II molecules in both cultured macrophages and in vivo mouse models, indicating that NE may disrupt antigen presentation and T-cell activation [119]. In contrast, NE cleaves and activates MMP-9, which may also have a destructive role in lung diseases [120]. Collectively, these data imply that NE may assist the dissemination of pneumococci by cleaving a variety of host immune proteins and inducing lung injury. Whether the inactivation of inflammatory cytokines also has a beneficial effect and prevents overwhelming leukocyte recruitment remains to be determined. Additionally, S. pneumoniae protects itself from NET killing by the incorporation of D-alanine into surface lipoteichoic acids (LTAs), which results in a positive charge of the bacterial membrane, blocking cationic peptides as histones [121]. In mice, lung infection with equal amounts of WT pneumococci and pneumococci lacking the dlt operon, encoding the respective enzymes for D-alanylation of LTA, displayed an increased dissemination of WT pneumococci in lungs and blood, indicating that D-alanylation supports NET evasion [121]. In summary, pneumococci successfully developed several strategies to evade clearance by neutrophils. As an unfortunate circumstance, capsules as part of this strategy also trigger additional NET formation, which additionally contributes to disease severity, finally resulting in increased mortality.
3.2. Staphylococcus Aureus
S. aureus emerged to an important cause of CAP with severe complications requiring intensive care and resulting in high mortality [122]. The rise of methicillin-resistant S. aureus (MRSA) even increased the threat of this pathogen. Preceding influenza infections increased the risk for mortality [123].
Especially known for the production of toxins as well as the generation of biofilms, both contribute to the successful invasion of the host while evading the hosts immune response [124,125,126]. The virulence depends on several factors, influencing the transition from colonization to infection, adhesion, iron acquisition, immune evasion, expression of pore-forming toxins, and metabolic regulators, as extensively reviewed elsewhere [127].
S. aureus infection of the lung leads to the recruitment of neutrophils combating infection. S. aureus has evolved multiple mechanisms of inhibiting neutrophil phagocytosis by interfering with complement activation and preventing Fc receptor binding [128,129,130,131]. Even if S. aureus is phagocytosed, it can survive inside neutrophils. Therefore, a rapid intervention to combat S. aureus infection might be more effective. Interestingly, S. aureus is able to induce the rapid, non-lytic, NADPH-independent NET formation [24]. Following DNA extrusion, cytoplasts are still able to migrate and phagocytose in vitro. Pilsczek and colleagues [22] revealed that Panton–Valentine leukocidin (PVL) is a potent NET inducer secreted by S. aureus. PVL is further able to lyse neutrophils, which can in turn neutralize PVL by α-defensins, which are part of NETs [132].
S. aureus expresses nucleases that are able to degrade NETs, conferring resistance to NET-mediated killing. In a murine mouse model, nuc-deficient S. aureus were significantly more susceptible to extracellular killing by neutrophils, whereas nuclease expression resulted in delayed bacterial clearance and increased mortality [133]. Additionally, in vitro assays indicated that the NET degradation product 2‘-deoxyadenosine (dAdo) is able to induce apoptosis in macrophages, further corroborating immune cell evasion of S. aureus [134]. The balance between the appropriate defense against pathogens and the destruction of lung barrier function is fragile, which was also demonstrated in murine pneumonia experiments investigating the impact of different levels of NETs during lung infection with S. aureus [135]. A decreased amount of NETs reduced lung injury and improved survival after DNase I treatment or with partial protein arginine deiminase 4 deficiency (PAD4+/–). Complete PAD4 deficiency (PAD4–/–) reduced NETs and lung injury but was counterbalanced by an increased bacterial load and inflammation. In line with this, mice deficient in the lipoxin receptor (Fpr2–/–) produce excessive NETs resulting in increased lung injury and mortality. In this context, samples from critically ill patients with increased plasma NETs were associated with ARDS severity and mortality, and lower plasma DNase I levels were associated with the development of sepsis-induced ARDS [135].
Several studies indicate a role of both PVL as well as nucleases during biofilm formation. Biofilm formation is a very effective measure to evade several immune defense mechanisms and enables long persistence of bacteria resulting in chronic disease.
During the early stages of biofilm formation, S. aureus produces immune modulators, such as staphylococcal complement inhibitor (SCIN), chemotaxis inhibitory protein of staphylococci (CHIPS), and formyl peptide receptor-like 1 inhibitor (FLIPr), as well as early production of thermonuclease [136,137]. These immunomodulators facilitate the defense of developing biofilms against the host early immune responses. To further corroborate these mechanisms, S. aureus biofilms have been shown to foster NET formation at the expense of other neutrophil defense mechanisms by releasing PVL and γ-hemolysin AB [138]. Competing with nuclease secretion by S. aureus, NETs are not able to efficiently combat infection. Additionally, long persisting S. aureus has been shown to be able to adapt to neutrophil-rich environments by increasing nuclease expression to evade NET killing, as it was demonstrated in airway isolates of cystic fibrosis patients [139].
The plethora of evasion mechanisms of S. aureus to the immune response underlines the importance of a fast and efficient treatment in the early phase of disease. Once S. aureus has manifested, the positive function of NETs declines and their deleterious role worsens disease outcome.
3.3. Cystic Fibrosis and Pseudomonas Aeruginosa
Cystic fibrosis (CF) is a genetic disease caused by mutations of the cystic fibrosis transmembrane regulator (CFTR) gene, a protein member of the ATP-binding cassette (ABC) transporter superfamily. It functions as a chloride channel that controls the transport of ions and water across epithelial tissues. The clinical manifestations of the CF are predominantly chronic airway infection and inflammation, which lead to a progressive decrement in lung function, pancreatic insufficiency, malnutrition, and hepatobiliary symptoms [140]. Pseudomonas aeruginosa is the major pathogen involved, causing thick mucus and reduced mucociliary clearance favoring chronic bacterial infections, including neutrophil-rich airway inflammation followed by increased rates of morbidity and mortality [141,142]. Further, P. aeruginosa induces neutrophil recruitment. Once migrated to the site of infection, neutrophils perform their classical anti-bacterial functions, including phagocytosis, ROS production, degranulation, and NET formation [143,144]. However, in CF patients, neutrophils fail to eliminate P. aeruginosa invasion but rather contribute to tissue damage [145,146], which is further supported by decreased apoptosis [147,148]. NETs have been shown to be present in the sputum, contributing to an increased mucus viscosity [149,150] and, additionally, to tissue damage with decreased pulmonary function [143,144]. When cell-free DNA is enriched in the sputum, patients display diminished lung function compared to patients showing mild symptoms, indicating that the airway obstruction is a result of accumulated NETs [3,151,152,153]. Moreover, it has been reported that NET components, such as MPO, NE and histones, can damage epithelial, endothelial, and connective tissues, worsening the lung pathology [32,149,154].
An impaired clearance of NETs from the airways might also contribute to pathogenesis and depends on several factors, including mucociliary clearance, DNase activity, and phagocytosis by macrophages. The phagocytosis is facilitated by pre-degradation by DNase and does not result in pro-inflammatory cytokine secretion [47,155]. CFTR-deficient macrophages have an abnormally high intraphagolysosomal pH, which was shown to alter bactericidal activity and might also impair NET resolution [156].
P. aeruginosa developed strategies to evade neutrophils and their respective immune responses. Upon infection of the patients’ lung, this pathogen can migrate to areas with low oxygen concentrations where only few immune cells can exert their defense function [157]. Additionally, it has been shown that biofilm formation promotes excessive production of alginate that allows the escape of bacteria to neutrophil degranulation and phagocytosis [158].
In order to attenuate NET production and resist NET-mediated killing, P. aeruginosa can express surface sialic acids that are capable of binding and inducing signaling through neutrophil Siglec-9, further suppressing the oxidative burst and, thus, NET formation [159]. The authors in this study showed that treatment with sialidase or the use of P. aeruginosa strains lacking sialic acids led to increased NET production compared with sialic acid-positive strains. Furthermore, it was demonstrated that paired P. aeruginosa isolates from patients with CF at early and late stages of disease developed resistance to NET-mediated killing over time, which corresponded to the development of the mucoid, alginate-rich phenotype [157]. Nevertheless, the same study revealed that alginate overexpression did not increase survival upon incubation with PMA-treated neutrophils. This might be due to additional yet unknown defense mechanisms conferring NET evasion. Alternatively, this might also indicate that PMA-induced NETs may differ in their capability to combat pathogens compared to NETs that were formed in response to physiological stimuli.
Additionally, P. aeruginosa can overexpress genes controlled by the two component systems PhoPQ and PmrAB that sense Mg2+ limitation and, at the same time, encode mechanisms to effectively obtain the ion so that it cannot be complexed by NET structures [158,160,161]. Furthermore, bacteria are capable of regulating genes that allow them to tolerate the toxicity of extracellular DNA and their components [162].
Taken together, NETs during CF have only a limited anti-bacterial effect. Additionally, the prolonged survival of neutrophils in addition to the reduced resolution of NET structures essentially contribute to exacerbated inflammation. Further studies are required to elucidate the interplay between NETs and pathogens during CF.
3.4. Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease (COPD) is one of the leading causes of death worldwide and a major cause of mortality in adults [163]. It is characterized by airflow limitation by narrowing of the small airways combined with emphysematous destruction of the alveoli. Chronic exposure to cigarette smoke contributes to COPD pathogenesis, where it can induce neutrophil retention within the airways [164]. In general, the degree of neutrophilia correlates with COPD severity [165,166], exacerbations [167], and disease progression [168]. Neutrophil recruitment into the sputum accounts for approximately one-third on CXCL8 [169], but also other CXCR2 ligands, such as CXCL1 and CXCL5, are elevated in COPD sputa, airway fluids, and bronchial tissues [166,170,171]. CXCR2 is upregulated in exacerbations of COPD where its expression co-localizes with the accumulation of airway mucosal neutrophils [170].
Several studies [172,173,174] observed NET formation in the sputum from both stable and exacerbated COPD patients using qPCR, ELISAs, and confocal fluorescent and electron microscopy, respectively. Elevated levels of sputum NETs were negatively associated with lung function and, additionally, COPD symptoms and PAD4 gene expression were found to be upregulated in neutrophilic compared to non-neutrophilic COPD patients [173]. Furthermore, increased NET formation in the airways of COPD patients was associated with disease severity [175]. This study suggested a relationship between sputum-enriched NETs and non-eosinophilic COPD exacerbations and reduced bacterial diversity accompanied by an abundance of Haemophilus species. Interestingly, the phagocytic capacity of neutrophils to engulf bacteria ex vivo was impaired in cells derived from patients with high sputum NET complexes or in the neutrophils of healthy donors incubated with the soluble sputum of COPD patients [175]. Since phagocytosis is more efficient in bacterial clearance, its suppression might also contribute to exaggerated inflammation and recurrent infections. This is further underlined by a study that demonstrated that COPD patients are highly susceptible to recurrent bacterial infection following infection with respiratory viruses, which are also known to induce NETs [176].
Taken together, these findings indicate a strong negative impairment of NETs on COPD and, therefore, also identify them as a promising therapeutic target. For a more detailed view on NETs in COPD, please see the recently published review by Trivedi and colleagues [177].
4. Pathogenic Fungal Lung Infection—Aspergillosis
Infection with pulmonary fungal pathogens is a severe clinical problem, especially in patients with compromised immune functions. Opportunistic fungi, including Aspergillus with invasive aspergillosis [178,179,180], Cryptococcus with cryptococcosis [181,182,183], Pneumocystis with pneumonia [184], and endemic fungi [185,186], are the main sources of fungal infections in the lungs of humans. Aspergillus mold is one of the most common fungal species, which is able to sporulate with released airborne conidia. With a size of 2–3 µm, they are small enough to infiltrate human airways and pulmonary alveoli, causing a spectrum of diseases [178,187]. Within the early phase of infection in healthy individuals, it is assumed that neutrophils restrict the tissue invasion of hyphae [188], whereas inhaled conidia are eliminated by alveolar macrophages [189,190,191]. Immunocompromised individuals exhibit tissue invasion by fungal hyphae due to the incomplete killing of inhaled fungal conidia [192]. Upon recognition of fungal pathogens, innate immune cells are activated by pathogen-associated molecular patterns (PAMPs) via specific pattern recognition receptors (PRRs) on the surface to trigger further intracellular signaling cascades. The PRRs involved in fungal detection identified to date include TLRs, C-type lectin receptors and NOD-like receptors [193,194].
As described before, neutrophils were suggested to sense microbe size and selectively release NETs in response to large pathogens such as A. fumigatus hyphae or large aggregated conidia but not in response to small single conidia [27], most probably to compensate for the inefficient phagocytosis of larger pathogens.
Indeed, investigating a murine model of pulmonary aspergillosis revealed that neutrophils are able to form NETs in response to A. fumigatus, in particular close to [195] developing clusters of fungi with outgrowing hyphae, whereas conidia are rather engulfed by neutrophils [195]. In vitro, NETs display fungistatic activity and are hypothesized to prevent fungal dissemination [29,195]. In response to Aspergillus, the inhibitory function of NETs has been shown to be mediated by calprotectin [29] and the release of long pentraxin 3, a pattern recognition receptor that activates complement and facilitates pathogen recognition [196]. The protective function of NETs was further underlined by investigating pulmonary aspergillosis in p47phox−/− mice, which failed to generate NETs and developed progressive pneumonia [197]. In contrast, another study induced invasive pulmonary aspergillosis in mice and demonstrated that Pad4−/− mice revealed a lower fungal burden in the lungs, accompanied by a reduced acute lung injury, and less TNFα and citH3 compared to wild-type controls [198]. These findings suggest a detrimental role of NETs contributing to tissue damage and limiting the control of fungal outgrowth. However, the NET-mediated ability to combat fungal invasion remains controversial [2,199], but it should be noted that the exact mechanisms of fungal killing in mice can actually be different from those observed in humans [200].
Patients with neutropenia or hematologic malignancy, as well as those who suffer from chronic granulomatous disease (CGD), are predisposed to Aspergillus infection [201]. CGD patients have impaired phox function, resulting in poor NET production and reduced neutrophil activity [202]. In this context, A. nidulans emerges as a major pathogen, often resulting in refractory, disseminated disease [203]. In a clinical study involving a patient with CGD suffering from refractory invasive A. nidulans infection, Bianchi and colleagues suggested a link between the production of NETs and the resolution of invasive aspergillosis [204]. This idea was further underlined by in vitro experiments that demonstrated that phox-deficient neutrophils lack activity against A. nidulans conidia and hyphae. Genetically complementing phox function restored both NET production and antifungal activity. Furthermore, administration of gene therapy providing phox activity rapidly cured the patient with treatment-refractory A. nidulans infection [205].
NET evasion by A. fumigatus was linked to the expression of galactosaminogalactan (GAG), an α-1,4-linked linear heteroglycan composed of various combinations of galactose and N-acetyl-galactosamine [206,207,208]. Disruption of GAG attenuates both biofilm formation and virulence [208,209]. Its protective role against NETs was suggested to be mediated by its positive charge, which is able to bind to the cationic antimicrobial peptides or histones on NETs [210].
Taken together, the current knowledge about the role of NETs during fungi-induced lung diseases is rather scarce. Additionally, some contradictory studies concerning human and murine neutrophils exist. Nonetheless, investigating exactly these differences will probably generate valuable information about NET formation as a host defense mechanism combatting pathogenic fungi.
5. NET-Targeting Therapies
As described above in detail, the abundance of NETs is a fragile balance, which tends to tilt over to a rather deleterious influence during several infectious diseases. To date, several attempts were taken to keep NETs in the right frame. Nevertheless, the combination of DNA with potentially damaging molecules makes it difficult to find the perfect treatment. Here, we summarize the most relevant and promising treatment strategies.
5.1. DNase1
Extracellular chromatin and NETs can be digested by naturally occurring DNase1. It dismantles the DNA structure and liberates entangled components, which has to be calculated as a significant risk factor since, e.g., NE or MPO are capable of perpetuating inflammation. However, DNase is the only NET-targeting therapy already in clinical use. It is used for the treatment of virus-associated bronchiolitis [211], as well as cystic fibrosis, in order to improve lung function and reduce the occurrence of infectious exacerbations [212,213]. Similarly, NET DNA in COVID-19 contributes to mucus accumulation, rigidity, and airway occlusion, indicating that severe cases of COVID-19 might also benefit from DNase treatment. A single-center case study was published recently, suggesting that nebulized dornase (recombinant human DNase) reduced supplemental oxygen requirements [214]. Further clinical trials are currently underway investigating the effect of nebulized dornase-α during COVID-19 [215].
5.2. Histones
One possible drawback of disentangling DNA fibers could be the subsequent release of histones or proteases, potentially causing cytotoxicity. Recently, a study suggested that the synergy between histone and DNA is critical for sub-lethal signaling [216]. Accordingly, another study proposes a mechanism whereby aggNETs contribute to the detoxification of histones. Neutralization of histones might be a promising target in future, as demonstrated in different murine disease models [217,218]. The C1 esterase inhibitor (C1INH), a serine protease inhibitor, is capable of targeting multiple pathways [219,220] and can bind and neutralize histones due to its glycosylation-dependent overall negative charge. Additional studies revealed that C1INH treatment reduced neutrophil activation and improved inflammation and survival in sepsis patients [221,222]. However, additional preclinical testing and investigation of different disease models is needed to further validate this promising inhibitor as a therapeutic agent during inflammation. Furthermore, a recent study suggests a promising role of an anti-citrullinated protein antibody (tACPA), which prevented NET-associated disease symptoms in different inflammatory pathologies in mice by inhibiting NET formation and increasing NET degradation through macrophages [218]. Accordingly, another study demonstrated that neutralizing citH3 attenuates endothelial damage in vitro and has the capability to improve survival rates and inflammatory responses during LPS-induced sepsis in mice [223].
5.3. Neutrophil Elastase
Similar to the abovementioned release of histones, the liberation of NE might also contribute to tissue damage and inflammation since it is capable of disturbing the lung barrier, inducing the release of inflammatory cytokines, thereby fostering a cytokine storm which is often a life-threatening event during ARDS. Small-molecule inhibitors, such as sivelestat [224], alvelestat and Bay-8550, are possible therapeutics directed against NE and currently under investigation. Different studies and clinical trials with ARDS/SIRS patients indicate that sivelestat improves pulmonary function, and shortens the duration of mechanical ventilation and the length of ICU care [225,226], most likely through inhibition of the exaggerated signaling pathways and neutrophil chemotaxis [227,228,229].
5.4. Other Treatments
There are several molecules that are able to influence NET formation. Aspirin treatment decreases NET formation in the lung microcirculation and plasma [230] and also decreases the deposition of platelets with neutrophils on the lungs’ vascular walls [231]. TLR-mediated NET formation can be inhibited by the use of blocking antibodies, such as anti-CLEC or the bispecific anti-CLEC5A/TLR2 [232]. Additionally, hydroxychloroquine, also known as an anti-malarial and anti-inflammatory drug, inhibits the stimulation of pDCs by NETs via TLR9 [230]. The antidiabetic drug metformin directly binds the alarmin HMGB1, resulting in increased NET clearance, and attenuates the pro-inflammatory activity of NETs [233,234]. Glucocorticoids, such as dexamethasone, belong to a class of drugs with anti-NET formation activity [235]. Additionally, NET-inhibitory factors have been identified. They specifically inhibit NET formation in vitro and in vivo, thereby suggesting them to be a potential therapeutic agent [236]. Further treatment options exist that do not directly target NET formation but rather neutrophil recruitment. For example, a CXCR2 antagonist reduced neutrophil influx into the airways following an LPS challenge in humans [237]. Nonetheless, blocking neutrophil recruitment always harbors the risk of impairing the innate immune response. In regard to this, a promising therapy might be the use of the CD40 antibody M7, which was shown to limit inflammation without affecting the protective host defense in mice [238]. A summary of possible interventions that are targeted against NETs or their components is listed in Table 1.
6. Summary
NETs seem to play a fundamental role in the pathogenesis of several respiratory diseases often attributed to worse outcomes, but the treatment options are scarce. However, in the last years, the knowledge about NET formation increased, and it became certainly clear that there has to be a consensus about the definition of NETs, their stimulation, and their components [239]. Results from experiments with non-physiologic triggers, such as PMA, are most likely not able to represent the in vivo situation. In addition, the detection of NETs should follow certain rules, since only staining of extracellular DNA does not necessarily detect NETs but also other necrotic cell remnants. Inconsistent methods of published studies complicate interpretation of their data. However, much more information about the underlying signaling pathways is required to establish potential therapies. With regard to respiratory diseases, it appears that NETs have a beneficial role in the early phase of disease. They often participate in capturing pathogens and prevent ongoing infection, secondary infections, and dissemination. A number of evasion mechanisms evolved by different bacteria support the importance of this preventive role. Nonetheless, in a later phase, where the disease has manifested, the anti-microbial components of NETs are no longer able to fight infection, but rather contribute to pathology due to their cytotoxic properties: this fragile balance complicates an effective treatment. Studies investigating NETs during different phases of disease are rare. The differentiation of the impact of NETs during onset, progression, and resolution of disease is of great interest and will provide essential contributions to the development of possible therapeutic interventions.
Author Contributions
H.B. and A.Z. wrote, reviewed, and edited the manuscript. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG)-CRC 1450-431460824, ZA428/17-1 and ZA428/18-1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef] [Green Version]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Biol. 2017, 216, 4073–4090. [Google Scholar] [CrossRef]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 2958. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Diaz-Godinez, C.; Fonseca, Z.; Nequiz, M.; Laclette, J.P.; Rosales, C.; Carrero, J.C. Entamoeba histolytica Trophozoites Induce a Rapid Non-classical NETosis Mechanism Independent of NOX2-Derived Reactive Oxygen Species and PAD4 Activity. Front. Cell Infect. Microbiol. 2018, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Avondt, K.; van der Linden, M.; Naccache, P.H.; Egan, D.A.; Meyaard, L. Signal Inhibitory Receptor on Leukocytes-1 Limits the Formation of Neutrophil Extracellular Traps, but Preserves Intracellular Bacterial Killing. J. Immunol. 2016, 196, 3686–3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Roos, D.; Voetman, A.A.; Meerhof, L.J. Functional activity of enucleated human polymorphonuclear leukocytes. J. Cell Biol. 1983, 97, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Douda, D.N.; Bruggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes-Costa, A.B.; Nascimento, M.T.; Froment, G.S.; Soares, R.P.; Morgado, F.N.; Conceicao-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.; Heesemann, L.; Wagener, J.; Marcos, V.; Hartl, D.; Loeffler, J.; Heesemann, J.; Ebel, F. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. 2010, 12, 928–936. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Kollnberger, M.; Wakefield, T.W.; Lammle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Doring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Sporn, L.A.; Marder, V.J.; Wagner, D.D. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell 1986, 46, 185–190. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 2000, 275, 6819–6823. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [Green Version]
- Apel, F.; Andreeva, L.; Knackstedt, L.S.; Streeck, R.; Frese, C.K.; Goosmann, C.; Hopfner, K.P.; Zychlinsky, A. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci. Signal. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Knopf, J.; Leppkes, M.; Schett, G.; Herrmann, M.; Munoz, L.E. Aggregated NETs Sequester and Detoxify Extracellular Histones. Front. Immunol. 2019, 10, 2176. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudit, G.Y.; Crackower, M.A.; Backx, P.H.; Penninger, J.M. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc. Med. 2003, 13, 93–101. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, C.Y.; Zhou, P.; Yue, H.; Du, R. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, 173, 324. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Kanthi, Y.; Knight, J.S.; Kim, A.H.J. The interplay between neutrophils, complement, and microthrombi in COVID-19. Best Pr. Res. Clin. Rheumatol. 2021, 35, 101661. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef] [Green Version]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Joppich, M.; Hoffknecht, M.L.; Gold, C.; Engel, A.; Polewka, V.; Muenchhoff, M.; et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2021, 19, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Lo, M.W.; Kemper, C.; Woodruff, T.M. COVID-19: Complement, Coagulation, and Collateral Damage. J. Immunol. 2020, 205, 1488–1495. [Google Scholar] [CrossRef]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arter. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Gerretsen, J.; Boeltz, S.; Munoz, L.E.; Herrmann, M.; Pickkers, P.; Hilbrands, L.B.; van der Vlag, J. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann. Rheum. Dis. 2018, 77, 1790–1798. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; da Silva, L.F.F.; de Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Mejias, A.; Chavez-Bueno, S.; Jafri, H.S.; Ramilo, O. Respiratory syncytial virus infections: Old challenges and new opportunities. Pediatr. Infect. Dis. J. 2005, 24, S189–S196. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.A.; Bueno, S.M.; Carreno, L.J.; Riedel, C.A.; Kalergis, A.M. Respiratory syncytial virus infection and immunity. Rev. Med. Virol. 2012, 22, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Lay, M.K.; Gonzalez, P.A.; Leon, M.A.; Cespedes, P.F.; Bueno, S.M.; Riedel, C.A.; Kalergis, A.M. Advances in understanding respiratory syncytial virus infection in airway epithelial cells and consequential effects on the immune response. Microbes Infect. 2013, 15, 230–242. [Google Scholar] [CrossRef]
- Jaovisidha, P.; Peeples, M.E.; Brees, A.A.; Carpenter, L.R.; Moy, J.N. Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J. Immunol. 1999, 163, 2816–2820. [Google Scholar]
- Lindemans, C.A.; Coffer, P.J.; Schellens, I.M.; de Graaff, P.M.; Kimpen, J.L.; Koenderman, L. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J. Immunol. 2006, 176, 5529–5537. [Google Scholar] [CrossRef] [Green Version]
- Cortjens, B.; de Boer, O.J.; de Jong, R.; Antonis, A.F.; Sabogal Pineros, Y.S.; Lutter, R.; van Woensel, J.B.; Bem, R.A. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef]
- Rewar, S.; Mirdha, D.; Rewar, P. Treatment and Prevention of Pandemic H1N1 Influenza. Ann. Glob. Health 2015, 81, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 697–708. [Google Scholar] [CrossRef]
- Perrone, L.A.; Plowden, J.K.; Garcia-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, M.D.; Deng, Y.M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Feng, K.; Wang, Y.C.; Mei, J.J.; Ning, R.T.; Zheng, H.W.; Wang, J.J.; Worthen, G.S.; Wang, X.; Song, J.; et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. 2017, 10, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Aloe, C.; Wilson, N.; Bozinovski, S. G-CSFR antagonism reduces neutrophilic inflammation during pneumococcal and influenza respiratory infections without compromising clearance. Sci. Rep. 2019, 9, 17732. [Google Scholar] [CrossRef]
- Wareing, M.D.; Shea, A.L.; Inglis, C.A.; Dias, P.B.; Sarawar, S.R. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral. Immunol. 2007, 20, 369–378. [Google Scholar] [CrossRef]
- Fujisawa, H. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J. Virol. 2008, 82, 2772–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malachowa, N.; Freedman, B.; Sturdevant, D.E.; Kobayashi, S.D.; Nair, V.; Feldmann, F.; Starr, T.; Steele-Mortimer, O.; Kash, J.C.; Taubenberger, J.K.; et al. Differential Ability of Pandemic and Seasonal H1N1 Influenza A Viruses To Alter the Function of Human Neutrophils. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 2013, 154, 197–212. [Google Scholar] [CrossRef] [Green Version]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef] [Green Version]
- Rudd, J.M.; Pulavendran, S.; Ashar, H.K.; Ritchey, J.W.; Snider, T.A.; Malayer, J.R.; Marie, M.; Chow, V.T.K.; Narasaraju, T. Neutrophils Induce a Novel Chemokine Receptors Repertoire During Influenza Pneumonia. Front. Cell. Infect. Microbiol. 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.L.Y.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L.; Chan, M.C.W.; Chan, R.W.Y. Host DNA released by NETosis in neutrophils exposed to seasonal H1N1 and highly pathogenic H5N1 influenza viruses. Respir. Res. 2020, 21, 160. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Salvatore, M.; Garcia-Sastre, A.; Ruchala, P.; Lehrer, R.I.; Chang, T.; Klotman, M.E. alpha-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J. Infect. Dis. 2007, 196, 835–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandell, L.A. Community-acquired pneumonia: An overview. Postgrad. Med. 2015, 127, 607–615. [Google Scholar] [CrossRef]
- Liu, N.; Zheng, Z.; Chen, P.; Hou, P.; Wang, X.; Li, H.; Chen, R. Detection of aspiration of nasopharyngeal secretion and the relationship between the aspiration of nasopharyngeal secretion and the incidence of pneumonia. Zhonghua Jie He He Hu Xi Za Zhi 2015, 38, 511–515. [Google Scholar] [PubMed]
- Standish, A.J.; Weiser, J.N. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J. Immunol. 2009, 183, 2602–2609. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Yamaguchi, M.; Terao, Y.; Hamada, S.; Ooshima, T.; Kawabata, S. alpha-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J. Biol. Chem. 2012, 287, 10472–10481. [Google Scholar] [CrossRef] [Green Version]
- Hyams, C.; Camberlein, E.; Cohen, J.M.; Bax, K.; Brown, J.S. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 2010, 78, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, A.N.; Rai, P.; Jiao, H.; Wang, S.; Tan, K.B.; Qin, L.; Watanabe, H.; Zhang, Y.; Teluguakula, N.; Chow, V.T. Capsules of virulent pneumococcal serotypes enhance formation of neutrophil extracellular traps during in vivo pathogenesis of pneumonia. Oncotarget 2016, 7, 19327–19340. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, F.; Giaglis, S.; Hahn, S.; Blum, C.A.; Baumgartner, C.; Kutz, A.; van Breda, S.V.; Mueller, B.; Schuetz, P.; Christ-Crain, M.; et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: Secondary analysis of a randomised controlled trial. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Kuang, Z.; Wilson, B.A.; Lau, G.W. Competence-independent activity of pneumococcal EndA [corrected] mediates degradation of extracellular dna and nets and is important for virulence. PLoS ONE 2013, 8, e70363. [Google Scholar] [CrossRef] [Green Version]
- Jhelum, H.; Sori, H.; Sehgal, D. A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci. Rep. 2018, 8, 7985. [Google Scholar] [CrossRef] [PubMed]
- Janusz, M.J.; Doherty, N.S. Degradation of cartilage matrix proteoglycan by human neutrophils involves both elastase and cathepsin G. J. Immunol. 1991, 146, 3922–3928. [Google Scholar] [PubMed]
- Taylor, S.; Dirir, O.; Zamanian, R.T.; Rabinovitch, M.; Thompson, A.A.R. The Role of Neutrophils and Neutrophil Elastase in Pulmonary Arterial Hypertension. Front. Med. 2018, 5, 217. [Google Scholar] [CrossRef] [PubMed]
- Domon, H.; Oda, M.; Maekawa, T.; Nagai, K.; Takeda, W.; Terao, Y. Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Sci. Rep. 2016, 6, 38013. [Google Scholar] [CrossRef] [PubMed]
- Mizikova, I.; Ruiz-Camp, J.; Steenbock, H.; Madurga, A.; Vadasz, I.; Herold, S.; Mayer, K.; Seeger, W.; Brinckmann, J.; Morty, R.E. Collagen and elastin cross-linking is altered during aberrant late lung development associated with hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1145–L1158. [Google Scholar] [CrossRef] [Green Version]
- Polverino, E.; Rosales-Mayor, E.; Dale, G.E.; Dembowsky, K.; Torres, A. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedow, O.; Meyer-Hoffert, U. Neutrophil serine proteases: Potential key regulators of cell signalling during inflammation. J. Intern. Med. 2005, 257, 319–328. [Google Scholar] [CrossRef]
- Van den Berg, C.W.; Tambourgi, D.V.; Clark, H.W.; Hoong, S.J.; Spiller, O.B.; McGreal, E.P. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 2014, 192, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Domon, H.; Nagai, K.; Maekawa, T.; Oda, M.; Yonezawa, D.; Takeda, W.; Hiyoshi, T.; Tamura, H.; Yamaguchi, M.; Kawabata, S.; et al. Neutrophil Elastase Subverts the Immune Response by Cleaving Toll-Like Receptors and Cytokines in Pneumococcal Pneumonia. Front. Immunol. 2018, 9, 732. [Google Scholar] [CrossRef] [Green Version]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [Green Version]
- Domon, H.; Maekawa, T.; Isono, T.; Furuta, K.; Kaito, C.; Terao, Y. Proteolytic cleavage of HLA class II by human neutrophil elastase in pneumococcal pneumonia. Sci. Rep. 2021, 11, 2432. [Google Scholar] [CrossRef]
- Jackson, P.L.; Xu, X.; Wilson, L.; Weathington, N.M.; Clancy, J.P.; Blalock, J.E.; Gaggar, A. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: Implications to cystic fibrosis proteolytic dysfunction. Mol. Med. 2010, 16, 159–166. [Google Scholar] [CrossRef]
- Wartha, F.; Beiter, K.; Albiger, B.; Fernebro, J.; Zychlinsky, A.; Normark, S.; Henriques-Normark, B. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 2007, 9, 1162–1171. [Google Scholar] [CrossRef]
- Self, W.H.; Wunderink, R.G.; Williams, D.J.; Zhu, Y.; Anderson, E.J.; Balk, R.A.; Fakhran, S.S.; Chappell, J.D.; Casimir, G.; Courtney, D.M.; et al. Staphylococcus aureus Community-acquired Pneumonia: Prevalence, Clinical Characteristics, and Outcomes. Clin. Infect. Dis. 2016, 63, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.S.; Greenberg, J.A.; McNulty, M.C.; Gregg, K.S.; Riddell, J.t.; Mangino, J.E.; Weber, D.M.; Hebert, C.L.; Marzec, N.S.; Barron, M.A.; et al. Bacterial and viral co-infections complicating severe influenza: Incidence and impact among 507 U.S. patients, 2013–2014. J. Clin. Virol. 2016, 80, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Balasubramanian, D.; Tam, K.; Askenazi, M.; Copin, R.; Shopsin, B.; Torres, V.J.; Ueberheide, B.M. Using Quantitative Spectrometry to Understand the Influence of Genetics and Nutritional Perturbations On the Virulence Potential of Staphylococcus aureus. Mol. Cell. Proteom. 2017, 16, S15–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Grousd, J.A.; Rich, H.E.; Alcorn, J.F. Host-Pathogen Interactions in Gram-Positive Bacterial Pneumonia. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Nizet, V. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J. Allergy Clin. Immunol. 2007, 120, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; van Kessel, K.P.; van Strijp, J.A. Staphylococcal innate immune evasion. Trends Microbiol. 2005, 13, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.; Loughman, A.; van Kessel, K.P.; van Strijp, J.A.; Foster, T.J. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol. Lett. 2006, 258, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Agniswamy, J.; Lei, B.; Musser, J.M.; Sun, P.D. Insight of host immune evasion mediated by two variants of group a Streptococcus Mac protein. J. Biol. Chem. 2004, 279, 52789–52796. [Google Scholar] [CrossRef] [Green Version]
- Cardot-Martin, E.; Casalegno, J.S.; Badiou, C.; Dauwalder, O.; Keller, D.; Prevost, G.; Rieg, S.; Kern, W.V.; Cuerq, C.; Etienne, J.; et al. alpha-Defensins partially protect human neutrophils against Panton-Valentine leukocidin produced by Staphylococcus aureus. Lett. Appl. Microbiol. 2015, 61, 158–164. [Google Scholar] [CrossRef]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Kockritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. Jci Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, A.R.; Swierstra, J.W.; Lemmens-den Toom, N.A.; Snijders, S.V.; Hansenova Manaskova, S.; Verbon, A.; van Wamel, W.J.B. Production of Staphylococcal Complement Inhibitor (SCIN) and Other Immune Modulators during the Early Stages of Staphylococcus aureus Biofilm Formation in a Mammalian Cell Culture Medium. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.R.; Hoppenbrouwers, T.; Lemmens-den Toom, N.A.; Snijders, S.V.; van Neck, J.W.; Verbon, A.; de Maat, M.P.M.; van Wamel, W.J.B. During the Early Stages of Staphylococcus aureus Biofilm Formation, Induced Neutrophil Extracellular Traps Are Degraded by Autologous Thermonuclease. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Berends, E.T.M.; Chan, R.; Schwab, E.; Roy, S.; Sen, C.K.; Torres, V.J.; Wozniak, D.J. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc. Natl. Acad. Sci. USA 2018, 115, 7416–7421. [Google Scholar] [CrossRef] [Green Version]
- Herzog, S.; Dach, F.; de Buhr, N.; Niemann, S.; Schlagowski, J.; Chaves-Moreno, D.; Neumann, C.; Goretzko, J.; Schwierzeck, V.; Mellmann, A.; et al. High Nuclease Activity of Long Persisting Staphylococcus aureus Isolates Within the Airways of Cystic Fibrosis Patients Protects Against NET-Mediated Killing. Front. Immunol. 2019, 10, 2552. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Durie, P.R. What is cystic fibrosis? N. Engl. J. Med. 2002, 347, 439–442. [Google Scholar] [CrossRef]
- Regamey, N.; Tsartsali, L.; Hilliard, T.N.; Fuchs, O.; Tan, H.L.; Zhu, J.; Qiu, Y.S.; Alton, E.W.; Jeffery, P.K.; Bush, A.; et al. Distinct patterns of inflammation in the airway lumen and bronchial mucosa of children with cystic fibrosis. Thorax 2012, 67, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, L.; McClean, S. Bacterial Adaptation during Chronic Respiratory Infections. Pathogens 2015, 4, 66–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.G.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef]
- Yoo, D.G.; Winn, M.; Pang, L.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L.; Rada, B. Release of cystic fibrosis airway inflammatory markers from Pseudomonas aeruginosa-stimulated human neutrophils involves NADPH oxidase-dependent extracellular DNA trap formation. J. Immunol. 2014, 192, 4728–4738. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Bruce, K.D. The cystic fibrosis airway microbiome. Cold Spring Harb. Perspect. Med. 2013, 3, a009738. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeon, D.J.; Condliffe, A.M.; Cowburn, A.S.; Cadwallader, K.C.; Farahi, N.; Bilton, D.; Chilvers, E.R. Prolonged survival of neutrophils from patients with Delta F508 CFTR mutations. Thorax 2008, 63, 660–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 2012, 11, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Aleman, S.R.; Campos-Garcia, L.; Palma-Nicolas, J.P.; Hernandez-Bello, R.; Gonzalez, G.M.; Sanchez-Gonzalez, A. Understanding the Entanglement: Neutrophil Extracellular Traps (NETs) in Cystic Fibrosis. Front. Cell. Infect. Microbiol. 2017, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef]
- Dubois, A.V.; Gauthier, A.; Brea, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 80–86. [Google Scholar] [CrossRef]
- Marcos, V.; Zhou-Suckow, Z.; Onder Yildirim, A.; Bohla, A.; Hector, A.; Vitkov, L.; Krautgartner, W.D.; Stoiber, W.; Griese, M.; Eickelberg, O.; et al. Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediat. Inflamm. 2015, 2015, 408935. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kinsella, M.G.; Wight, T.N. Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am. J. Pathol. 1993, 143, 907–917. [Google Scholar]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell. Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]
- Young, R.L.; Malcolm, K.C.; Kret, J.E.; Caceres, S.M.; Poch, K.R.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Randell, S.H.; Vasil, M.L.; et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: Evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 2011, 6, e23637. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatua, B.; Bhattacharya, K.; Mandal, C. Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through siglec-9. J. Leukoc. Biol. 2012, 91, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.N.; Mutharasan, R. A cantilever biosensor-based assay for toxin-producing cyanobacteria Microcystis aeruginosa using 16S rRNA. Environ. Sci. Technol. 2013, 47, 12333–12341. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Mulcahy, H.; Kanevets, U.; Shi, Y.; Lewenza, S. Surface-localized spermidine protects the Pseudomonas aeruginosa outer membrane from antibiotic treatment and oxidative stress. J. Bacteriol. 2012, 194, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Halverson, T.W.; Wilton, M.; Poon, K.K.; Petri, B.; Lewenza, S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- MacNee, W.; Wiggs, B.; Belzberg, A.S.; Hogg, J.C. The effect of cigarette smoking on neutrophil kinetics in human lungs. N. Engl. J. Med. 1989, 321, 924–928. [Google Scholar] [CrossRef]
- Quint, J.K.; Wedzicha, J.A. The neutrophil in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2007, 119, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Bristot, L.; Capelli, A.; Ricciardolo, F.L.; Magno, F.; D’Anna, S.E.; Zanini, A.; et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 2009, 64, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaron, S.D.; Angel, J.B.; Lunau, M.; Wright, K.; Fex, C.; Le Saux, N.; Dales, R.E. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Beeh, K.M.; Kornmann, O.; Buhl, R.; Culpitt, S.V.; Giembycz, M.A.; Barnes, P.J. Neutrophil chemotactic activity of sputum from patients with COPD: Role of interleukin 8 and leukotriene B4. Chest 2003, 123, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhu, J.; Bandi, V.; Atmar, R.L.; Hattotuwa, K.; Guntupalli, K.K.; Jeffery, P.K. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 168, 968–975. [Google Scholar] [CrossRef]
- Chung, K.F. Inflammatory mediators in chronic obstructive pulmonary disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 619–625. [Google Scholar] [CrossRef]
- Pedersen, F.; Marwitz, S.; Holz, O.; Kirsten, A.; Bahmer, T.; Waschki, B.; Magnussen, H.; Rabe, K.F.; Goldmann, T.; Uddin, M.; et al. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir. Med. 2015, 109, 1360–1362. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.K.; Gibson, P.G.; Simpson, J.L.; McDonald, V.M.; Wood, L.G.; Baines, K.J. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 2016, 21, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G.; et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Williams, N.P.; Coombs, N.A.; Johnson, M.J.; Josephs, L.K.; Rigge, L.A.; Staples, K.J.; Thomas, M.; Wilkinson, T.M. Seasonality, risk factors and burden of community-acquired pneumonia in COPD patients: A population database study using linked health care records. Int. J. Chron. Obs. Pulmon. Dis. 2017, 12, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, A.; Khan, M.A.; Bade, G.; Talwar, A. Orchestration of Neutrophil Extracellular Traps (Nets), a Unique Innate Immune Function during Chronic Obstructive Pulmonary Disease (COPD) Development. Biomedicines 2021, 9, 53. [Google Scholar] [CrossRef]
- Latge, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Liao, W. Fungal respiratory disease. Curr. Opin. Pulm. Med. 2006, 12, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Cunha, C.; Bistoni, F.; Romani, L. Immunotherapy of aspergillosis. Clin. Microbiol. Infect. 2012, 18, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chayakulkeeree, M.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. N. Am. 2006, 20, 507–544. [Google Scholar] [CrossRef]
- Kronstad, J.W.; Attarian, R.; Cadieux, B.; Choi, J.; D’Souza, C.A.; Griffiths, E.J.; Geddes, J.M.; Hu, G.; Jung, W.H.; Kretschmer, M.; et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 2011, 9, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.G.; Perfect, J.R. Cryptococcosis in the era of AIDS--100 years after the discovery of Cryptococcus neoformans. Clin. Microbiol. Rev. 1995, 8, 515–548. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Doshi, A.V.; Bhaduri, A.S. Pneumocystis pneumonia. N. Engl. J. Med. 2004, 351, 1262–1263. [Google Scholar]
- Lortholary, O.; Denning, D.W.; Dupont, B. Endemic mycoses: A treatment update. J. Antimicrob. Chemother. 1999, 43, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheat, J. Endemic mycoses in AIDS: A clinical review. Clin. Microbiol. Rev. 1995, 8, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, T. The genus Aspergillus. Med. Mycol. J. 2011, 52, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Mircescu, M.M.; Lipuma, L.; van Rooijen, N.; Pamer, E.G.; Hohl, T.M. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J. Infect. Dis. 2009, 200, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim-Granet, O.; Philippe, B.; Boleti, H.; Boisvieux-Ulrich, E.; Grenet, D.; Stern, M.; Latge, J.P. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect. Immun. 2003, 71, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Mouyna, I.; Sarfati, J.; Recco, P.; Fontaine, T.; Henrissatz, B.; Latge, J.P. Molecular characterization of a cell wall-associated beta(1-3)endoglucanase of Aspergillus fumigatus. Med. Mycol. 2002, 40, 455–464. [Google Scholar] [CrossRef]
- Bernard, M.; Mouyna, I.; Dubreucq, G.; Debeaupuis, J.P.; Fontaine, T.; Vorgias, C.; Fuglsang, C.; Latge, J.P. Characterization of a cell-wall acid phosphatase (PhoAp) in Aspergillus fumigatus. Microbiology 2002, 148, 2819–2829. [Google Scholar] [CrossRef] [Green Version]
- Latge, J.P. The pathobiology of Aspergillus fumigatus. Trends Microbiol. 2001, 9, 382–389. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywissen, A.; Jeron, A.; Latge, J.P.; Brakhage, A.A.; Gunzer, M. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010, 6, e1000873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, S.; Peri, G.; Delneste, Y.; Fremaux, I.; Doni, A.; Moalli, F.; Garlanda, C.; Romani, L.; Gascan, H.; Bellocchio, S.; et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 2007, 204, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Rohm, M.; Grimm, M.J.; D’Auria, A.C.; Almyroudis, N.G.; Segal, B.H.; Urban, C.F. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect. Immun. 2014, 82, 1766–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alflen, A.; Aranda Lopez, P.; Hartmann, A.K.; Maxeiner, J.; Bosmann, M.; Sharma, A.; Platten, J.; Ries, F.; Beckert, H.; Ruf, W.; et al. Neutrophil extracellular traps impair fungal clearance in a mouse model of invasive pulmonary aspergillosis. Immunobiology 2020, 225, 151867. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; Hoogenboezem, M.; van den Berg, J.M.; Prins, J.M.; Vitkov, L.; van de Veerdonk, F.L.; van den Berg, T.K.; Roos, D.; et al. Human Neutrophils Use Different Mechanisms To Kill Aspergillus fumigatus Conidia and Hyphae: Evidence from Phagocyte Defects. J. Immunol. 2016, 196, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Segal, B.H.; Holland, S.M.; Miller, G.F.; Kwon-Chung, K.J. Virulence of catalase-deficient aspergillus nidulans in p47(phox)-/- mice. Implications for fungal pathogenicity and host defense in chronic granulomatous disease. J. Clin. Invest. 1998, 101, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, D.E.; Gifford, M.A.; Li, L.L.; Doerschuk, C.M.; Dinauer, M.C. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 1997, 185, 207–218. [Google Scholar] [CrossRef]
- Henriet, S.S.; Hermans, P.W.; Verweij, P.E.; Simonetti, E.; Holland, S.M.; Sugui, J.A.; Kwon-Chung, K.J.; Warris, A. Human leukocytes kill Aspergillus nidulans by reactive oxygen species-independent mechanisms. Infect. Immun. 2011, 79, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.; Niemiec, M.J.; Siler, U.; Urban, C.F.; Reichenbach, J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 2011, 127, 1243–1252.e1247. [Google Scholar] [CrossRef] [PubMed]
- Loussert, C.; Schmitt, C.; Prevost, M.C.; Balloy, V.; Fadel, E.; Philippe, B.; Kauffmann-Lacroix, C.; Latge, J.P.; Beauvais, A. In vivo biofilm composition of Aspergillus fumigatus. Cell. Microbiol. 2010, 12, 405–410. [Google Scholar] [CrossRef]
- Fontaine, T.; Delangle, A.; Simenel, C.; Coddeville, B.; van Vliet, S.J.; van Kooyk, Y.; Bozza, S.; Moretti, S.; Schwarz, F.; Trichot, C.; et al. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 2011, 7, e1002372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Geller, A.M.; Bamford, N.C.; Liu, H.; Gravelat, F.N.; Snarr, B.D.; Le Mauff, F.; Chabot, J.; Ralph, B.; Ostapska, H.; et al. Deacetylation of Fungal Exopolysaccharide Mediates Adhesion and Biofilm Formation. mBio 2016, 7, e00252-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravelat, F.N.; Beauvais, A.; Liu, H.; Lee, M.J.; Snarr, B.D.; Chen, D.; Xu, W.; Kravtsov, I.; Hoareau, C.M.; Vanier, G.; et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog. 2013, 9, e1003575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Liu, H.; Barker, B.M.; Snarr, B.D.; Gravelat, F.N.; Al Abdallah, Q.; Gavino, C.; Baistrocchi, S.R.; Ostapska, H.; Xiao, T.; et al. The Fungal Exopolysaccharide Galactosaminogalactan Mediates Virulence by Enhancing Resistance to Neutrophil Extracellular Traps. PLoS Pathog. 2015, 11, e1005187. [Google Scholar] [CrossRef]
- Nasr, S.Z.; Strouse, P.J.; Soskolne, E.; Maxvold, N.J.; Garver, K.A.; Rubin, B.K.; Moler, F.W. Efficacy of recombinant human deoxyribonuclease I in the hospital management of respiratory syncytial virus bronchiolitis. Chest 2001, 120, 203–208. [Google Scholar] [CrossRef]
- Hodson, M.E.; McKenzie, S.; Harms, H.K.; Koch, C.; Mastella, G.; Navarro, J.; Strandvik, B.; Investigators of the Epidemiologic Registry of Cystic Fibrosis. Dornase alfa in the treatment of cystic fibrosis in Europe: A report from the Epidemiologic Registry of Cystic Fibrosis. Pediatr. Pulmonol. 2003, 36, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, B.; Pressler, T.; Hansen, A.; Koch, C.; Hoiby, N. Effect of aerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatr. 2006, 95, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.G.; Chau, A.S.; Egeblad, M.; Barnes, B.J.; Janowitz, T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: A case series. Mol. Med. 2020, 26, 91. [Google Scholar] [CrossRef] [PubMed]
- Desilles, J.P.; Gregoire, C.; Le Cossec, C.; Lambert, J.; Mophawe, O.; Losser, M.R.; Lambiotte, F.; Le Tacon, S.; Cantier, M.; Engrand, N.; et al. Efficacy and safety of aerosolized intra-tracheal dornase alfa administration in patients with SARS-CoV-2-induced acute respiratory distress syndrome (ARDS): A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 548. [Google Scholar] [CrossRef]
- Tsourouktsoglou, T.D.; Warnatsch, A.; Ioannou, M.; Hoving, D.; Wang, Q.; Papayannopoulos, V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020, 31, 107602. [Google Scholar] [CrossRef]
- Wygrecka, M.; Kosanovic, D.; Wujak, L.; Reppe, K.; Henneke, I.; Frey, H.; Didiasova, M.; Kwapiszewska, G.; Marsh, L.M.; Baal, N.; et al. Antihistone Properties of C1 Esterase Inhibitor Protect against Lung Injury. Am. J. Respir. Crit. Care Med. 2017, 196, 186–199. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef] [Green Version]
- Caliezi, C.; Wuillemin, W.A.; Zeerleder, S.; Redondo, M.; Eisele, B.; Hack, C.E. C1-Esterase inhibitor: An anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharm. Rev. 2000, 52, 91–112. [Google Scholar] [PubMed]
- Liu, D.; Cai, S.; Gu, X.; Scafidi, J.; Wu, X.; Davis, A.E., 3rd. C1 inhibitor prevents endotoxin shock via a direct interaction with lipopolysaccharide. J. Immunol. 2003, 171, 2594–2601. [Google Scholar] [CrossRef] [Green Version]
- Zeerleder, S.; Caliezi, C.; van Mierlo, G.; Eerenberg-Belmer, A.; Sulzer, I.; Hack, C.E.; Wuillemin, W.A. Administration of C1 inhibitor reduces neutrophil activation in patients with sepsis. Clin. Diagn. Lab. Immunol. 2003, 10, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Igonin, A.A.; Protsenko, D.N.; Galstyan, G.M.; Vlasenko, A.V.; Khachatryan, N.N.; Nekhaev, I.V.; Shlyapnikov, S.A.; Lazareva, N.B.; Herscu, P. C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Crit. Care Med. 2012, 40, 770–777. [Google Scholar] [CrossRef]
- Deng, Q.; Pan, B.; Alam, H.B.; Liang, Y.; Wu, Z.; Liu, B.; Mor-Vaknin, N.; Duan, X.; Williams, A.M.; Tian, Y.; et al. Citrullinated Histone H3 as a Therapeutic Target for Endotoxic Shock in Mice. Front. Immunol. 2019, 10, 2957. [Google Scholar] [CrossRef] [PubMed]
- Hagio, T.; Matsumoto, S.; Nakao, S.; Matsuoka, S.; Kawabata, K. Sivelestat, a specific neutrophil elastase inhibitor, prevented phorbol myristate acetate-induced acute lung injury in conscious rabbits. Pulm. Pharm. 2005, 18, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Okayama, N.; Kakihana, Y.; Setoguchi, D.; Imabayashi, T.; Omae, T.; Matsunaga, A.; Kanmura, Y. Clinical effects of a neutrophil elastase inhibitor, sivelestat, in patients with acute respiratory distress syndrome. J. Anesth. 2006, 20, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Tamakuma, S.; Ogawa, M.; Aikawa, N.; Kubota, T.; Hirasawa, H.; Ishizaka, A.; Taenaka, N.; Hamada, C.; Matsuoka, S.; Abiru, T. Relationship between neutrophil elastase and acute lung injury in humans. Pulm. Pharm. 2004, 17, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Okayama, Y.; Shime, N.; Kimura, A.; Funakoshi, Y.; Kawabata, K.; Ishizaka, A.; Amaya, F. Neutrophil elastase activity in acute lung injury and respiratory distress syndrome. Respirology 2008, 13, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Tosa, R.; Omura, M.; Fukushima, H.; Kaneko, T.; Endo, T.; Rinka, H.; Murai, A.; Yamaguchi, J.; Yoshikawa, K.; et al. Effect of a selective neutrophil elastase inhibitor on mortality and ventilator-free days in patients with increased extravascular lung water: A post hoc analysis of the PiCCO Pulmonary Edema Study. J. Intensive Care 2014, 2, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, S.; Hamada, H.; Ito, R.; Katayama, H.; Irifune, K.; Suwaki, T.; Nakanishi, N.; Kanematsu, T.; Dote, K.; Aibiki, M.; et al. Usefulness of a selective neutrophil elastase inhibitor, sivelestat, in acute lung injury patients with sepsis. Drug Des. Devel. 2013, 7, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharm. Exp. 2013, 345, 430–437. [Google Scholar] [CrossRef]
- Hirose, T.; Hamaguchi, S.; Matsumoto, N.; Irisawa, T.; Seki, M.; Tasaki, O.; Hosotsubo, H.; Yamamoto, N.; Yamamoto, K.; Akeda, Y.; et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS ONE 2014, 9, e111755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamphier, M.; Zheng, W.; Latz, E.; Spyvee, M.; Hansen, H.; Rose, J.; Genest, M.; Yang, H.; Shaffer, C.; Zhao, Y.; et al. Novel small molecule inhibitors of TLR7 and TLR9: Mechanism of action and efficacy in vivo. Mol. Pharm. 2014, 85, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Gregoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Vargas, A.; Boivin, R.; Cano, P.; Murcia, Y.; Bazin, I.; Lavoie, J.P. Neutrophil extracellular traps are downregulated by glucocorticosteroids in lungs in an equine model of asthma. Respir. Res. 2017, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.C.; Schwertz, H.; Cody, M.J.; Wallace, J.A.; Campbell, R.A.; Vieira-de-Abreu, A.; Araujo, C.V.; Schubert, S.; Harris, E.S.; Rowley, J.W.; et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J. Clin. Invest. 2016, 126, 3783–3798. [Google Scholar] [CrossRef]
- Leaker, B.R.; Barnes, P.J.; O’Connor, B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir. Res. 2013, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Anto-Michel, N.; Blankenbach, H.; Wiedemann, A.; Buscher, K.; Hohmann, J.D.; Lim, B.; Bauml, M.; Marki, A.; Mauler, M.; et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat. Commun. 2018, 9, 525. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic illustration of the essential steps of NET formation. Several pathogens are capable of inducing NET formation directly or via release of peptides or damage associated molecular patterns (DAMPs). Receptors like TLR, FcR or GPCR transmit signals into the cell and activate predominantly the NADPH-oxidase complex (NOX), which subsequently catalyzes production of reactive oxygen species (ROS). In azurophilic granules, NE gets released from the membranes in a ROS-dependent manner and translocates into the nucleus and in parallel degrades actin. NE activity results in the decondensation of chromatin, which is further supported by the PAD4-dependent citrullination of histones. The chromatin, decorated with microbicidal molecules like histones, MPO and NE, is released in the environment. This occurs either in a non-lytic procedure, where DNA fibers are suggested to be released via vesicles, or in a lytic process, followed by the breakdown of the nuclear envelope and the cell membrane, ending with cell death.
Figure 1.
Schematic illustration of the essential steps of NET formation. Several pathogens are capable of inducing NET formation directly or via release of peptides or damage associated molecular patterns (DAMPs). Receptors like TLR, FcR or GPCR transmit signals into the cell and activate predominantly the NADPH-oxidase complex (NOX), which subsequently catalyzes production of reactive oxygen species (ROS). In azurophilic granules, NE gets released from the membranes in a ROS-dependent manner and translocates into the nucleus and in parallel degrades actin. NE activity results in the decondensation of chromatin, which is further supported by the PAD4-dependent citrullination of histones. The chromatin, decorated with microbicidal molecules like histones, MPO and NE, is released in the environment. This occurs either in a non-lytic procedure, where DNA fibers are suggested to be released via vesicles, or in a lytic process, followed by the breakdown of the nuclear envelope and the cell membrane, ending with cell death.

Figure 2.
Schematic illustration of how NETs might contribute to vaso-occlusion and endothelial damage. NETs are able to promote vaso-occlusion, initiated by von Willebrand production in platelets, which induces the upregulation of intercellular adhesion molecule-1 (ICAM-1), further strengthening neutrophil–endothelium interactions. Platelet-dependent NET formation requires the heterodimerized CXCL4 and CCL5, as well as the simultaneous stimulation of GPCRs and integrins. NET-derived NE cleaves the coagulation-inhibiting tissue factor pathway inhibitor (TFPI) and, in parallel, activates platelet receptors to increase platelet accumulation. NET-decorated proteins further contribute to tissue damage. Histones especially exhibit cytotoxic effects, disturbing the endothelial integrity.
Figure 2.
Schematic illustration of how NETs might contribute to vaso-occlusion and endothelial damage. NETs are able to promote vaso-occlusion, initiated by von Willebrand production in platelets, which induces the upregulation of intercellular adhesion molecule-1 (ICAM-1), further strengthening neutrophil–endothelium interactions. Platelet-dependent NET formation requires the heterodimerized CXCL4 and CCL5, as well as the simultaneous stimulation of GPCRs and integrins. NET-derived NE cleaves the coagulation-inhibiting tissue factor pathway inhibitor (TFPI) and, in parallel, activates platelet receptors to increase platelet accumulation. NET-decorated proteins further contribute to tissue damage. Histones especially exhibit cytotoxic effects, disturbing the endothelial integrity.


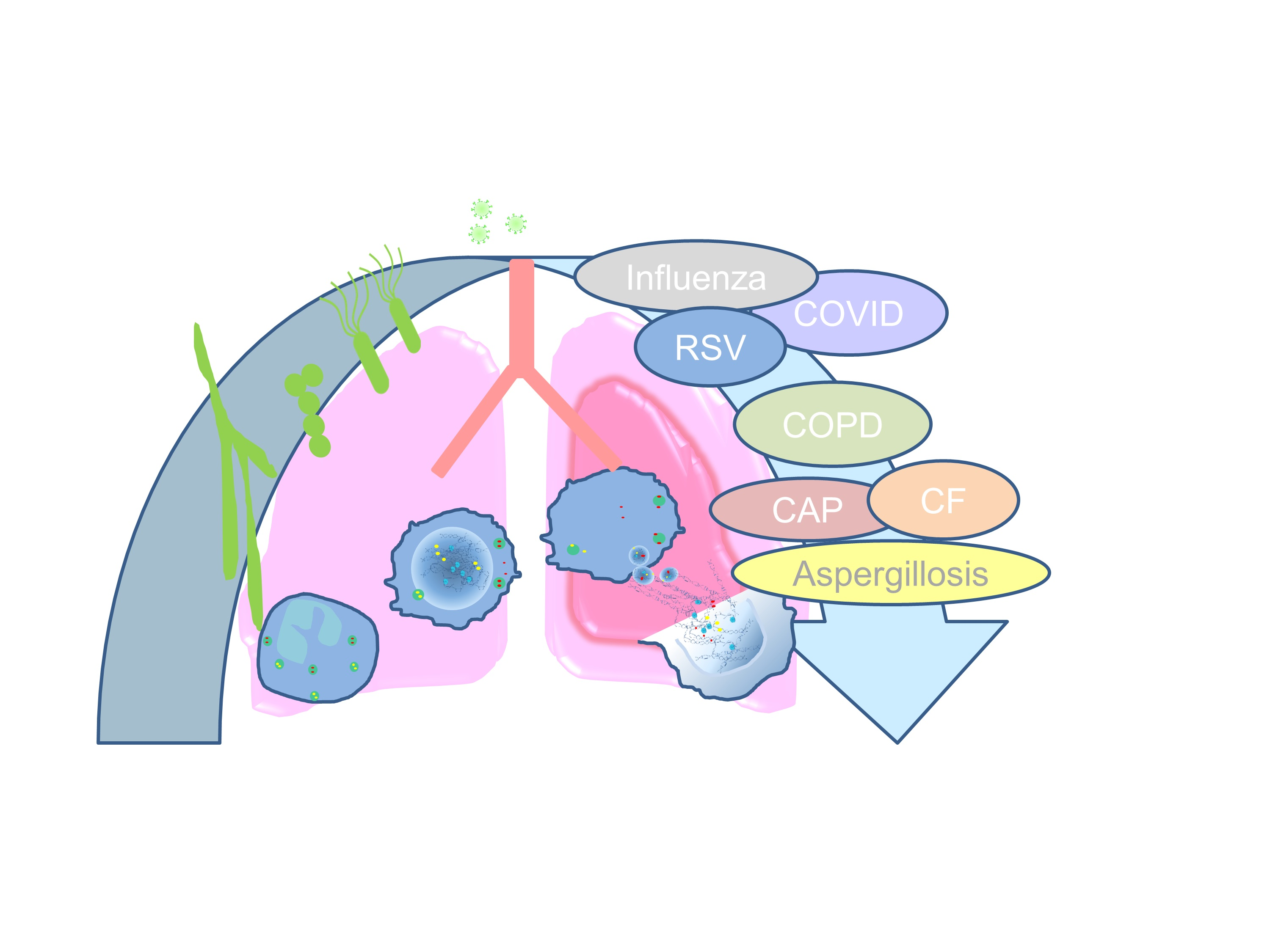{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of NET-targeting therapeutics or compounds in preclinical and clinical applications.
Table 1.
Summary of NET-targeting therapeutics or compounds in preclinical and clinical applications.
| Compound | Target | Application | Reference |
|---|---|---|---|
| Dornase Alfa/DNase | DNA | Bronchiolitis Cystic fibrosis | [211,212,213,214,215] |
| Clinical Phase 2 study: COVID19 | NCT04359654 | ||
| C1 esterase inhibitor | Histones | Sepsis patients | [219,220,221,222] |
| tACPA α-H3-cit | Citrullinated Histones | Inflammatory murine disease models | [218,223] |
| Sivelestat | Neutrophil elastase | ARDS patients ALI patients | [225,226,227,228,229] |
| Clinical phase 4 study: ARDS | NCT00036062 | ||
| Aspirin αCLEC Glucocorticoids NET-inhibiting factors | Inhibition of NET formation | Critically ill patients Inflammatory murine disease models | [230,231,232,236] |
| Metformin | HMGB/NET clearance | Diabetes patients | [233,234] |
| CXCR2 antagonist | Neutrophil recruitment | LPS-challenged humans | [237] |
| CD40L-M7 | Mac1 | Inflammatory murine disease models | [238] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Block, H.; Zarbock, A. A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression? Cells 2021, 10, 1932. https://doi.org/10.3390/cells10081932
AMA Style
Block H, Zarbock A. A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression? Cells. 2021; 10(8):1932. https://doi.org/10.3390/cells10081932
Chicago/Turabian StyleBlock, Helena, and Alexander Zarbock. 2021. "A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression?" Cells 10, no. 8: 1932. https://doi.org/10.3390/cells10081932
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.