
Mitochondrial Ca2+ Signaling in Health, Disease and Therapy

 , , , and
, , , and
Abstract
:1. Introduction
2. Mitochondrial Calcium Homeostasis
2.1. Mitochondrial Ca2+ Influx
2.2. Mitochondrial Ca2+ Efflux
2.3. Physiological Role of Mitochondrial Ca2+
3. Mitochondrial Calcium Dyshomeostasis
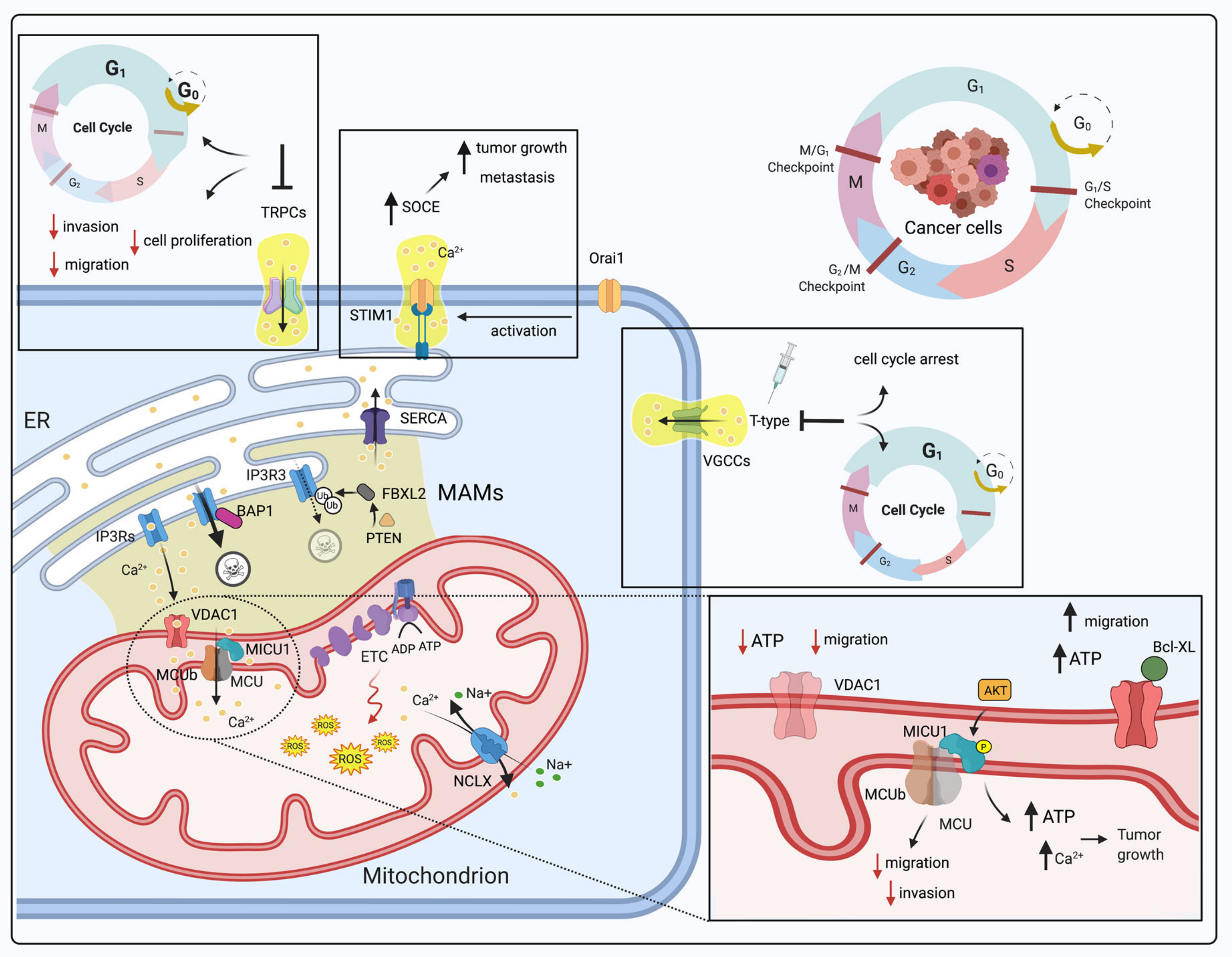
3.1. Dysregulation of Mitochondrial Ca2+ Signaling in Cancer and the Cell Cycle
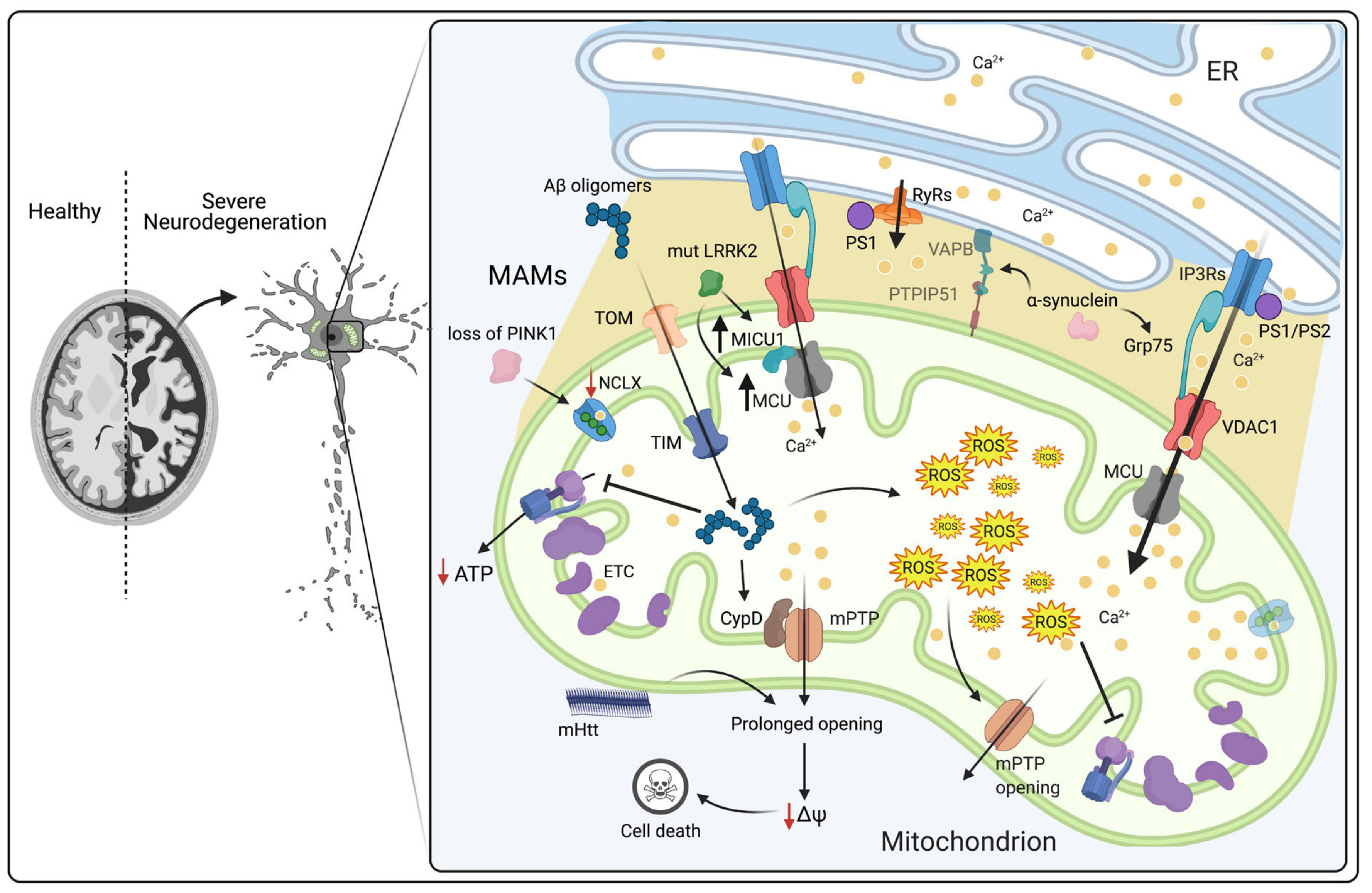
3.2. Dysregulation of Mitochondrial Ca2+ Signaling in Neurodegenerative Diseases
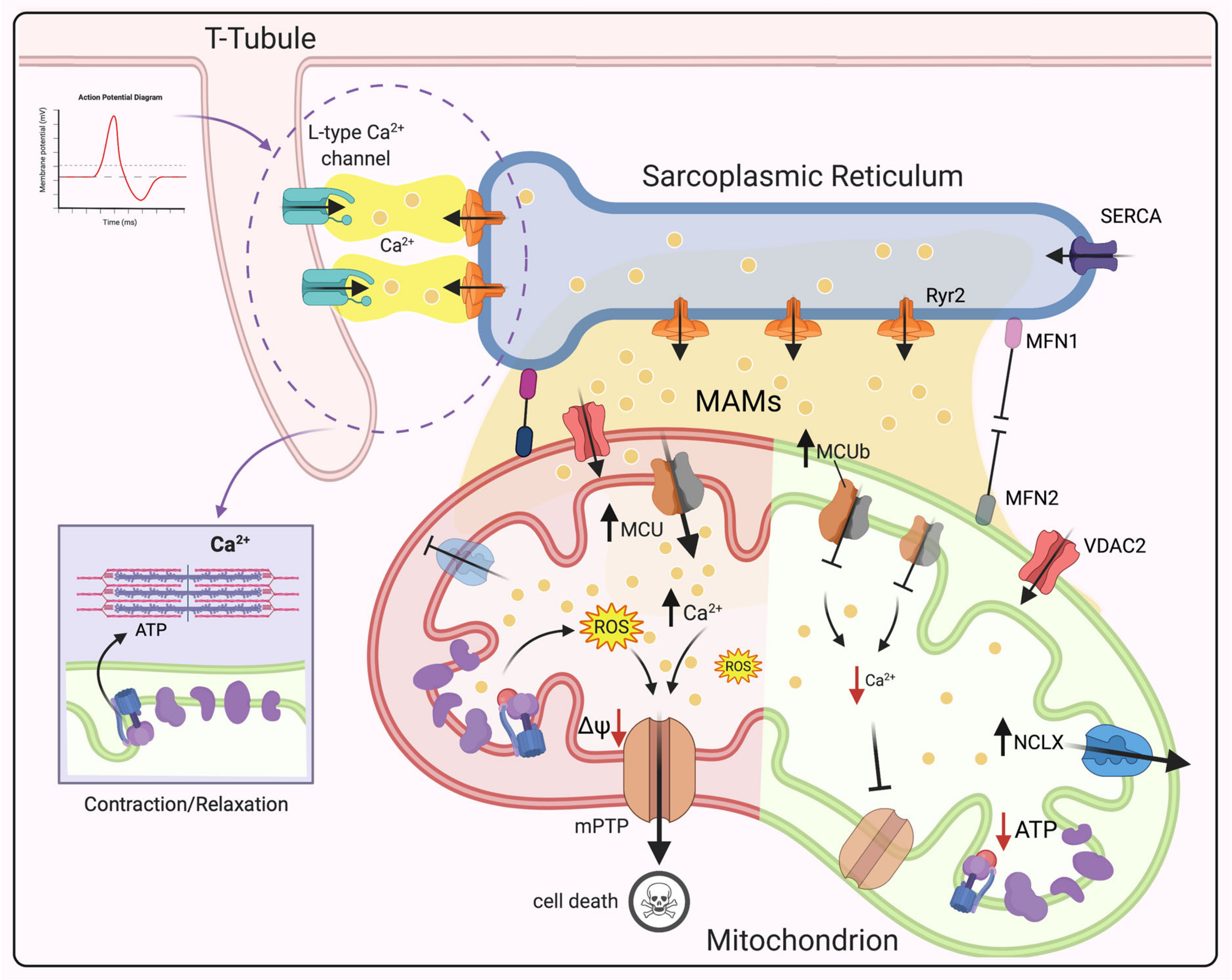
3.3. Dysregulation of Mitochondrial Ca2+ Signaling in CVDs
4. Targeting Mitochondrial Ca2+ Signaling as a Promising Therapeutic Approach
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer disease |
| APP | Amyloid precursor protein |
| Aβ | Amyloid β-peptide |
| APOE | Apolipoprotein E |
| BAP1 | BRCA1-associated protein-1 |
| CaMKI | Ca2+/calmodulin-dependent protein kinase |
| Ca2+ | Calcium |
| CaM | Calmodulin |
| CVDs | Cardiovascular diseases |
| CDKs | Cyclin-dependent kinases |
| CypD | Cyclophilin D |
| cytCa2+ | Cytosolic calcium |
| ETC | Electron transport chain |
| ER | Endoplasmic reticulum |
| EMRE | Essential MCU regulator |
| FAD | Familial AD |
| HCX | H+/Ca2+ exchangers |
| HF | Heart failure |
| HD | Huntington disease |
| IMM | Inner mitochondrial membrane |
| IP3R3 | Inositol 1,4,5-trisphosphate (IP3) receptor type 3 |
| IP3Rs | Inositol 1,4,5-trisphosphate (IP3) receptors |
| IMS | Intermembrane mitochondrial space |
| IRI | Ischemia/reperfusion injury |
| IDH | Isocitrate dehydrogenase |
| KGDH | Ketoglutarate dehydrogenase |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAMs | Mitochondrial associated membranes |
| MCUC | Mitochondrial calcium Uniporter Complex |
| MCU | Mitochondrial calcium Uniporter |
| MICU1 | Mitochondrial calcium uptake 1 |
| mCa2+ | Mitochondrial calcium |
| ΔΨm | Mitochondrial membrane potential |
| mPTP | Mitochondrial permeability transition pore |
| mROS | Mitochondrial ROS |
| MFN2 | Mitofusin 2 |
| MI | Myocardial infarction |
| NCLX | Na+/Ca2+ exchangers |
| OMM | Outer mitochondrial membrane |
| OXPHOS | Oxidative phosphorylation |
| PD | Parkinson disease |
| PTEN | Phosphatase and tensin homolog |
| PS1 | Presenilin-1 |
| PS2 | Presenilin-2 |
| PINK1 | PTEN-induced kinase 1 |
| PDH | Pyruvate dehydrogenase |
| ROS | Reactive oxygen species |
| Ru360 | Ruthenium 360 |
| RuR | Ruthenium red |
| RyR2 | Ryanodine receptor (RyR) type 2 |
| RyR | Ryanodine receptor |
| SERCA | Sarco-Endoplasmic Reticulum Calcium ATPase |
| SR | Sarcoplasmic reticulum |
| SAD | Sporadic AD |
| SOCE | Store-operated calcium entry |
| TRP | Transient receptor potential |
| VDAC | Voltage dependent anion channel |
| VGCC | Voltage-gated calcium channel |
References
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial dynamics in health and disease. FEBS Lett. 2021, 595, 1184–1204. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, E.J.; Stathopulos, P.B.; Madesh, M. Regulation of Ca2+ exchanges and signaling in mitochondria. Curr. Opin. Physiol. 2020, 17, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Marchi, S.; Vitto, V.A.M.; Patergnani, S.; Pinton, P. High mitochondrial Ca2+ content increases cancer cell proliferation upon inhibition of mitochondrial permeability transition pore (mPTP). Cell Cycle 2019, 18, 914–916. [Google Scholar] [CrossRef] [Green Version]
- Sebag, S.C.; Koval, O.M.; Paschke, J.D.; Winters, C.J.; Comellas, A.P.; Grumbach, I.M. Inhibition of the mitochondrial calcium uniporter prevents IL-13 and allergen-mediated airway epithelial apoptosis and loss of barrier function. Exp. Cell Res. 2018, 362, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Oropeza-Almazán, Y.; Vázquez-Garza, E.; Chapoy-Villanueva, H.; Torre-Amione, G.; García-Rivas, G. Small Interfering RNA Targeting Mitochondrial Calcium Uniporter Improves Cardiomyocyte Cell Viability in Hypoxia/Reoxygenation Injury by Reducing Calcium Overload. Oxid. Med. Cell. Longev. 2017, 2017, 5750897. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatiero, V.; De Stefani, D.; Rizzuto, R. Mitochondrial Calcium Handling in Physiology and Disease. Adv. Exp. Med. Biol. 2017, 982, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflugers Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [Green Version]
- Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cell Dev. Biol. 2019, 94, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Simamura, E.; Shimada, H.; Hatta, T.; Hirai, K.-I. Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J. Bioenerg. Biomembr. 2008, 40, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Rizzuto, R. Molecular structure and pathophysiological roles of the Mitochondrial Calcium Uniporter. Biochim. Biophys. Acta 2016, 1863, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; Perez, S.D.L.F.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Gherardi, G.; Rizzuto, R. Structure, Activity Regulation, and Role of the Mitochondrial Calcium Uniporter in Health and Disease. Front. Oncol. 2017, 7, 139. [Google Scholar] [CrossRef] [Green Version]
- Pozzan, T.; Bragadin, M.; Azzone, G.F. Disequilibrium between steady-state Ca2+ accumulation ratio and membrane potential in mitochondria. Pathway and role of Ca2+ efflux. Biochemistry 1977, 16, 5618–5625. [Google Scholar] [CrossRef]
- Crompton, M.; Künzi, M.; Carafoli, E. The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur. J. Biochem. 1977, 79, 549–558. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowikovsky, K.; Pozzan, T.; Rizzuto, R.; Scorrano, L.; Bernardi, P. Perspectives on: SGP symposium on mitochondrial physiology and medicine: The pathophysiology of LETM1. J. Gen. Physiol. 2012, 139, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 2010, 120, 3680–3687. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kwong, J.Q.; Molkentin, J.D.; Bers, D.M. Individual Cardiac Mitochondria Undergo Rare Transient Permeability Transition Pore Openings. Circ. Res. 2016, 118, 834–841. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, E.; Bonora, M.; Giorgi, C.; Pinton, P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 2014, 56, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.B.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bodhinathan, K.; Foster, T.C. Susceptibility to Calcium Dysregulation during Brain Aging. Front. Aging Neurosci. 2009, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Danese, A.; Marchi, S.; Vitto, V.A.M.; Modesti, L.; Leo, S.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Cancer-Related Increases and Decreases in Calcium Signaling at the Endoplasmic Reticulum-Mitochondria Interface (MAMs). Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenerg. 2017, 1858, 615–627. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomer, J.; López-Girona, A.; Agell, N.; Bachs, O. Calmodulin regulates the expression of cdks, cyclins and replicative enzymes during proliferative activation of human T lymphocytes. Biochem. Biophys. Res. Commun. 1994, 200, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Kahl, C.R.; Means, A.R. Calcineurin regulates cyclin D1 accumulation in growth-stimulated fibroblasts. Mol. Biol. Cell 2004, 15, 1833–1842. [Google Scholar] [CrossRef] [Green Version]
- Takuwa, N.; Zhou, W.; Kumada, M.; Takuwa, Y. Ca2+-dependent stimulation of retinoblastoma gene product phosphorylation and p34cdc2 kinase activation in serum-stimulated human fibroblasts. J. Biol. Chem. 1993, 268, 138–145. [Google Scholar] [CrossRef]
- Yuan, K.; Wang, X.; Dong, H.; Min, W.; Hao, H.; Yang, P. Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm. Sin. B 2021, 11, 30–54. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. Alterations of calcium homeostasis in cancer cells. Curr. Opin. Pharmacol. 2016, 29, 1–6. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Chen, Y.-F.; Chen, Y.-T.; Chiu, W.-T.; Shen, M.-R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbioni, S.; Veronese, A.; Trubia, M.; Taramelli, R.; Barbanti-Brodano, G.; Croce, C.M.; Negrini, M. Exon structure and promoter identification of STIM1 (alias GOK), a human gene causing growth arrest of the human tumor cell lines G401 and RD. Cytogenet. Cell Genet. 1999, 86, 214–218. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphaël, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karacicek, B.; Erac, Y.; Tosun, M. Functional consequences of enhanced expression of STIM1 and Orai1 in Huh-7 hepatocellular carcinoma tumor-initiating cells. BMC Cancer 2019, 19, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouriou, Y.; Bijlenga, P.; Demaurex, N. Mitochondrial Ca2+ uptake from plasma membrane Cav3.2 protein channels contributes to ischemic toxicity in PC12 cells. J. Biol. Chem. 2013, 288, 12459–12468. [Google Scholar] [CrossRef] [Green Version]
- Barceló, C.; Sisó, P.; Maiques, O.; de la Rosa, I.; Martí, R.M.; Macià, A. T-Type Calcium Channels: A Potential Novel Target in Melanoma. Cancers 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Pushparaj, C.; Bahí, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Martí, R.M.; Cantí, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment Cell Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. T-type Ca2+ channel inhibition induces p53-dependent cell growth arrest and apoptosis through activation of p38-MAPK in colon cancer cells. Mol. Cancer Res. 2014, 12, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, S.-L.; Wang, N.; Zhang, B.-B.; Li, M. Blockade of T-type Ca2+ channels inhibits human ovarian cancer cell proliferation. Cancer Invest. 2011, 29, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Liu, F.; Ruan, J.; Li, A.; Chen, J.; Li, R.; Shen, J.; Zheng, D.; Luo, R. Silencing TRPC1 expression inhibits invasion of CNE2 nasopharyngeal tumor cells. Oncol. Rep. 2012, 27, 1548–1554. [Google Scholar] [CrossRef] [PubMed]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The Role of TRPC1 in Modulating Cancer Progression. Cells 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Monet, M.; Gkika, D.; Lehen’kyi, V.; Pourtier, A.; Vanden Abeele, F.; Bidaux, G.; Juvin, V.; Rassendren, F.; Humez, S.; Prevarsakaya, N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim. Biophys. Acta 2009, 1793, 528–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Lago, C.U.; Sung, H.J.; Ma, W.; Wang, P.; Hwang, P.M. p53, aerobic metabolism, and cancer. Antioxid. Redox Signal. 2011, 15, 1739–1748. [Google Scholar] [CrossRef]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic functions of the p53 tumor suppressor: Regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol. 2009, 19, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Galli, S.; Antico Arciuch, V.G.; Poderoso, C.; Converso, D.P.; Zhou, Q.; Bal de Kier Joffé, E.; Cadenas, E.; Boczkowski, J.; Carreras, M.C.; Poderoso, J.J. Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PLoS One 2008, 3, e2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgering, B.M.T.; Kops, G.J.P.L. Cell cycle and death control: Long live Forkheads. Trends Biochem. Sci. 2002, 27, 352–360. [Google Scholar] [CrossRef]
- Dijkers, P.F.; Medema, R.H.; Pals, C.; Banerji, L.; Thomas, N.S.; Lam, E.W.; Burgering, B.M.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 2000, 20, 9138–9148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Vitto, V.A.M.; Danese, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle 2019, 18, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corrà, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudent, J.; Popgeorgiev, N.; Gadet, R.; Deygas, M.; Rimokh, R.; Gillet, G. Mitochondrial Ca2+ uptake controls actin cytoskeleton dynamics during cell migration. Sci. Rep. 2016, 6, 36570. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef]
- Koval, O.M.; Nguyen, E.K.; Santhana, V.; Fidler, T.P.; Sebag, S.C.; Rasmussen, T.P.; Mittauer, D.J.; Strack, S.; Goswami, P.C.; Abel, E.D.; et al. Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Han, M.; Xu, Y.; Zhang, X.; Zhang, C.; Zhang, D.; Ji, J.; Wei, Y.; Wang, S.; Huang, B.; et al. Coiled-coil domain containing 109B is a HIF1α-regulated gene critical for progression of human gliomas. J. Transl. Med. 2017, 15, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Fouqué, A.; Lepvrier, E.; Debure, L.; Gouriou, Y.; Malleter, M.; Delcroix, V.; Ovize, M.; Ducret, T.; Li, C.; Hammadi, M.; et al. The apoptotic members CD95, BclxL, and Bcl-2 cooperate to promote cell migration by inducing Ca2+ flux from the endoplasmic reticulum to mitochondria. Cell Death Differ. 2016, 23, 1702–1716. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezprozvanny, I.B. Calcium signaling and neurodegeneration. Acta Naturae 2010, 2, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.-K. Dysfunction of Mitochondrial Ca2+ Regulatory Machineries in Brain Aging and Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Zou, F.; Chai, H.S.; Younkin, C.S.; Crook, J.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; Nair, A.A.; Middha, S.; et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 2012, 79, 221–228. [Google Scholar] [CrossRef]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in Alzheimer disease. Int. J. Alzheimers. Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Donev, R.; Kolev, M.; Millet, B.; Thome, J. Neuronal death in Alzheimer’s disease and therapeutic opportunities. J. Cell. Mol. Med. 2009, 13, 4329–4348. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of Alzheimer’s disease -A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. The role of calcium regulation in brain aging: Reexamination of a hypothesis. Aging 1989, 1, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirashima, N.; Etcheberrigaray, R.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Govoni, S.; Alkon, D.L. Calcium responses in human fibroblasts: A diagnostic molecular profile for Alzheimer’s disease. Neurobiol. Aging 1996, 17, 549–555. [Google Scholar] [CrossRef]
- Cheung, K.-H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.-Y.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [Green Version]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Rybalchenko, V.; Hwang, S.-Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Bacskai, B.J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 2021, 44, 136–151. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Arndt, J.W.; Cancilla, M.T.; Cao, K.; Elling, R.A.; English, N.; Friedman, J.; Hansen, S.K.; Hession, C.; Joseph, I.; et al. Discovery of a potent and highly selective PDK1 inhibitor via fragment-based drug discovery. Bioorg. Med. Chem. Lett. 2011, 21, 3078–3083. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2018, 2, NS20180061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S. Du Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef]
- Du, H.; Yan, S.S. Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim. Biophys. Acta 2010, 1802, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.K.; Carlson, E.A.; Yan, S.S. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 2014, 1842, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Pérez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Greotti, E.; Wong, A.; Pozzan, T.; Pendin, D.; Pizzo, P. Characterization of the ER-Targeted Low Affinity Ca2+ Probe D4ER. Sensors 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; de Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, B.; Hedskog, L.; Wiehager, B.; Ankarcrona, M. Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers. Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roze, E.; Bonnet, C.; Betuing, S.; Caboche, J. Huntington’s disease. Adv. Exp. Med. Biol. 2010, 685, 45–63. [Google Scholar]
- Brustovetsky, N. Mutant Huntingtin and Elusive Defects in Oxidative Metabolism and Mitochondrial Calcium Handling. Mol. Neurobiol. 2016, 53, 2944–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247. [Google Scholar] [CrossRef]
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [CrossRef]
- Guedes-Dias, P.; Pinho, B.R.; Soares, T.R.; de Proença, J.; Duchen, M.R.; Oliveira, J.M.A. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol. Dis. 2016, 90, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Johnson, G.V.W. Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res. Bull. 2009, 80, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef] [Green Version]
- Choo, Y.S.; Johnson, G.V.W.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Jia, K.; Du, H. Mitochondrial Permeability Transition: A Pore Intertwines Brain Aging and Alzheimer’s Disease. Cells 2021, 10. [Google Scholar] [CrossRef]
- Panov, A.V.; Gutekunst, C.A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Pellman, J.J.; Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Ca2+ handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington’s disease. J. Neurochem. 2015, 134, 652–667. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.M.A.; Jekabsons, M.B.; Chen, S.; Lin, A.; Rego, A.C.; Gonçalves, J.; Ellerby, L.M.; Nicholls, D.G. Mitochondrial dysfunction in Huntington’s disease: The bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 2007, 101, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinás, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+ signaling in Parkinson’s disease. Dis. Model. Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calì, T.; Ottolini, D.; Brini, M. Calcium signaling in Parkinson’s disease. Cell Tissue Res. 2014, 357, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 100. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Muqit, M.M.K.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef]
- Ferreira, M.; Massano, J. An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 2017, 135, 273–284. [Google Scholar] [CrossRef]
- Chen, Y.P.; Song, W.; Huang, R.; Chen, K.; Zhao, B.; Li, J.; Yang, Y.; Shang, H.-F. GAK rs1564282 and DGKQ rs11248060 increase the risk for Parkinson’s disease in a Chinese population. J. Clin. Neurosci. 2013, 20, 880–883. [Google Scholar] [CrossRef]
- Ghanbari, M.; Darweesh, S.K.L.; de Looper, H.W.J.; van Luijn, M.M.; Hofman, A.; Ikram, M.A.; Franco, O.H.; Erkeland, S.J.; Dehghan, A. Genetic Variants in MicroRNAs and Their Binding Sites Are Associated with the Risk of Parkinson Disease. Hum. Mutat. 2016, 37, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Parisiadou, L.; Sgobio, C.; Liu, G.; Yu, J.; Sun, L.; Shim, H.; Gu, X.-L.; Luo, J.; Long, C.-X.; et al. Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 2012, 32, 9248–9264. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Dawson, V.L.; Dawson, T.M. Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genomics Hum. Genet. 2011, 12, 301–325. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s disease: Genetics and pathogenesis. Annu. Rev. Pathol. 2011, 6, 193–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wissemann, W.T.; Hill-Burns, E.M.; Zabetian, C.P.; Factor, S.A.; Patsopoulos, N.; Hoglund, B.; Holcomb, C.; Donahue, R.J.; Thomson, G.; Erlich, H.; et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet. 2013, 93, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The Close Encounter Between Alpha-Synuclein and Mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Yang, R.; Guo, J.-C.; Ren, H.-M.; Zha, X.-L.; Cheng, J.-S.; Cai, D.-F. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 2007, 18, 1543–1546. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [Green Version]
- Calì, T.; Ottolini, D.; Vicario, M.; Catoni, C.; Vallese, F.; Cieri, D.; Barazzuol, L.; Brini, M. splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by α-Synuclein A53T and A30P Mutants. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef]
- Akundi, R.S.; Huang, Z.; Eason, J.; Pandya, J.D.; Zhi, L.; Cass, W.A.; Sullivan, P.G.; Büeler, H. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS ONE 2011, 6, e16038. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Fon, E.A. The three ’P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.-F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.J.; Balaska, D.; Cheng, W.H.Y. The ups and downs of mitochondrial calcium signalling in the heart. Biochim. Biophys. Acta 1797, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.L.; Adaniya, S.M.; Cypress, M.W.; Suzuki, Y.; Kusakari, Y.; Jhun, B.S.; O-Uchi, J. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch. Biochem. Biophys. 2019, 663, 276–287. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Cao, Y.-P.; Zheng, M. Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch. Biochem. Biophys. 2019, 663, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Horita, K.; Murakami, M.; Ogura, R. Morphological studies of different mitochondrial populations in monkey myocardial cells. Cell Tissue Res. 1984, 238, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Giorgi, C.; Pinton, P.; Rizzuto, R. Controlling metabolism and cell death: At the heart of mitochondrial calcium signalling. J. Mol. Cell. Cardiol. 2009, 46, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabiato, A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983, 245, C1–C14. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M. The role of Ca2+ in the function and dysfunction of heart mitochondria. In Calcium Hear and the Heart; Langer, G.A., Ed.; Raven Press: New York, NY, USA, 1990; pp. 167–198. [Google Scholar]
- O’Rourke, B.; Blatter, L.A. Mitochondrial Ca2+ uptake: Tortoise or hare? J. Mol. Cell. Cardiol. 2009, 46, 767–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedkova, E.N.; Blatter, L.A. Calcium signaling in cardiac mitochondria. J. Mol. Cell. Cardiol. 2013, 58, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Fuente, S.; Sheu, S.-S. SR-mitochondria communication in adult cardiomyocytes: A close relationship where the Ca2+ has a lot to say. Arch. Biochem. Biophys. 2019, 663, 259–268. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Sharma, V.K.; Ramesh, V.; Franzini-Armstrong, C.; Sheu, S.S. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 2000, 32, 97–104. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.A.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef] [Green Version]
- Buntinas, L.; Gunter, K.K.; Sparagna, G.C.; Gunter, T.E. The rapid mode of calcium uptake into heart mitochondria (RaM): Comparison to RaM in liver mitochondria. Biochim. Biophys. Acta 2001, 1504, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Beutner, G.; Sharma, V.K.; Giovannucci, D.R.; Yule, D.I.; Sheu, S.S. Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 2001, 276, 21482–21488. [Google Scholar] [CrossRef] [Green Version]
- Morganti, C.; Bonora, M.; Sbano, L.; Morciano, G.; Aquila, G.; Campo, G.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. The Mitochondrial Permeability Transition Pore. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P.J., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 47–73. [Google Scholar]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Morciano, G.; Bonora, M.; Giorgi, C.; Pinton, P. Other bricks for the correct construction of the mitochondrial permeability transition pore complex. Cell Death Dis. 2017, 8, e2698. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; Burchell, V.; Schiavone, M.; Bassot, C.; Minervini, G.; Petronilli, V.; Argenton, F.; Forte, M.; Tosatto, S.; Lippe, G.; et al. Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Rep. 2017, 18, 1065–1076. [Google Scholar] [CrossRef]
- Bonora, M.; Pinton, P. A New Current for the Mitochondrial Permeability Transition. Trends Biochem. Sci. 2019, 44, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, K.S.; Kiser, R.F.; Weislow, O.S. Magnesium counteracts nickel-induced suppression of T lymphocyte response to concanavalin A. Magnesium 1988, 7, 166–172. [Google Scholar] [PubMed]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.-L.A.; Hall, D.D.; Chen, B.; Luczak, E.D.; Wang, Q.; Rokita, A.G.; Wehrens, X.H.T.; et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081. [Google Scholar] [CrossRef] [Green Version]
- Huo, J.; Lu, S.; Kwong, J.Q.; Bround, M.J.; Grimes, K.M.; Sargent, M.A.; Brown, M.E.; Davis, M.E.; Bers, D.M.; Molkentin, J.D. MCUb Induction Protects the Heart From Postischemic Remodeling. Circ. Res. 2020, 127, 379–390. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Ramaccini, D.; Montoya-Uribe, V.; Aan, F.J.; Modesti, L.; Potes, Y.; Wieckowski, M.R.; Krga, I.; Glibetić, M.; Pinton, P.; Giorgi, C.; et al. Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. cell Dev. Biol. 2020, 8, 624216. [Google Scholar] [CrossRef]
- Min, C.K.; Yeom, D.R.; Lee, K.-E.; Kwon, H.-K.; Kang, M.; Kim, Y.-S.; Park, Z.Y.; Jeon, H.; Kim, D.H. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 2012, 447, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [Green Version]
- Terentyev, D.; Györke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiferaw, Y.; Aistrup, G.L.; Wasserstrom, J.A. Intracellular Ca2+ waves, afterdepolarizations, and triggered arrhythmias. Cardiovasc. Res. 2012, 95, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Pozzoni, L.; Verzoni, A.; Lomuscio, A.; Guffanti, E.; Croce, L. Activity of a new long-acting anti-arrhythmic drug on ventricular extrasystole. Study with Holter continuous electrocardiographic monitoring. G. Ital. Cardiol. 1981, 11, 657–662. [Google Scholar] [PubMed]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Csordás, G.; Jowdy, C.; Schneider, T.G.; Csordás, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, J.J.; Wilson, J.J. Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr. Opin. Chem. Biol. 2020, 55, 9–18. [Google Scholar] [CrossRef]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxid. Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Sharipov, R.R.; Boyarkin, D.P.; Belosludtseva, N.V.; Dubinin, M.V.; Krasilnikova, I.A.; Bakaeva, Z.V.; Zgodova, A.E.; Pinelis, V.G.; Surin, A.M. The effect of DS16570511, a new inhibitor of mitochondrial calcium uniporter, on calcium homeostasis, metabolism, and functional state of cultured cortical neurons and isolated brain mitochondria. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129847. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.; Li, C.; Fernandez-Garcia, E.; Vais, H.; Foskett, K. The MCU Inhibitor Ds16570511 has Off-Target Effects on Mitochondrial Membrane Potential. Biophys. J. 2019, 116, 270a. [Google Scholar] [CrossRef] [Green Version]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Nägele, H.; Castel, M.A.; Deutsch, O.; Wagner, F.M.; Reichenspurner, H. Heart transplantation in a patient with multiple sclerosis and mitoxantrone-induced cardiomyopathy. J. Heart Lung Transplant. 2004, 23, 641–643. [Google Scholar] [CrossRef]
- Bittremieux, M.; La Rovere, R.M.; Akl, H.; Martines, C.; Welkenhuyzen, K.; Dubron, K.; Baes, M.; Janssens, A.; Vandenberghe, P.; Laurenti, L.; et al. Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ. 2019, 26, 531–547. [Google Scholar] [CrossRef]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.-P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood 2011, 117, 2924–2934. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, N.T.Q.; Paulsen, E.S.; Sehgal, P.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Madreiter-Sokolowski, C.T.; Gottschalk, B.; Parichatikanond, W.; Eroglu, E.; Klec, C.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Resveratrol Specifically Kills Cancer Cells by a Devastating Increase in the Ca2+ Coupling Between the Greatly Tethered Endoplasmic Reticulum and Mitochondria. Cell. Physiol. Biochem. 2016, 39, 1404–1420. [Google Scholar] [CrossRef] [PubMed]
- Francioso, A.; Mastromarino, P.; Masci, A.; D’Erme, M.; Mosca, L. Chemistry, stability and bioavailability of resveratrol. Med. Chem. 2014, 10, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.-Y.; Bai, H.-H.; Cai, J.-Y.; Deng, S.-P. The mechanism of kaempferol induced apoptosis and inhibited proliferation in human cervical cancer SiHa cell: From macro to nano. Scanning 2016, 38, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Nandakumar, N.; Palaniswami, R.; Nishigaki, Y.; Nishigaki, I. Kaempferol, a potential cytostatic and cure for inflammatory disorders. Eur. J. Med. Chem. 2014, 86, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Park, J.H.Y. Kaempferol Induces Cell Cycle Arrest in HT-29 Human Colon Cancer Cells. J. Cancer Prev. 2013, 18, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, M.; Lobatón, C.D.; Hernández-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wang, X.; Zhao, Y.; Wang, Z.; Du, L. The uptake behaviors of kaempferol and quercetin through rat primary cultured cortical neurons. Biomed. Chromatogr. 2006, 20, 1178–1184. [Google Scholar] [CrossRef]
- Bermont, F.; Hermant, A.; Benninga, R.; Chabert, C.; Jacot, G.; Santo-Domingo, J.; Kraus, M.R.-C.; Feige, J.N.; De Marchi, U. Targeting Mitochondrial Calcium Uptake with the Natural Flavonol Kaempferol, to Promote Metabolism/Secretion Coupling in Pancreatic β-cells. Nutrients 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Liao, Z.; Huang, L.; Liu, D.; Yin, D.; He, M. Kaempferol protects cardiomyocytes against anoxia/reoxygenation injury via mitochondrial pathway mediated by SIRT1. Eur. J. Pharmacol. 2015, 761, 245–253. [Google Scholar] [CrossRef]
- Montero, M.; Lobaton, C.D.; Moreno, A.; Alvarez, J. A novel regulatory mechanism of the mitochondrial Ca2+ uniporter revealed by the p38 mitogen-activated protein kinase inhibitor SB202190. FASEB J. 2002, 16, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Macle, L.; Nattel, S. Arrhythmias in 2015: Advances in drug, ablation, and device therapy for cardiac arrhythmias. Nat. Rev. Cardiol. 2016, 13, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Magee, W.P.; Deshmukh, G.; Deninno, M.P.; Sutt, J.C.; Chapman, J.G.; Tracey, W.R. Differing cardioprotective efficacy of the Na+/Ca2+ exchanger inhibitors SEA0400 and KB-R7943. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H903–H910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namekata, I.; Shimada, H.; Kawanishi, T.; Tanaka, H.; Shigenobu, K. Reduction by SEA0400 of myocardial ischemia-induced cytoplasmic and mitochondrial Ca2+ overload. Eur. J. Pharmacol. 2006, 543, 108–115. [Google Scholar] [CrossRef]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, S.M.; Egea, J.; López, M.G.; García, A.G. Mitochondrial Na+/Ca2+ exchanger, a new target for neuroprotection in rat hippocampal slices. Biochem. Biophys. Res. Commun. 2010, 400, 140–144. [Google Scholar] [CrossRef]
- Ruiz, A.; Alberdi, E.; Matute, C. CGP37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, protects neurons from excitotoxicity by blocking voltage-gated Ca2+ channels. Cell Death Dis. 2014, 5, e1156. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Nishimaru, K.; Aikawa, T.; Hirayama, W.; Tanaka, Y.; Shigenobu, K. Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br. J. Pharmacol. 2002, 135, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Kho, C.; Lee, A.; Hajjar, R.J. Altered sarcoplasmic reticulum calcium cycling--targets for heart failure therapy. Nat. Rev. Cardiol. 2012, 9, 717–733. [Google Scholar] [CrossRef] [Green Version]
- Motegi, K.; Tanonaka, K.; Takenaga, Y.; Takagi, N.; Takeo, S. Preservation of mitochondrial function may contribute to cardioprotective effects of Na+/Ca2+ exchanger inhibitors in ischaemic/reperfused rat hearts. Br. J. Pharmacol. 2007, 151, 963–978. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, O.; Akiyama, D.; Hara, T.; Cho, S.; Tomiyasu, S.; Sumikawa, K. Cardioprotective effects of KB-R7943, a novel inhibitor of Na+/Ca2+ exchanger, on stunned myocardium in anesthetized dogs. J. Anesth. 2005, 19, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Storozhevykh, T.P.; Senilova, Y.E.; Brustovetsky, T.; Pinelis, V.G.; Brustovetsky, N. Neuroprotective effect of KB-R7943 against glutamate excitotoxicity is related to mild mitochondrial depolarization. Neurochem. Res. 2010, 35, 323–335. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Vay, L.; Hernández-Sanmiguel, E.; Lobatón, C.D.; Moreno, A.; Montero, M.; Alvarez, J. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br. J. Pharmacol. 2007, 151, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Wiczer, B.M.; Marcu, R.; Hawkins, B.J. KB-R7943, a plasma membrane Na+/Ca2+ exchanger inhibitor, blocks opening of the mitochondrial permeability transition pore. Biochem. Biophys. Res. Commun. 2014, 444, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marco, G.; Vallese, F.; Jourde, B.; Bergsdorf, C.; Sturlese, M.; De Mario, A.; Techer-Etienne, V.; Haasen, D.; Oberhauser, B.; Schleeger, S.; et al. A High-Throughput Screening Identifies MICU1 Targeting Compounds. Cell Rep. 2020, 30, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Walters, A.M.; Porter, G.A.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant 2016, 31, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Cung, T.-T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Wang, W.; Karamanlidis, G.; Tian, R. Novel targets for mitochondrial medicine. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, G.; Preti, D.; Pedriali, G.; Aquila, G.; Missiroli, S.; Fantinati, A.; Caroccia, N.; Pacifico, S.; Bonora, M.; Talarico, A.; et al. Discovery of Novel 1,3,8-Triazaspiro[4.5]decane Derivatives That Target the c Subunit of F1/FO-Adenosine Triphosphate (ATP) Synthase for the Treatment of Reperfusion Damage in Myocardial Infarction. J. Med. Chem. 2018, 61, 7131–7143. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca2+-Related Proteins and Channels | Genetic Alteration/Protein Modification | Cellular Model | Ca2+-Related Mechanism | References |
|---|---|---|---|---|
| MCU | KD | Prostate and colon cancer, TNBC, HCC | Low mCa2+ uptake | [86,88,89] |
| Upregulation | AD | High mCa2+ uptake and mPTP opening | [119] | |
| MCUb | Cardiac specific KO | Mouse cardiomyocyte | Low mCa2+ uptake and mPTP opening reduction | [204,205] |
| OE | Mouse cardiomyocytes | Low mCa2+ uptake and mPTP opening inhibition | [217] | |
| MICU1 | Akt-mediated phosphorylation | Renal, ovarian, breast, and lung cancer | High mCa2+ uptake | [91] |
| NCLX | downregulation | AD mouse and human brains | High mCa2+ uptake | [130] |
| Tamoxifen-induced deletion | Adult mouse hearts | High mCa2+ uptake and mPTP opening | [220] | |
| MFN2 | KO | Mouse cardiac myocytes | Low mCa2+ uptake and mPTP opening reduction | [227] |
| BAP1 | IP3R3 | Mesothelioma | Low ER- Ca2+ release | [98] |
| deubiquitylation | ||||
| and stabilization | ||||
| PTEN | downregulation | Prostate and lung cancer | Low ER- Ca2+ release | [97] |
| PS1 | Mutation | Animal model of AD | High ER- Ca2+ release | [117,118] |
| PS1/PS2 | PS1 (mutation M146L) and PS2 (mutation N141I) | FAD | High ER- Ca2+ release | [116] |
| LRKK2 | mutation | Fibroblasts from PD patients | High mCa2+ uptake | [182] |
| PINK1 | Mutation | Dopaminergic neurons | High mCa2+ levels and increased ER–mitochondria contact sites | [174] |
| Parkin | Deficiency or mutation | PD patient fibroblasts | Low mCa2+ uptake and reduced ER–mitochondria contact sites | [180] |
| Therapeutic Target | Compound | Side Effects | Cell Permeability | In Vivo Applicability | References |
|---|---|---|---|---|---|
| MCU | anti-miR25 | Not observed | Yes | ND | [86] |
| Ru265 | Not observed | Yes | ND | [231] | |
| DS16570511 | ΔΨm loss and cell death | Yes | No | [232,234,235] | |
| Mitoxantrone | Cardiotoxicity | Yes | Limited | [236,237] | |
| Kaempferol | Not observed | Yes | Yes | [249,250] | |
| SB202190 | Not observed | Yes | ND | [249] | |
| KB-R7943 | Not selective | Yes | ND | [265] | |
| MICU1 | MCU-i4 and MCU-i11 | Not observed | Yes | ND | [267] |
| NCLX | KB-R7943, | Not selective | Yes | ND | [255,262,264] |
| CGP-37157 | Not observed | Yes | ND | [257,259] | |
| SEA0400 | Not observed | Yes | ND | [255,256] | |
| mPTP | Cyclosporin A | Not observed | Yes | ND | [270] |
| 1,3,8-Triazaspiro[4.5]decane derivatives | Not observed | Yes | ND | [274] | |
| SERCA | Thapsigargin | Not selective | Yes | No | [241] |
| Mipsagargin G-202 | Not observed | Yes | Yes | [243] | |
| Resveratrol and piceatannol | Not observed | Yes | Limited bioavailability | [244] | |
| MAMs: BCl2-IP3R3 | BIRD-2 | Not observed | Yes | ND | [238,239] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. https://doi.org/10.3390/cells10061317
Modesti L, Danese A, Angela Maria Vitto V, Ramaccini D, Aguiari G, Gafà R, Lanza G, Giorgi C, Pinton P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells. 2021; 10(6):1317. https://doi.org/10.3390/cells10061317
Chicago/Turabian StyleModesti, Lorenzo, Alberto Danese, Veronica Angela Maria Vitto, Daniela Ramaccini, Gianluca Aguiari, Roberta Gafà, Giovanni Lanza, Carlotta Giorgi, and Paolo Pinton. 2021. "Mitochondrial Ca2+ Signaling in Health, Disease and Therapy" Cells 10, no. 6: 1317. https://doi.org/10.3390/cells10061317