
Cyclic AMP Signaling in Biliary Proliferation: A Possible Target for Cholangiocarcinoma Treatment?

 , ,
, ,
Abstract
:1. Introduction
2. cAMP Signaling
3. cAMP Signaling in Hepatocytes
3.1. General Signaling Effects
3.2. cAMP in Liver Diseases with Metabolic Impairment
4. cAMP Signaling in Cholangiocytes
4.1. cAMP and Biliary Secretion
4.2. cAMP and Biliary Proliferation
5. cAMP and Cancer
6. CCA and cAMP Signaling
6.1. CCA
6.2. Hormones/Neuropeptides Modulation of cAMP in CCA
6.3. BAs Modulation of cAMP in CCA
6.4. PKA Subunits Changes in CCA
7. Conclusions/Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
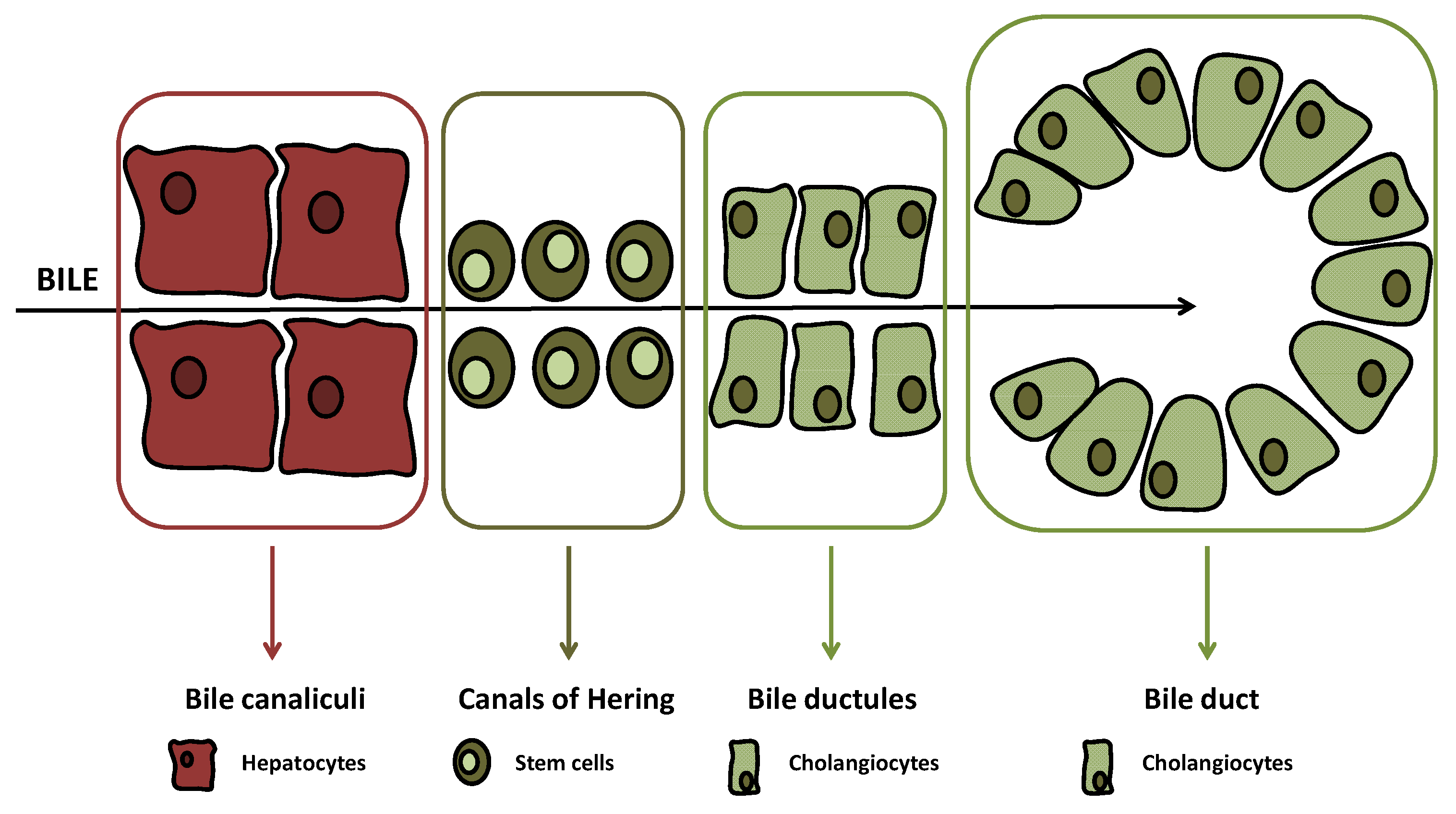
- Han, Y.; Glaser, S.; Meng, F.; Francis, H.; Marzioni, M.; McDaniel, K.; Alvaro, D.; Venter, J.; Carpino, G.; Onori, P.; et al. Recent advances in the morphological and functional heterogeneity of the biliary epithelium. Exp. Biol. Med. 2013, 238, 549–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliwal, J.K.; Ramesh, D.; Gupta, R.C. Synthesis and disposition of 14C-labelled 81/470, a new anthelminthic agent in rats. Int. J. Clin. Pharmacol. Res. 1997, 17, 23–30. [Google Scholar]
- Kanno, N.; LeSage, G.; Glaser, S.; Alvaro, D.; Alpini, G. Functional heterogeneity of the intrahepatic biliary epithelium. Hepatology 2000, 31, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Kanno, N.; LeSage, G.; Glaser, S.; Alpini, G. Regulation of cholangiocyte bicarbonate secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G612–G625. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Glaser, S.; Robertson, W.; Rodgers, R.E.; Phinizy, J.L.; Lasater, J.; LeSage, G.D. Large but not small intrahepatic bile ducts are involved in secretin-regulated ductal bile secretion. Am. J. Physiol. 1997, 272, G1064–G1074. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Baiocchi, L.; Zhou, T.; Kennedy, L.; Ceci, L.; Meng, F.; Sato, K.; Wu, C.; Ekser, B.; Kyritsi, K.; et al. Functional role of the secretin/secretin receptor signaling during cholestatic liver injury. Hepatology 2020, 72, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.; Glaser, S.; DeMorrow, S.; Gaudio, E.; Ueno, Y.; Venter, J.; Dostal, D.; Onori, P.; Franchitto, A.; Marzioni, M.; et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. Am. J. Physiol. Cell Physiol. 2008, 295, C499–C513. [Google Scholar] [CrossRef] [Green Version]
- Mancinelli, R.; Franchitto, A.; Gaudio, E.; Onori, P.; Glaser, S.; Francis, H.; Venter, J.; Demorrow, S.; Carpino, G.; Kopriva, S.; et al. After damage of large bile ducts by gamma-aminobutyric acid, small ducts replenish the biliary tree by amplification of calcium-dependent signaling and de novo acquisition of large cholangiocyte phenotypes. Am. J. Pathol. 2010, 176, 1790–1800. [Google Scholar] [CrossRef]
- LeSage, G.; Benedetti, A.; Glaser, S.; Marucci, L.; Tretjak, Z.; Caligiuri, A.; Rodgers, R.; Phinizy, J.L.; Baiocchi, L.; Francis, H.; et al. Acute carbon tetrachloride feeding selectively damages large, but not small, cholangiocytes from normal rat liver. Hepatology 1999, 29, 307–319. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular reaction in liver diseases: Pathological mechanisms and translational significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Guicciardi, M.E.; Trussoni, C.E.; LaRusso, N.F.; Gores, G.J. The spectrum of reactive cholangiocytes in primary sclerosing cholangitis. Hepatology 2020, 71, 741–748. [Google Scholar] [CrossRef]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef]
- Lee, S.J.; Park, J.B.; Kim, K.H.; Lee, W.R.; Kim, J.Y.; An, H.J.; Park, K.K. Immunohistochemical study for the origin of ductular reaction in chronic liver disease. Int. J. Clin. Exp. Pathol. 2014, 7, 4076–4085. [Google Scholar] [PubMed]
- LeSage, G.; Glaser, S.; Alpini, G. Regulation of cholangiocyte proliferation. Liver 2001, 21, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Gigliozzi, A.; Attili, A.F. Regulation and deregulation of cholangiocyte proliferation. J. Hepatol. 2000, 33, 333–340. [Google Scholar] [CrossRef]
- Sato, K.; Francis, H.; Zhou, T.; Meng, F.; Kennedy, L.; Ekser, B.; Baiocchi, L.; Onori, P.; Mancinelli, R.; Gaudio, E.; et al. Neuroendocrine changes in cholangiocarcinoma growth. Cells 2020, 9, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef]
- Hall, C.; Sato, K.; Wu, N.; Zhou, T.; Kyritsi, K.; Meng, F.; Glaser, S.; Alpini, G. Regulators of cholangiocyte proliferation. Gene Expr. 2017, 17, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, E.W.; Rall, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, K.V.; Yang, W.L.; Ravatn, R.; Kita, T.; Reitman, E.; Vettori, D.; Cvijic, M.E.; Shin, M.; Iacono, L. Reinventing the wheel of cyclic AMP: Novel mechanisms of cAMP signaling. Ann. N. Y. Acad. Sci. 2002, 968, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Kleinboelting, S.; Diaz, A.; Moniot, S.; van den Heuvel, J.; Weyand, M.; Levin, L.R.; Buck, J.; Steegborn, C. Crystal structures of human soluble adenylyl cyclase reveal mechanisms of catalysis and of its activation through bicarbonate. Proc. Natl. Acad. Sci. USA 2014, 111, 3727–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.S.; Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Sowadski, J.M. Structural framework for the protein kinase family. Annu. Rev. Cell Biol. 1992, 8, 429–462. [Google Scholar] [CrossRef] [PubMed]
- Tasken, K.; Skalhegg, B.S.; Solberg, R.; Andersson, K.B.; Taylor, S.S.; Lea, T.; Blomhoff, H.K.; Jahnsen, T.; Hansson, V. Novel isozymes of cAMP-dependent protein kinase exist in human cells due to formation of RI alpha-RI beta heterodimeric complexes. J. Biol. Chem. 1993, 268, 21276–21283. [Google Scholar] [CrossRef]
- Rich, T.C.; Fagan, K.A.; Tse, T.E.; Schaack, J.; Cooper, D.M.; Karpen, J.W. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA 2001, 98, 13049–13054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kong, Q.; Wang, J.; Jiang, Y.; Hua, H. Complex roles of cAMP-PKA-CREB signaling in cancer. Exp. Hematol. Oncol. 2020, 9, 32. [Google Scholar] [CrossRef]
- Akimoto, M.; VanSchouwen, B.; Melacini, G. The structure of the apo cAMP-binding domain of HCN4—A stepping stone toward understanding the cAMP-dependent modulation of the hyperpolarization-activated cyclic-nucleotide-gated ion channels. FEBS J. 2018, 285, 2182–2192. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, L. Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J. Mol. Endocrinol. 2016, 57, R93–R108. [Google Scholar] [CrossRef] [Green Version]
- Jitrapakdee, S. Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 2012, 44, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Lent, B.A.; Kim, K.H. Phosphorylation and activation of acetyl-coenzyme A Carboxylase kinase by the catalytic subunit of cyclic AMP-dependent protein kinase. Arch. Biochem. Biophys. 1983, 225, 972–978. [Google Scholar] [CrossRef]
- Barlas, N.; Mutchnick, M.G.; Grant, G.J.; Trainin, N. The effect of thymic humoral factor on intracellular lymphocyte cyclic AMP in alcoholic liver disease. Thymus 1983, 5, 433–437. [Google Scholar] [PubMed]
- Gouillon, Z.Q.; Miyamoto, K.; Donohue, T.M.; Wan, Y.J.; French, B.A.; Nagao, Y.; Fu, P.; Reitz, R.C.; Hagbjork, A.; Yap, C.; et al. Role of CYP2E1 in the pathogenesis of alcoholic liver disease: Modifications by cAMP and ubiquitin-proteasome pathway. Front. Biosci. 1999, 4, A16–A25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila, D.V.; Barker, D.F.; Zhang, J.; McClain, C.J.; Barve, S.; Gobejishvili, L. Dysregulation of hepatic cAMP levels via altered Pde4b expression plays a critical role in alcohol-induced steatosis. J. Pathol. 2016, 240, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Gobejishvili, L.; Barve, S.; Joshi-Barve, S.; Uriarte, S.; Song, Z.; McClain, C. Chronic ethanol-mediated decrease in cAMP primes macrophages to enhanced LPS-inducible NF-kappaB activity and TNF expression: Relevance to alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G681–G688. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Nelson, V.K.; Mukhopadhyay, S.; Bandhopadhyay, S.; Maganti, L.; Ghoshal, N.; Sen, G.; Biswas, T. 14-Deoxyandrographolide targets adenylate cyclase and prevents ethanol-induced liver injury through constitutive NOS dependent reduced redox signaling in rats. Food Chem. Toxicol. 2013, 59, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Roth, K.; Agarwal, M.; Liu, W.; Petriello, M.C. The transcription factors CREBH, PPARa, and FOXO1 as critical hepatic mediators of diet-induced metabolic dysregulation. J. Nutr. Biochem. 2021, 95, 108633. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, S.; Tan, M. bZIP transmembrane transcription factor CREBH: Potential role in non-alcoholic fatty liver disease (Review). Mol. Med. Rep. 2016, 13, 1455–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Fazia, M.A.; Servillo, G.; Sassone-Corsi, P. Cyclic AMP signalling and cellular proliferation: Regulation of CREB and CREM. FEBS Lett. 1997, 410, 22–24. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Xia, J.; Zhou, Y.; Lu, X.; Zhang, L.; Gou, M.; Li, L.; Zhang, X.; Ji, H.; Zhu, K.; et al. Loss of Gsalpha impairs liver regeneration through a defect in the crosstalk between cAMP and growth factor signaling. J. Hepatol. 2016, 64, 342–351. [Google Scholar] [CrossRef]
- Lenzen, R.; Alpini, G.; Tavoloni, N. Secretin stimulates bile ductular secretory activity through the cAMP system. Am. J. Physiol. 1992, 263, G527–G532. [Google Scholar] [CrossRef]
- Kato, A.; Gores, G.J.; LaRusso, N.F. Secretin stimulates exocytosis in isolated bile duct epithelial cells by a cyclic AMP-mediated mechanism. J. Biol. Chem. 1992, 267, 15523–15529. [Google Scholar] [CrossRef]
- Farouk, M.; Vigna, S.R.; McVey, D.C.; Meyers, W.C. Localization and characterization of secretin binding sites expressed by rat bile duct epithelium. Gastroenterology 1992, 102, 963–968. [Google Scholar] [CrossRef]
- Medina, J.F. Role of the anion exchanger 2 in the pathogenesis and treatment of primary biliary cirrhosis. Dig. Dis. 2011, 29, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Ulrich, C.; Roberts, S.; Phillips, J.O.; Ueno, Y.; Podila, P.V.; Colegio, O.; LeSage, G.D.; Miller, L.J.; LaRusso, N.F. Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation. Am. J. Physiol. 1997, 272, G289–G297. [Google Scholar] [CrossRef]
- LeSage, G.; Marucci, L.; Alvaro, D.; Glaser, S.S.; Benedetti, A.; Marzioni, M.; Patel, T.; Francis, H.; Phinizy, J.L.; Alpini, G. Insulin inhibits secretin-induced ductal secretion by activation of PKC alpha and inhibition of PKA activity. Hepatology 2002, 36, 641–651. [Google Scholar] [CrossRef]
- Francis, H.; Glaser, S.; Ueno, Y.; Lesage, G.; Marucci, L.; Benedetti, A.; Taffetani, S.; Marzioni, M.; Alvaro, D.; Venter, J.; et al. cAMP stimulates the secretory and proliferative capacity of the rat intrahepatic biliary epithelium through changes in the PKA/Src/MEK/ERK1/2 pathway. J. Hepatol. 2004, 41, 528–537. [Google Scholar] [CrossRef]
- Francis, H.L.; Demorrow, S.; Franchitto, A.; Venter, J.K.; Mancinelli, R.A.; White, M.A.; Meng, F.; Ueno, Y.; Carpino, G.; Renzi, A.; et al. Histamine stimulates the proliferation of small and large cholangiocytes by activation of both IP3/Ca2+ and cAMP-dependent signaling mechanisms. Lab. Investig. 2012, 92, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Alpini, G.; Ulrich, C.D., 2nd; Phillips, J.O.; Pham, L.D.; Miller, L.J.; LaRusso, N.F. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am. J. Physiol. 1994, 266, G922–G928. [Google Scholar] [CrossRef]
- Guerrier, M.; Attili, F.; Alpini, G.; Glaser, S. Prolonged administration of secretin to normal rats increases biliary proliferation and secretin-induced ductal secretory activity. Hepatobiliary Surg. Nutr. 2014, 3, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.; Lam, I.P.; Franchitto, A.; Gaudio, E.; Onori, P.; Chow, B.K.; Wise, C.; Kopriva, S.; Venter, J.; White, M.; et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 2010, 52, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Marzioni, M.; Alpini, G.; Saccomanno, S.; Candelaresi, C.; Venter, J.; Rychlicki, C.; Fava, G.; Francis, H.; Trozzi, L.; Glaser, S.; et al. Glucagon-like peptide-1 and its receptor agonist exendin-4 modulate cholangiocyte adaptive response to cholestasis. Gastroenterology 2007, 133, 244–255. [Google Scholar] [CrossRef]
- Glaser, S.; Benedetti, A.; Marucci, L.; Alvaro, D.; Baiocchi, L.; Kanno, N.; Caligiuri, A.; Phinizy, J.L.; Chowdury, U.; Papa, E.; et al. Gastrin inhibits cholangiocyte growth in bile duct-ligated rats by interaction with cholecystokinin-B/Gastrin receptors via D-myo-inositol 1,4,5-triphosphate-, Ca(2+)-, and protein kinase C alpha-dependent mechanisms. Hepatology 2000, 32, 17–25. [Google Scholar] [CrossRef]
- Tietz, P.S.; Alpini, G.; Pham, L.D.; Larusso, N.F. Somatostatin inhibits secretin-induced ductal hypercholeresis and exocytosis by cholangiocytes. Am. J. Physiol. 1995, 269, G110–G118. [Google Scholar] [CrossRef]
- Alpini, G.; Glaser, S.S.; Ueno, Y.; Pham, L.; Podila, P.V.; Caligiuri, A.; LeSage, G.; LaRusso, N.F. Heterogeneity of the proliferative capacity of rat cholangiocytes after bile duct ligation. Am. J. Physiol 1998, 274, G767–G775. [Google Scholar] [CrossRef] [PubMed]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef] [PubMed]
- LeSage, G.; Alvaro, D.; Benedetti, A.; Glaser, S.; Marucci, L.; Baiocchi, L.; Eisel, W.; Caligiuri, A.; Phinizy, J.L.; Rodgers, R.; et al. Cholinergic system modulates growth, apoptosis, and secretion of cholangiocytes from bile duct-ligated rats. Gastroenterology 1999, 117, 191–199. [Google Scholar] [CrossRef]
- Francis, H.; Franchitto, A.; Ueno, Y.; Glaser, S.; DeMorrow, S.; Venter, J.; Gaudio, E.; Alvaro, D.; Fava, G.; Marzioni, M.; et al. H3 histamine receptor agonist inhibits biliary growth of BDL rats by downregulation of the cAMP-dependent PKA/ERK1/2/ELK-1 pathway. Lab. Investig. 2007, 87, 473–487. [Google Scholar] [CrossRef] [Green Version]
- Baiocchi, L.; Zhou, T.; Liangpunsakul, S.; Ilaria, L.; Milana, M.; Meng, F.; Kennedy, L.; Kusumanchi, P.; Yang, Z.; Ceci, L.; et al. Possible application of melatonin treatment in human diseases of the biliary tract. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G651–G660. [Google Scholar] [CrossRef]
- Deutschmann, K.; Reich, M.; Klindt, C.; Droge, C.; Spomer, L.; Haussinger, D.; Keitel, V. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1319–1325. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Huang, B.Q.; Radtke, B.N.; Gajdos, G.B.; Splinter, P.L.; Masyuk, T.V.; Gradilone, S.A.; LaRusso, N.F. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1013–G1024. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Glaser, S.; Robertson, W.; Phinizy, J.L.; Rodgers, R.E.; Caligiuri, A.; LeSage, G. Bile acids stimulate proliferative and secretory events in large but not small cholangiocytes. Am. J. Physiol. 1997, 273, G518–G529. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Glaser, S.S.; Ueno, Y.; Rodgers, R.; Phinizy, J.L.; Francis, H.; Baiocchi, L.; Holcomb, L.A.; Caligiuri, A.; LeSage, G.D. Bile acid feeding induces cholangiocyte proliferation and secretion: Evidence for bile acid-regulated ductal secretion. Gastroenterology 1999, 116, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, D.; Rahal, H.; Jimenez, M.; Viramontes, M.; Choi, G.; Saab, S. Treatment of primary biliary cholangitis ursodeoxycholic acid non-responders: A systematic review. Liver Int. 2017, 37, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Baiocchi, L.; Glaser, S.; Ueno, Y.; Marzioni, M.; Francis, H.; Phinizy, J.L.; Angelico, M.; Lesage, G. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology 2002, 35, 1041–1052. [Google Scholar] [CrossRef]
- Caretta, A.; Mucignat-Caretta, C. Protein kinase a in cancer. Cancers 2011, 3, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Cho-Chung, Y.S.; Nesterova, M.; Becker, K.G.; Srivastava, R.; Park, Y.G.; Lee, Y.N.; Cho, Y.S.; Kim, M.K.; Neary, C.; Cheadle, C. Dissecting the circuitry of protein kinase A and cAMP signaling in cancer genesis: Antisense, microarray, gene overexpression, and transcription factor decoy. Ann. N. Y. Acad. Sci. 2002, 968, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Hinton, D.R.; Zidovetzki, R.; Hofman, F.M. Up-regulation of the cAMP/PKA pathway inhibits proliferation, induces differentiation, and leads to apoptosis in malignant gliomas. Lab. Investig. 1998, 78, 165–174. [Google Scholar]
- Moon, E.Y.; Lee, G.H.; Lee, M.S.; Kim, H.M.; Lee, J.W. Phosphodiesterase inhibitors control A172 human glioblastoma cell death through cAMP-mediated activation of protein kinase A and Epac1/Rap1 pathways. Life Sci. 2012, 90, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, D.; Zhou, Q.; Chen, T.; Ibe, J.C.; Raj, J.U.; Zhou, G. cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial-mesenchymal transition, migration, and invasion in lung cancer cells. Cell Signal. 2012, 24, 2396–2406. [Google Scholar] [CrossRef]
- Pollack, A.; Bae, K.; Khor, L.Y.; Al-Saleem, T.; Hammond, M.E.; Venkatesan, V.; Byhardt, R.W.; Asbell, S.O.; Shipley, W.U.; Sandler, H.M. The importance of protein kinase A in prostate cancer: Relationship to patient outcome in Radiation Therapy Oncology Group trial 92-02. Clin. Cancer Res. 2009, 15, 5478–5484. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Watkins, A.A.; Freeman, B.B.; Meyers, J.A.; Rifkin, I.R.; Lerner, A. Inhibition of type 4 cyclic nucleotide phosphodiesterase blocks intracellular TLR signaling in chronic lymphocytic leukemia and normal hematopoietic cells. J. Immunol. 2015, 194, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.R.; Resnick, D.Z.; Niewiadomski, P.; Dong, H.; Liau, L.M.; Waschek, J.A. Pituitary adenylyl cyclase activating polypeptide inhibits gli1 gene expression and proliferation in primary medulloblastoma derived tumorsphere cultures. BMC Cancer 2010, 10, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkle, D.; Hoffmann, R. Roles of cAMP and cAMP-dependent protein kinase in the progression of prostate cancer: Cross-talk with the androgen receptor. Cell Signal. 2011, 23, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Yokozaki, H.; Budillon, A.; Tortora, G.; Meissner, S.; Beaucage, S.L.; Miki, K.; Cho-Chung, Y.S. An antisense oligodeoxynucleotide that depletes RI alpha subunit of cyclic AMP-dependent protein kinase induces growth inhibition in human cancer cells. Cancer Res. 1993, 53, 868–872. [Google Scholar]
- Agrawal, S.; Kandimalla, E.R.; Yu, D.; Ball, R.; Lombardi, G.; Lucas, T.; Dexter, D.L.; Hollister, B.A.; Chen, S.F. GEM 231, a second-generation antisense agent complementary to protein kinase A RIalpha subunit, potentiates antitumor activity of irinotecan in human colon, pancreas, prostate and lung cancer xenografts. Int. J. Oncol. 2002, 21, 65–72. [Google Scholar]
- Goel, S.; Desai, K.; Macapinlac, M.; Wadler, S.; Goldberg, G.; Fields, A.; Einstein, M.; Volterra, F.; Wong, B.; Martin, R.; et al. A phase I safety and dose escalation trial of docetaxel combined with GEM231, a second generation antisense oligonucleotide targeting protein kinase A R1alpha in patients with advanced solid cancers. Investig. New Drugs 2006, 24, 125–134. [Google Scholar] [CrossRef]
- Rohlff, C.; Clair, T.; Cho-Chung, Y.S. 8-Cl-cAMP induces truncation and down-regulation of the RI alpha subunit and up-regulation of the RII beta subunit of cAMP-dependent protein kinase leading to type II holoenzyme-dependent growth inhibition and differentiation of HL-60 leukemia cells. J. Biol. Chem. 1993, 268, 5774–5782. [Google Scholar] [CrossRef]
- Katsaros, D.; Tortora, G.; Tagliaferri, P.; Clair, T.; Ally, S.; Neckers, L.; Robins, R.K.; Cho-Chung, Y.S. Site-selective cyclic AMP analogs provide a new approach in the control of cancer cell growth. FEBS Lett. 1987, 223, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Man, S.; Lu, Y.; Yin, L.; Cheng, X.; Ma, L. Potential and promising anticancer drugs from adenosine and its analogs. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. S1), 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. S1), 7–18. [Google Scholar] [CrossRef] [Green Version]
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef]
- Baiocchi, L.; Sato, K.; Ekser, B.; Kennedy, L.; Francis, H.; Ceci, L.; Lenci, I.; Alvaro, D.; Franchitto, A.; Onori, P.; et al. Cholangiocarcinoma: Bridging the translational gap from preclinical to clinical development and implications for future therapy. Expert Opin. Investig. Drugs 2021, 30, 365–375. [Google Scholar] [CrossRef]
- Munshi, M.K.; Priester, S.; Gaudio, E.; Yang, F.; Alpini, G.; Mancinelli, R.; Wise, C.; Meng, F.; Franchitto, A.; Onori, P.; et al. Regulation of biliary proliferation by neuroendocrine factors: Implications for the pathogenesis of cholestatic liver diseases. Am. J. Pathol. 2011, 178, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
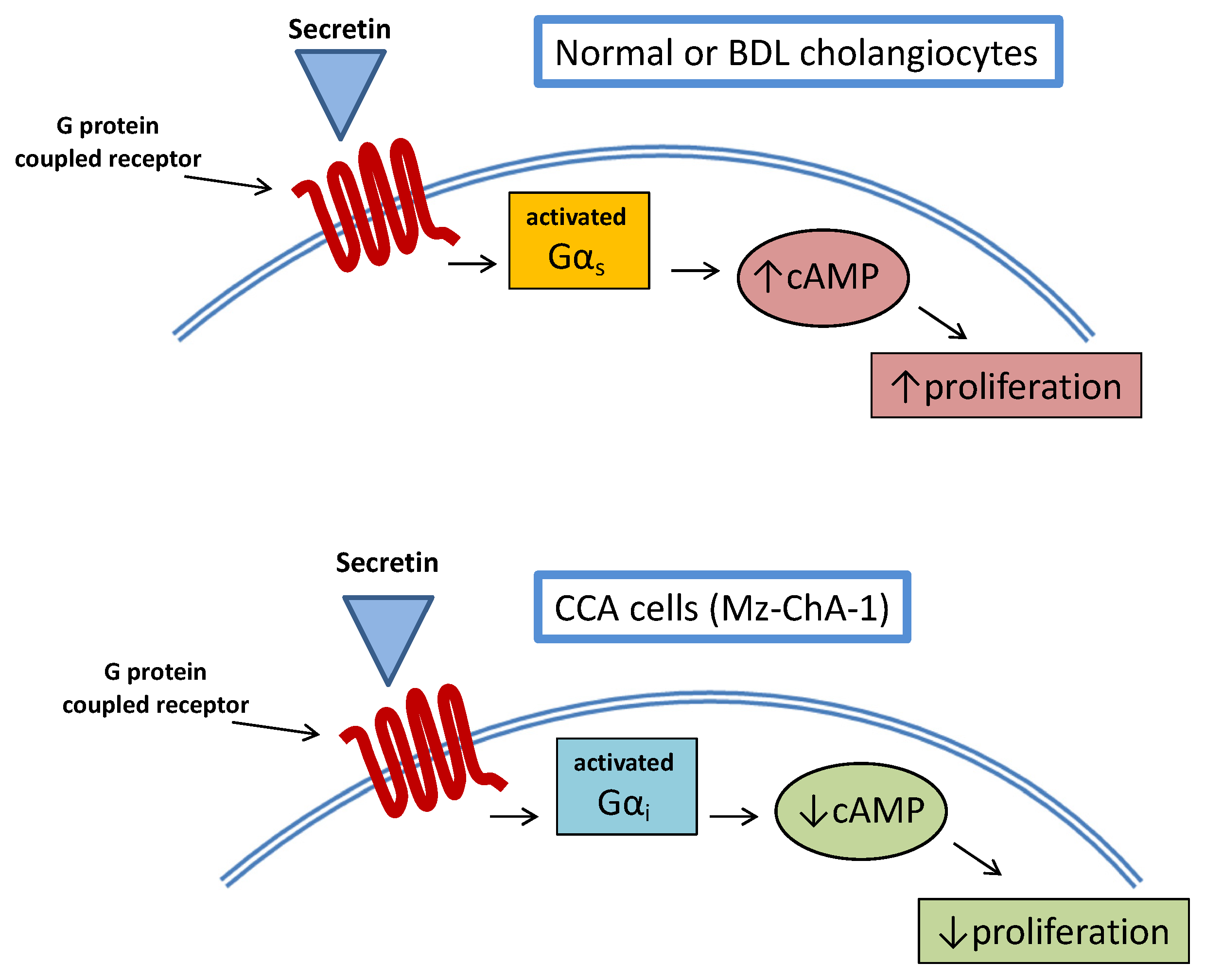
- Onori, P.; Wise, C.; Gaudio, E.; Franchitto, A.; Francis, H.; Carpino, G.; Lee, V.; Lam, I.; Miller, T.; Dostal, D.E.; et al. Secretin inhibits cholangiocarcinoma growth via dysregulation of the cAMP-dependent signaling mechanisms of secretin receptor. Int. J. Cancer 2010, 127, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Podila, P.V.; Taylor, J.E.; Nagorney, D.M.; Wiseman, G.A.; Gores, G.J.; LaRusso, N.F. Human cholangiocarcinomas express somatostatin receptors and respond to somatostatin with growth inhibition. Gastroenterology 1995, 108, 1908–1916. [Google Scholar] [CrossRef]
- Kanno, N.; Lesage, G.; Phinizy, J.L.; Glaser, S.; Francis, H.; Alpini, G. Stimulation of alpha2-adrenergic receptor inhibits cholangiocarcinoma growth through modulation of Raf-1 and B-Raf activities. Hepatology 2002, 35, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Fava, G.; Marucci, L.; Glaser, S.; Francis, H.; De Morrow, S.; Benedetti, A.; Alvaro, D.; Venter, J.; Meininger, C.; Patel, T.; et al. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005, 65, 11437–11446. [Google Scholar] [CrossRef] [Green Version]
- Reich, M.; Deutschmann, K.; Sommerfeld, A.; Klindt, C.; Kluge, S.; Kubitz, R.; Ullmer, C.; Knoefel, W.T.; Herebian, D.; Mayatepek, E.; et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut 2016, 65, 487–501. [Google Scholar] [CrossRef]
- Li, A.D.; Xie, X.L.; Qi, W.; Wang, W.B.; Ma, J.J.; Zhao, D.Q.; Jiang, X.Y.; Chen, L.; Bai, Y.; Jiang, H.Q. TGR5 promotes cholangiocarcinoma by interacting with mortalin. Exp. Cell Res. 2020, 389, 111855. [Google Scholar] [CrossRef] [PubMed]
- Erice, O.; Labiano, I.; Arbelaiz, A.; Santos-Laso, A.; Munoz-Garrido, P.; Jimenez-Aguero, R.; Olaizola, P.; Caro-Maldonado, A.; Martin-Martin, N.; Carracedo, A.; et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1335–1344. [Google Scholar] [CrossRef]
- Loilome, W.; Juntana, S.; Namwat, N.; Bhudhisawasdi, V.; Puapairoj, A.; Sripa, B.; Miwa, M.; Saya, H.; Riggins, G.J.; Yongvanit, P. PRKAR1A is overexpressed and represents a possible therapeutic target in human cholangiocarcinoma. Int. J. Cancer 2011, 129, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Panya, A.; Thepmalee, C.; Sawasdee, N.; Sujjitjoon, J.; Phanthaphol, N.; Junking, M.; Wongkham, S.; Yenchitsomanus, P.T. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol. Immunother. 2018, 67, 1579–1588. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. Emerging molecular therapeutic targets for cholangiocarcinoma. J. Hepatol. 2017, 67, 632–644. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Vyas, M.; Hechtman, J.F.; Zhang, Y.; Benayed, R.; Yavas, A.; Askan, G.; Shia, J.; Klimstra, D.S.; Basturk, O. DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod. Pathol. 2020, 33, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Wood, L.D.; Parks, E.; Torbenson, M.S.; Felsenstein, M.; Hruban, R.H.; Nikiforova, M.N.; Wald, A.I.; Kaya, C.; Nikiforov, Y.E.; et al. Recurrent rearrangements in PRKACA and PRKACB in intraductal oncocytic papillary neoplasms of the pancreas and bile duct. Gastroenterology 2020, 158, 573–582 e572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, J.W.; Borbath, I.; Khan, S.A.; Huguet, F.; Gruenberger, T.; Arnold, D.; Committee, E.G. Biliary cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v28–v37. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author Reference | Target | Experimental Setting | cAMP Levels | Results |
|---|---|---|---|---|
| Tan, C.K. [91] | Stimulation of somatostatin receptor (SSTR2) | CCA cell culture Human CCA specimens CCA xenotransplantation in nude mice | Unchanged | Reduced cancer growth |
| Kanno, N. [92] | Stimulation of α2-adrenergic receptor | CCA cell culture | Increased | Reduced cancer growth |
| Fava, G. [93] | Stimulation of γ-aminobutyric acid (GABA) receptor | CCA cell culture CCA xenotransplantation in nude mice | Increased | Reduced cancer growth and spread |
| Onori, P. [90] | Stimulation of secretin receptor | CCA cell culture Human CCA specimens CCA xenotransplantation in nude mice | Unchanged | Reduced cancer growth |
| Reich, M. [94] | Stimulation of Takeda G-protein-coupled receptor 5 (TGR5), receptor | CCA cell culture CCA xenotransplantation in nude mice | Increased | Enhanced cancer growth, reduced apoptosis |
| Loilome, W. [97] | PKA subunit RIα suppression | RIα knockdown in CCA cell culture | Not assessed | Reduced cancer growth |
| Panya, A. [98] | PKA subunit RIα suppression | T-cells against RIα subunit in CCA cell culture | Not assessed | Reduced cancer growth |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baiocchi, L.; Lenci, I.; Milana, M.; Kennedy, L.; Sato, K.; Zhang, W.; Ekser, B.; Ceci, L.; Meadows, V.; Glaser, S.; et al. Cyclic AMP Signaling in Biliary Proliferation: A Possible Target for Cholangiocarcinoma Treatment? Cells 2021, 10, 1692. https://doi.org/10.3390/cells10071692
Baiocchi L, Lenci I, Milana M, Kennedy L, Sato K, Zhang W, Ekser B, Ceci L, Meadows V, Glaser S, et al. Cyclic AMP Signaling in Biliary Proliferation: A Possible Target for Cholangiocarcinoma Treatment? Cells. 2021; 10(7):1692. https://doi.org/10.3390/cells10071692
Chicago/Turabian StyleBaiocchi, Leonardo, Ilaria Lenci, Martina Milana, Lindsey Kennedy, Keisaku Sato, Wenjun Zhang, Burcin Ekser, Ludovica Ceci, Vik Meadows, Shannon Glaser, and et al. 2021. "Cyclic AMP Signaling in Biliary Proliferation: A Possible Target for Cholangiocarcinoma Treatment?" Cells 10, no. 7: 1692. https://doi.org/10.3390/cells10071692