
Effects of FGFR Tyrosine Kinase Inhibition in OLN-93 Oligodendrocytes

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
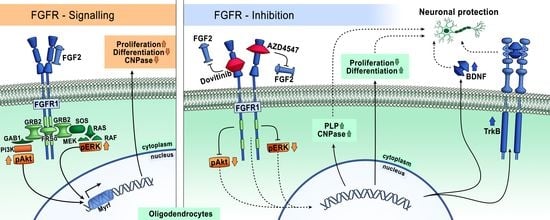
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Experimental Treatment of Cells
2.3. Proliferation Measurement
2.4. Cytotoxicity Assessment
2.5. Western Blotting and Antibodies
2.6. RNA Analysis
2.7. Immunofluorescence
2.8. Statistical Analysis
3. Results
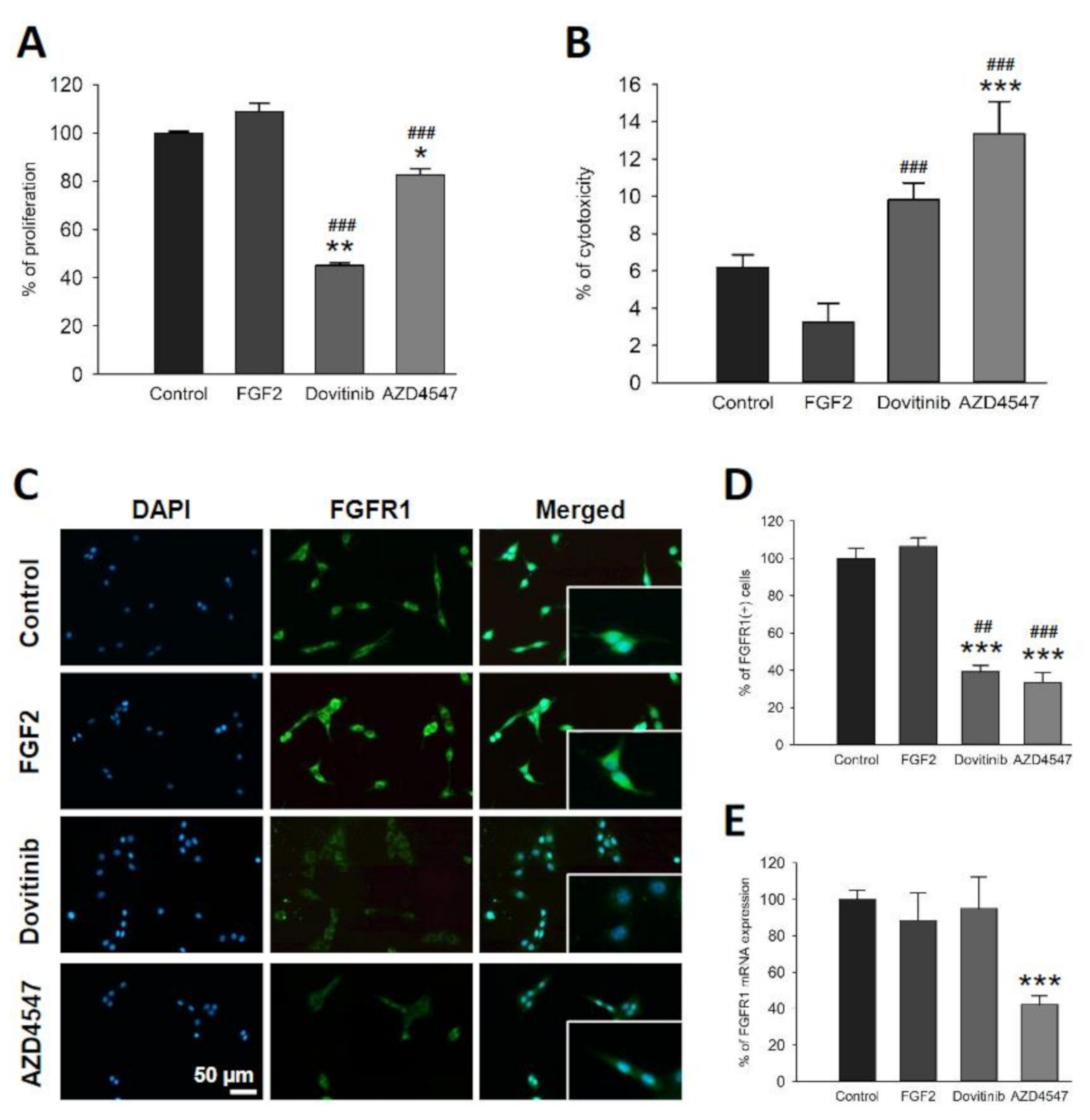
3.1. FGFR Inhibition Reduces Proliferation of OLN-93 Cells
3.2. Inhibition of FGFR Is Cytotoxic to OLN-93 Cells
3.3. FGFR1 Expression Is Reduced by FGFR Inhibition
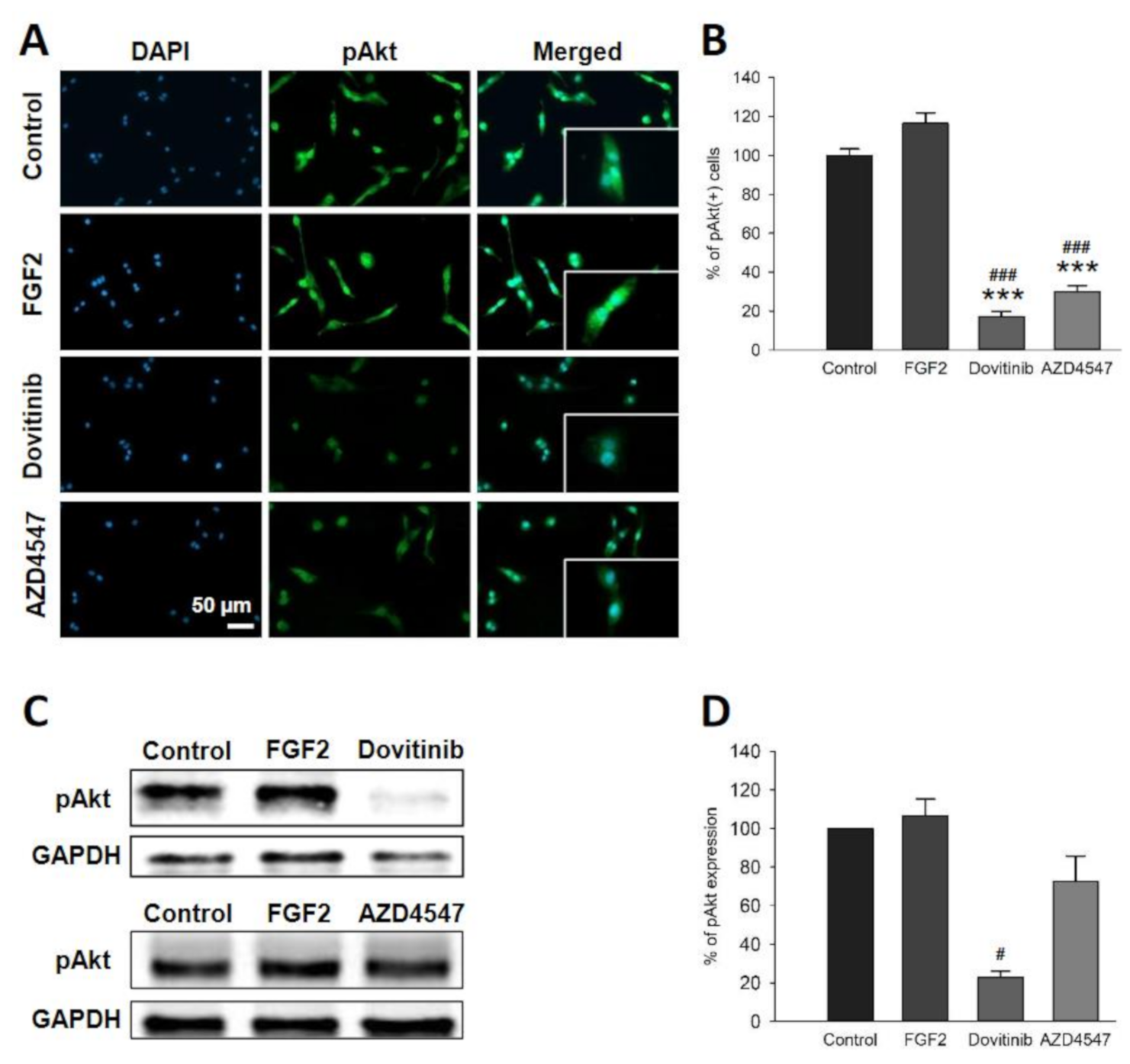
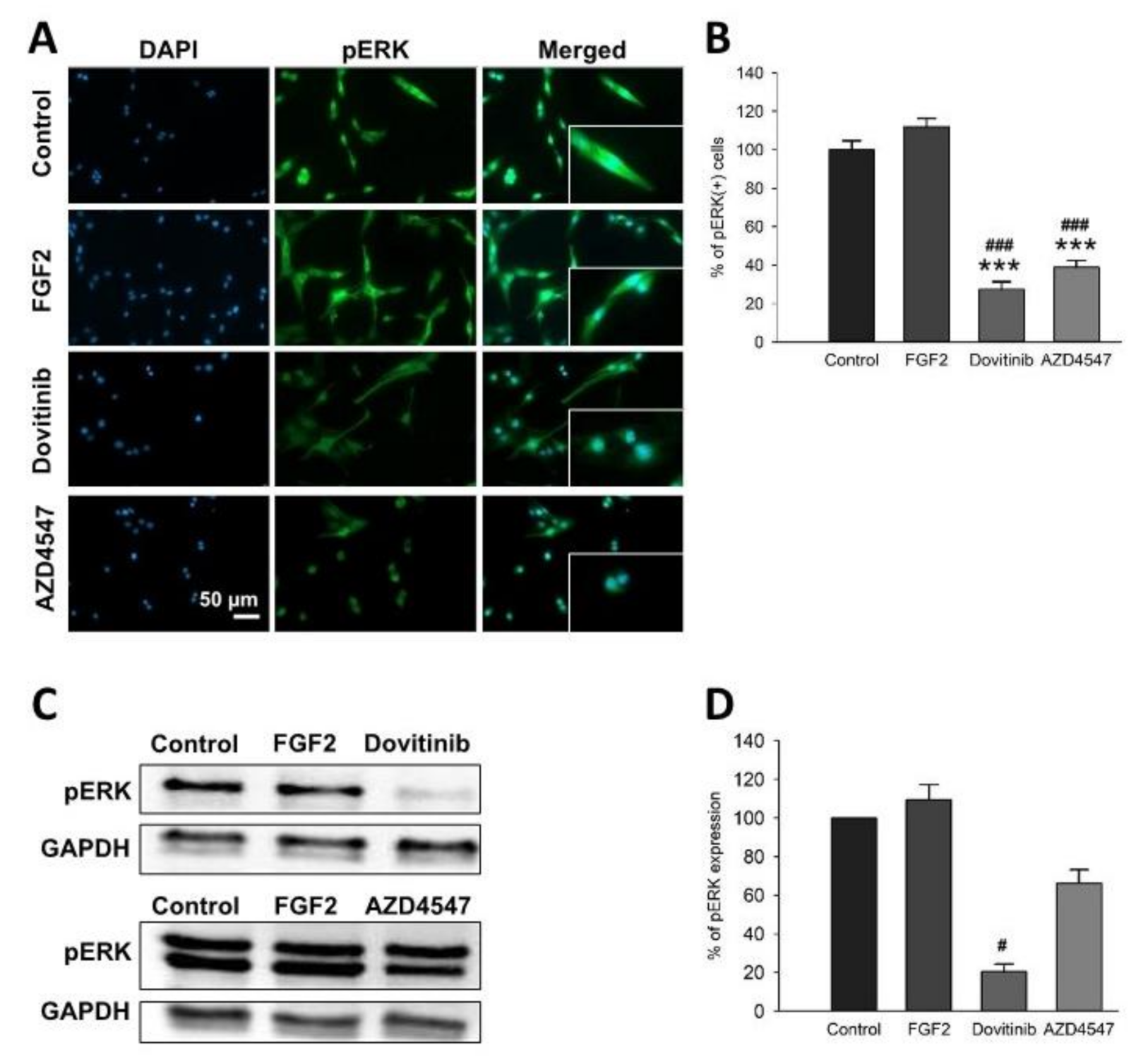
3.4. FGFR Inhibition Decreased Phosphorylation of FGFR Downstream Signaling via Akt and ERK
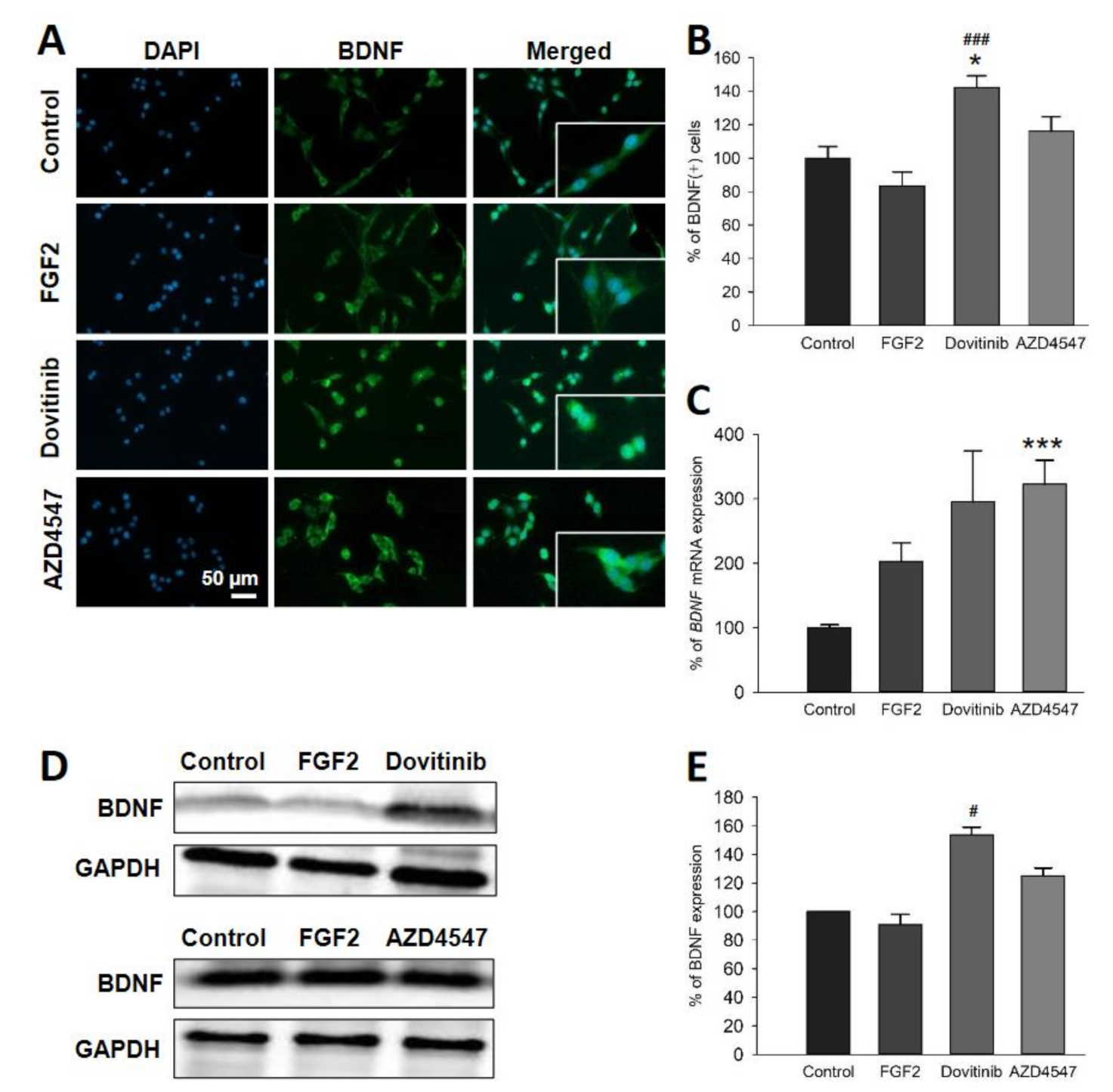
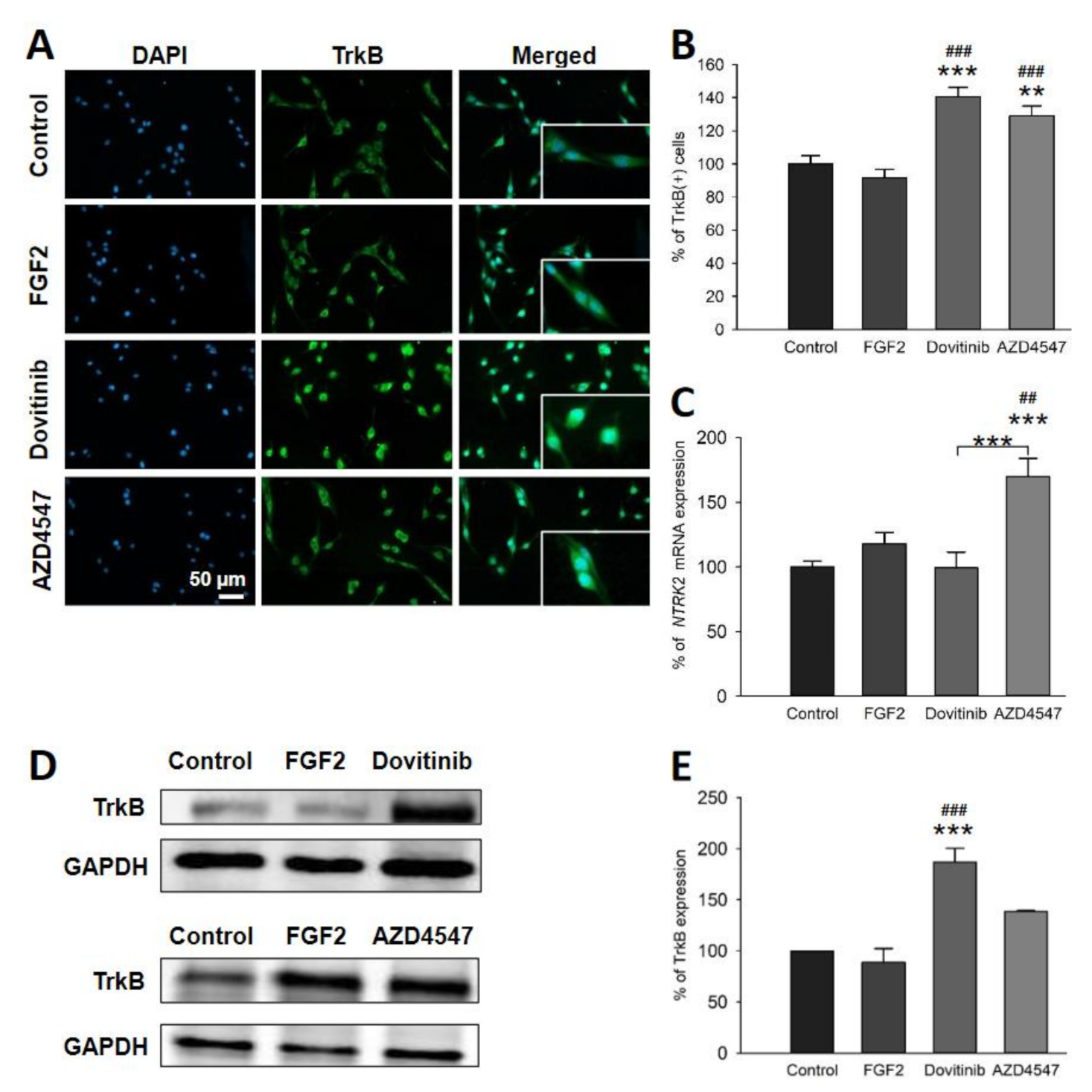
3.5. The Expression of Neuroprotective BDNF and Its Receptor TrkB Is Elevated Following Inhibition of FGFR Signaling
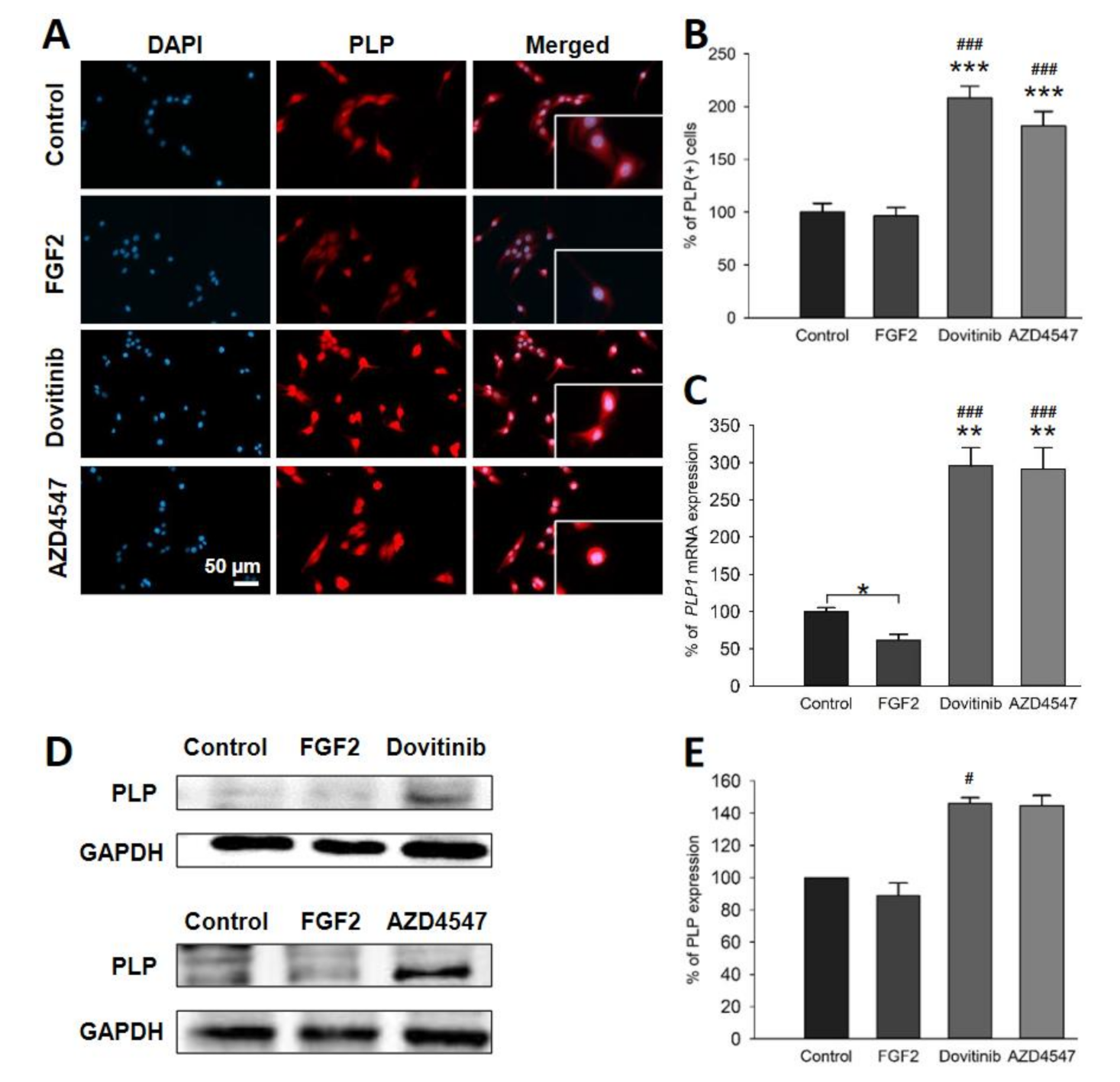
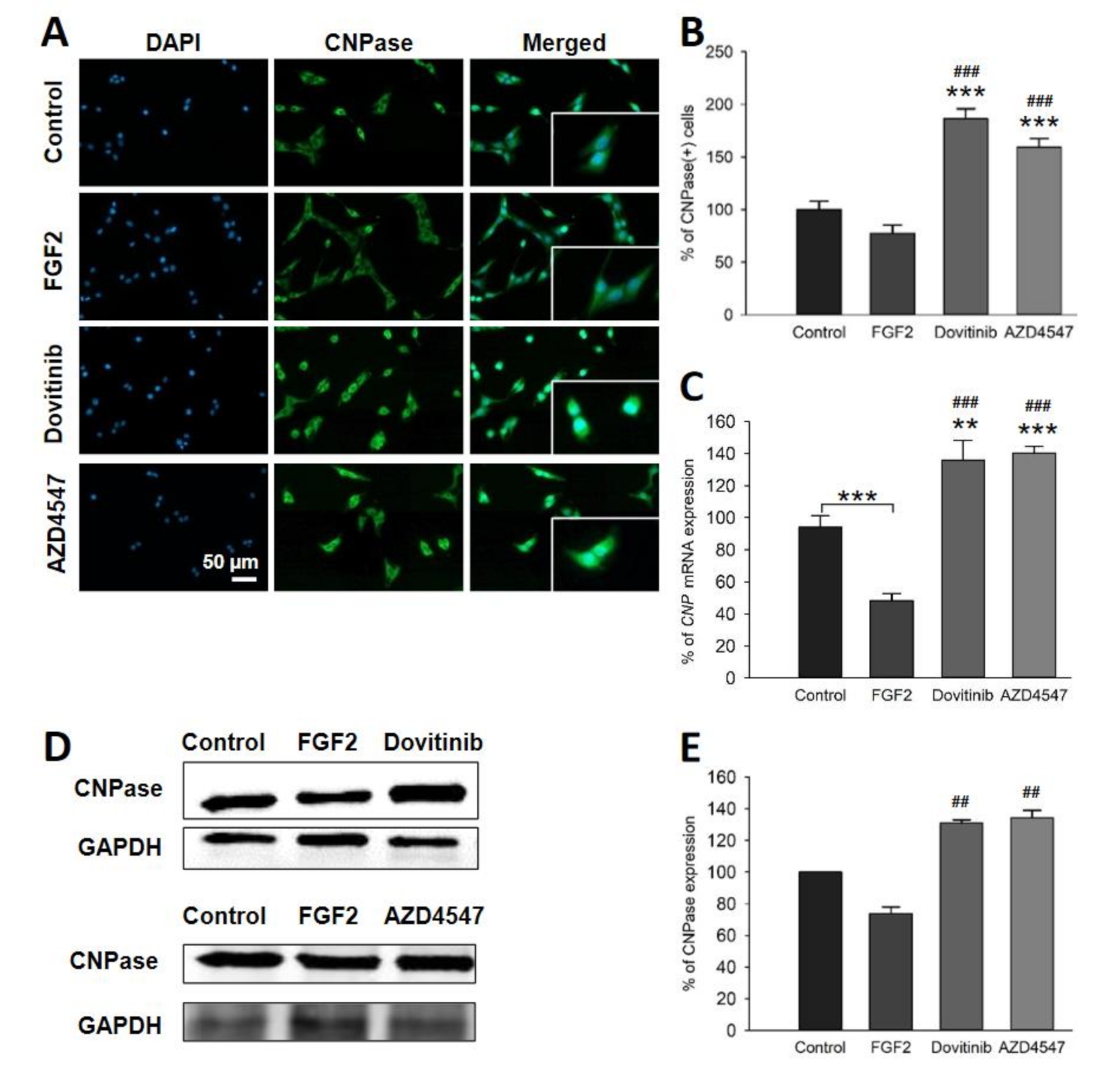
3.6. Expression of PLP and CNPase in OLN-93 Cells Is Increased by FGFR Inhibition
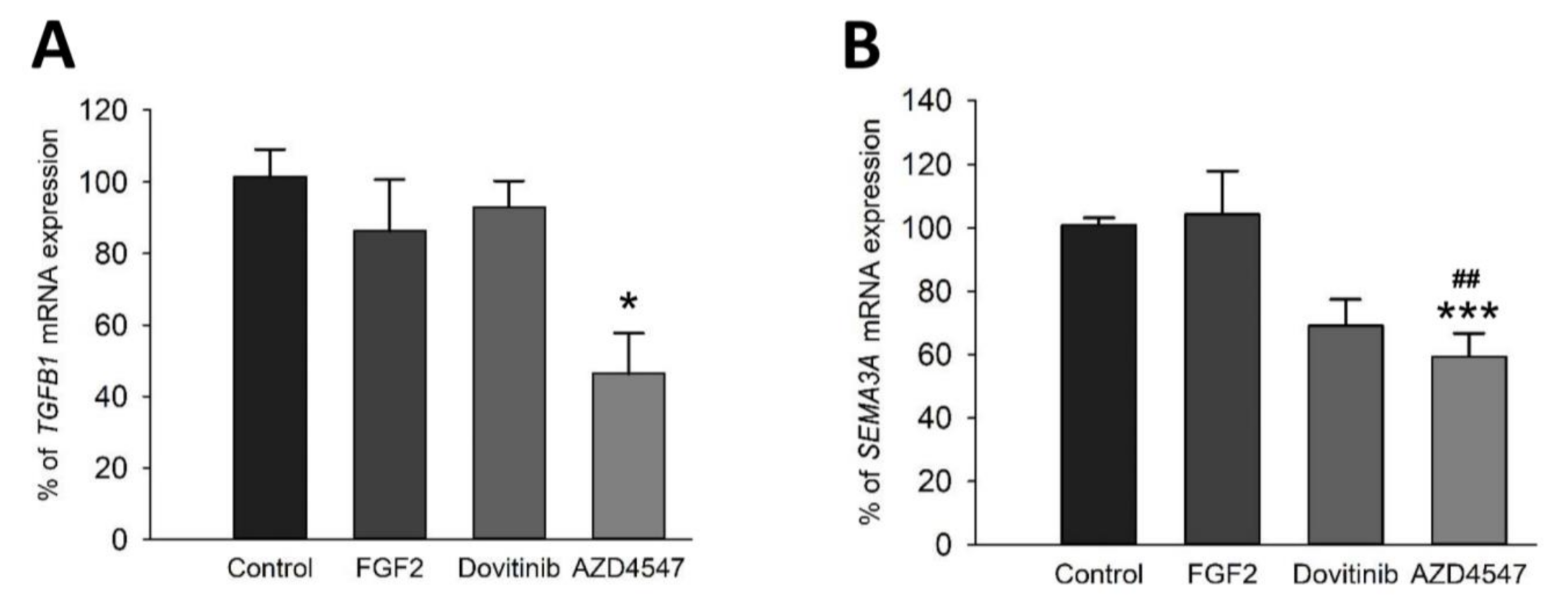
3.7. Expression of the Myelin Inhibitory Genes SEMA3A and TGFß Is Reduced after FGFR Inhibition by AZD4547
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BDNF | Brain derived neurotrophic factor |
| CNPase | 2′, 3′-cyclic-nucleotide 3′-phosphodiesterase |
| CNS | Central nervous system |
| CNTF | Ciliary neurotrophic factor |
| CSF | Cerebrospinal fluid |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl sulfoxide |
| EAE | Experimental autoimmune encephalomyelitis |
| (p)ERK | (Phosphorylated) extracellular signal-regulated kinase |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| IGF | Insulin-like growth factor |
| LDH | Lactate dehydrogenase |
| MS | Multiple sclerosis |
| OL | Oligodendrocyte |
| OPC | Oligodendrocyte progenitor cell |
| PI3K | Phosphoinositide 3-kinase |
| PLP | Proteolipid protein |
| PDGF | Platelet derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| SEM | Standard error of the mean |
| SEMA3A | Semaphorin 3A |
| TGFβ | Transforming growth factor β |
| TrkB | Tropomyosin receptor kinase B |
| VEGF | Vascular endothelial growth factor |
References
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin—from mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Nave, K.-A.; Werner, H.B. Myelination of the Nervous System: Mechanisms and Functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Kotter, M.R. The biology of CNS remyelination: The key to therapeutic advances. J. Neurol. 2008, 255, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, T.; Antel, J.; Konig, F.B.; Bruck, W.; Kuhlmann, T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 2009, 72, 1914–1921. [Google Scholar] [CrossRef]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, T.; Miron, V.; Cuo, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Crawford, A.H.; Chambers, C.; Franklin, R.J.M. Remyelination: The True Regeneration of the Central Nervous System. J. Comp. Pathol. 2013, 149, 242–254. [Google Scholar] [CrossRef]
- Acosta, C.M.; Cortes, C.; MacPhee, H.; Namaka, M.P. Exploring the role of nerve growth factor in multiple sclerosis: Implications in myelin repair. CNS Neurol. Disord. Drug Targets 2013, 12, 1242–1256. [Google Scholar] [CrossRef]
- Huang, Y.; Dreyfus, C.F. The role of growth factors as a therapeutic approach to demyelinating disease. Exp. Neurol. 2016, 283, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Mohan, H.; Friese, A.; Albrecht, S.; Krumbholz, M.; Elliott, C.L.; Arthur, A.; Menon, R.; Farina, C.; Junker, A.; Stadelmann, C.; et al. Transcript profiling of different types of multiple sclerosis lesions yields FGF1 as a promoter of remyelination. Acta Neuropathol. Commun. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Clemente, D.; Ortega, M.C.; Arenzana, F.J.; De Castro, F. FGF-2 and Anosmin-1 Are Selectively Expressed in Different Types of Multiple Sclerosis Lesions. J. Neurosci. 2011, 31, 14899–14909. [Google Scholar] [CrossRef] [Green Version]
- Lindner, M.; Thümmler, K.; Arthur, A.; Brunner, S.; Elliott, C.; McElroy, D.; Mohan, H.; Williams, A.; Edgar, J.M.; Schuh, C.; et al. Fibroblast growth factor signalling in multiple sclerosis: Inhibition of myelination and induction of pro-inflammatory environment by FGF9. Brain 2015, 138, 1875–1893. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Di Filippo, M.; Ercolani, M.V.; Chiasserini, D.; Mattioni, A.; Bonucci, M.; Tenaglia, S.; Eusebi, P.; Calabresi, P. Fibroblast growth factor-2 levels are elevated in the cerebrospinal fluid of multiple sclerosis patients. Neurosci. Lett. 2008, 435, 223–228. [Google Scholar] [CrossRef]
- Rajendran, R.; Giraldo-Velasquez, M.; Stadelmann, C.; Berghoff, M. Oligodendroglial fibroblast growth factor receptor 1 gene targeting protects mice from experimental autoimmune encephalomyelitis through ERK/AKT phosphorylation. Brain Pathol. 2018, 28, 212–224. [Google Scholar] [CrossRef]
- Kamali, S.; Rajendran, R.; Stadelmann, C.; Karnati, S.; Rajendran, V.; Giraldo-Velasquez, M.; Berghoff, M. Oligodendrocyte-specific deletion of FGFR2 ameliorates MOG(35–55) -induced EAE through ERK and Akt signalling. Brain Pathol. 2021, 31, 297–311. [Google Scholar] [CrossRef]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef] [Green Version]
- Delpuech, O.; Rooney, C.; Mooney, L.; Baker, D.; Shaw, R.; Dymond, M.; Wang, D.; Zhang, P.; Cross, S.; Veldman-Jones, M.; et al. Identification of Pharmacodynamic Transcript Biomarkers in Response to FGFR Inhibition by AZD4547. Mol. Cancer Ther. 2016, 15, 2802–2813. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, C.; Redman, M.W.; Lara, P.N.; Borghaei, H.; Hoffman, P.; Bradley, J.D.; Newman, A.J.; Feldman, M.J.; Minichiello, K.; Miao, J.; et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients With Fibroblast Growth Factor Pathway–Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J. Thorac. Oncol. 2019, 14, 1847–1852. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Mansoor, W.; Petty, R.D.; Chao, Y.; Cunningham, D.; Ferry, D.; Landers, D.; Stockman, P.; Smith, N.R.; et al. A randomized, open-label phase II study of AZD4547 (AZD) versus Paclitaxel (P) in previously treated patients with advanced gastric cancer (AGC) with Fibroblast Growth Factor Receptor 2 (FGFR2) polysomy or gene amplification (amp): SHINE study. J. Clin. Oncol. 2015, 33, 4014. [Google Scholar] [CrossRef]
- Kim, K.B.; Chesney, J.; Robinson, D.; Gardner, H.; Shi, M.M.; Kirkwood, J.M. Phase I/II and Pharmacodynamic Study of Dovitinib (TKI258), an Inhibitor of Fibroblast Growth Factor Receptors and VEGF Receptors, in Patients with Advanced Melanoma. Clin. Cancer Res. 2011, 17, 7451–7461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef]
- Facchinetti, F.; Hollebecque, A.; Bahleda, R.; Loriot, Y.; Olaussen, K.A.; Massard, C.; Friboulet, L. Facts and New Hopes on Selective FGFR Inhibitors in Solid Tumors. Clin. Cancer Res. 2020, 26, 764–774. [Google Scholar] [CrossRef] [Green Version]
- André, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with dovitinib (TKI258): Preclinical and clinical data in breast cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef] [Green Version]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Xiao, J.; Wong, A.W.; Willingham, M.M.; Van Den Buuse, M.; Kilpatrick, T.J.; Murray, S.S. Brain-Derived Neurotrophic Factor Promotes Central Nervous System Myelination via a Direct Effect upon Oligodendrocytes. Neurosignals 2010, 18, 186–202. [Google Scholar] [CrossRef]
- Fletcher, J.L.; Wood, R.J.; Nguyen, J.; Norman, E.M.L.; Jun, C.M.K.; Prawdiuk, A.R.; Biemond, M.; Nguyen, H.T.H.; Northfield, S.E.; Hughes, R.A.; et al. Targeting TrkB with a Brain-Derived Neurotrophic Factor Mimetic Promotes Myelin Repair in the Brain. J. Neurosci. 2018, 38, 7088–7099. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Myllykoski, M.; Ruskamo, S.; Wang, C.; Kursula, P. Myelin-specific proteins: A structurally diverse group of membrane-interacting molecules. BioFactors 2013, 39, 233–241. [Google Scholar] [CrossRef]
- Eixarch, H.; Gutiérrez-Franco, A.; Montalban, X.; Espejo, C. Semaphorins 3A and 7A: Potential immune and neuroregenerative targets in multiple sclerosis. Trends Mol. Med. 2013, 19, 157–164. [Google Scholar] [CrossRef]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.H.; Kidd, G.; Avila, R.L.; Kirschner, D.A.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt Signals through the Mammalian Target of Rapamycin Pathway to Regulate CNS Myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef]
- Ishii, A.; Fyffe-Maricich, S.L.; Furusho, M.; Miller, R.H.; Bansal, R. ERK1/ERK2 MAPK Signaling is Required to Increase Myelin Thickness Independent of Oligodendrocyte Differentiation and Initiation of Myelination. J. Neurosci. 2012, 32, 8855–8864. [Google Scholar] [CrossRef] [Green Version]
- Furusho, M.; Ishii, A.; Bansal, R. Signaling by FGF Receptor 2, Not FGF Receptor 1, Regulates Myelin Thickness through Activation of ERK1/2-MAPK, Which Promotes mTORC1 Activity in an Akt-Independent Manner. J. Neurosci. 2017, 37, 2931–2946. [Google Scholar] [CrossRef] [Green Version]
- Tsiperson, V.; Gruber, R.C.; Goldberg, M.F.; Jordan, A.; Weinger, J.G.; Macian, F.; Shafit-Zagardo, B. Suppression of Inflammatory Responses during Myelin Oligodendrocyte Glycoprotein-Induced Experimental Autoimmune Encephalomyelitis Is Regulated by AKT3 Signaling. J. Immunol. 2013, 190, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Furusho, M.; Dupree, J.L.; Nave, K.A.; Bansal, R. Fibroblast Growth Factor Receptor Signaling in Oligodendrocytes Regulates Myelin Sheath Thickness. J. Neurosci. 2012, 32, 6631–6641. [Google Scholar] [CrossRef] [Green Version]
- Ishii, A.; Furusho, M.; Dupree, J.L.; Bansal, R. Role of ERK1/2 MAPK Signaling in the Maintenance of Myelin and Axonal Integrity in the Adult CNS. J. Neurosci. 2014, 34, 16031–16045. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Martinez Santos, J.L.; Alshareef, M.; Porto, G.B.F.; Infinger, L.K.; Vandergrift, W.A., 3rd; Lindhorst, S.M.; Varma, A.K.; Patel, S.J.; Cachia, D. In Vitro Effect of Dovitinib (TKI258), a Multi-Target Angiokinase Inhibitor on Aggressive Meningioma Cells. Cancer Investig. 2020, 38, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Chow, P.K.H.; Tai, W.M.; Choo, S.P.; Chung, A.Y.F.; Ong, H.S.; Soo, K.C.; Ong, R.; Linnartz, R.; Shi, M.M. Dovitinib demonstrates antitumor and antimetastatic activities in xenograft models of hepatocellular carcinoma. J. Hepatol. 2012, 56, 595–601. [Google Scholar] [CrossRef]
- Zhang, H.; Hylander, B.L.; Levea, C.; Repasky, E.A.; Straubinger, R.M.; Adjei, A.A.; Ma, W.W. Enhanced FGFR signalling predisposes pancreatic cancer to the effect of a potent FGFR inhibitor in preclinical models. Br. J. Cancer 2014, 110, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzhauser, S.; Lukoseviciute, M.; Andonova, T.; Ursu, R.G.; Dalianis, T.; Wickström, M.; Kostopoulou, O.N. Targeting Fibroblast Growth Factor Receptor (FGFR) and Phosphoinositide 3-kinase (PI3K) Signaling Pathways in Medulloblastoma Cell Lines. Anticancer Res. 2020, 40, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Nakagama, H. FGF Receptors: Cancer Biology and Therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Gospodarowicz, D.; Neufeld, G.; Schweigerer, L. Molecular and biological characterization of fibroblast growth factor, an angiogenic factor which also controls the proliferation and differentiation of mesoderm and neuroectoderm derived cells. Cell Differ. 1986, 19, 1–17. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Baron, W.; Metz, B.; Bansal, R.; Hoekstra, D.; de Vries, H. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: Regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol. Cell. Neurosci. 2000, 15, 314–329. [Google Scholar] [CrossRef]
- Fortin, D. Distinct Fibroblast Growth Factor (FGF)/FGF Receptor Signaling Pairs Initiate Diverse Cellular Responses in the Oligodendrocyte Lineage. J. Neurosci. 2005, 25, 7470–7479. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.H.; Demir, S.; Wiese, S.; Kruse, N.; Siglienti, I.; Gerhardt, E.; Neumann, H.; Sendtner, M.; Luhder, F.; et al. Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: Therapeutic implications in a model of multiple sclerosis. Brain 2010, 133, 2248–2263. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Geyer, E.; Flach, A.C.; Jung, K.; Gold, R.; Flügel, A.; Linker, R.A.; Lühder, F. Central nervous system rather than immune cell-derived BDNF mediates axonal protective effects early in autoimmune demyelination. Acta Neuropathol. 2012, 123, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Kerschensteiner, M.; Misgeld, T.; Brück, W.; Hohlfeld, R.; Lassmann, H. BDNF and gp145trkB in multiple sclerosis brain lesions: Neuroprotective interactions between immune and neuronal cells? Brain 2002, 125, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makar, T.K.; Nimmagadda, V.K.; Singh, I.S.; Lam, K.; Mubariz, F.; Judge, S.I.; Trisler, D.; Bever, C.T., Jr. TrkB agonist, 7,8-dihydroxyflavone, reduces the clinical and pathological severity of a murine model of multiple sclerosis. J. Neuroimmunol. 2016, 292, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, F.; Zimmer, C. From cradle to grave: The multiple roles of fibroblast growth factors in neural development. Neuron 2011, 71, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Wieghofer, P.; Müller, P.F.; Wolf, Y.; Varol, D.; Yona, S.; Brendecke, S.M.; Kierdorf, K.; Staszewski, O.; Datta, M.; et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 2013, 16, 1618–1626. [Google Scholar] [CrossRef]
- Zöller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFβ signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Kuçi, S.; Kuçi, Z.; Schmid, S.; Seitz, G.; Müller, I.; Dufke, A.; Leimig, T.; Murti, G.; Jurecic, R.; Schumm, M.; et al. Efficient in vitro generation of adult multipotent cells from mobilized peripheral blood CD133+ cells. Cell Prolif. 2008, 41, 12–27. [Google Scholar] [CrossRef]
- Yamasaki, K.; Setoguchi, T.; Takenouchi, T.; Yone, K.; Komiya, S. Stem cell factor prevents neuronal cell apoptosis after acute spinal cord injury. Spine 2009, 34, 323–327. [Google Scholar] [CrossRef]
- Sentilhes, L.; Marret, S.; Leroux, P.; Gonzalez, B.J.; Laquerrière, A. Vascular-endothelial growth factor and its high affinity receptor VEGFR-2 in the normal versus destructive lesions human forebrain during development: An immuno-histochemical comparative study. Brain Res. 2011, 1385, 77–86. [Google Scholar] [CrossRef]
- Funa, K.; Sasahara, M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol. 2014, 9, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Zhao, X.; Zheng, K.; Li, H.; Huang, H.; Zhang, Z.; Mastracci, T.; Wegner, M.; Chen, Y.; Sussel, L.; et al. Genetic evidence that Nkx2.2 and Pdgfra are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development 2014, 141, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Shao, C.Y.; Xie, Y.J.; Wang, N.; Xu, S.M.; Luo, B.Y.; Wu, Z.Y.; Ke, Y.H.; Qiu, M.; Shen, Y. Gab1 mediates PDGF signaling and is essential to oligodendrocyte differentiation and CNS myelination. eLife 2020, 9. [Google Scholar] [CrossRef]
- Shen, J.; Ishii, Y.; Xu, G.; Dang, T.C.; Hamashima, T.; Matsushima, T.; Yamamoto, S.; Hattori, Y.; Takatsuru, Y.; Nabekura, J.; et al. PDGFR-β as a positive regulator of tissue repair in a mouse model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2012, 32, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.C.; Le, T.Q.; Flint, N.C.; Vana, A.C.; Zhou, Y.-X. Endogenous Cell Repair of Chronic Demyelination. J. Neuropathol. Exp. Neurol. 2006, 65, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Messersmith, D.J.; Murtie, J.C.; Le, T.Q.; Frost, E.E.; Armstrong, R.C. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J. Neurosci. Res. 2000, 62, 241–256. [Google Scholar] [CrossRef]
- Bansal, R.; Kumar, M.; Murray, K.; Morrison, R.S.; Pfeiffer, S.E. Regulation of FGF receptors in the oligodendrocyte lineage. Mol. Cell. Neurosci. 1996, 7, 263–275. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajendran, R.; Böttiger, G.; Dentzien, N.; Rajendran, V.; Sharifi, B.; Ergün, S.; Stadelmann, C.; Karnati, S.; Berghoff, M. Effects of FGFR Tyrosine Kinase Inhibition in OLN-93 Oligodendrocytes. Cells 2021, 10, 1318. https://doi.org/10.3390/cells10061318
Rajendran R, Böttiger G, Dentzien N, Rajendran V, Sharifi B, Ergün S, Stadelmann C, Karnati S, Berghoff M. Effects of FGFR Tyrosine Kinase Inhibition in OLN-93 Oligodendrocytes. Cells. 2021; 10(6):1318. https://doi.org/10.3390/cells10061318
Chicago/Turabian StyleRajendran, Ranjithkumar, Gregor Böttiger, Niklas Dentzien, Vinothkumar Rajendran, Bischand Sharifi, Süleyman Ergün, Christine Stadelmann, Srikanth Karnati, and Martin Berghoff. 2021. "Effects of FGFR Tyrosine Kinase Inhibition in OLN-93 Oligodendrocytes" Cells 10, no. 6: 1318. https://doi.org/10.3390/cells10061318