
Assessing the Anti-Inflammatory Activity of the Anxiolytic Drug Buspirone Using CRISPR-Cas9 Gene Editing in LPS-Stimulated BV-2 Microglial Cells

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Knockout Cell Lines Using CRISPR-Cas9 Gene Editing
2.2. Cell Culture
2.3. Cell Viability
2.4. Nitric Oxide (Griess Reagent Assay)
2.5. RNA Extraction and cDNA Synthesis
2.6. Real-Time Quantitative Polymerase Chain Reaction
2.7. Protein Extraction and Western Blot
2.8. Statistical Analysis
3. Results
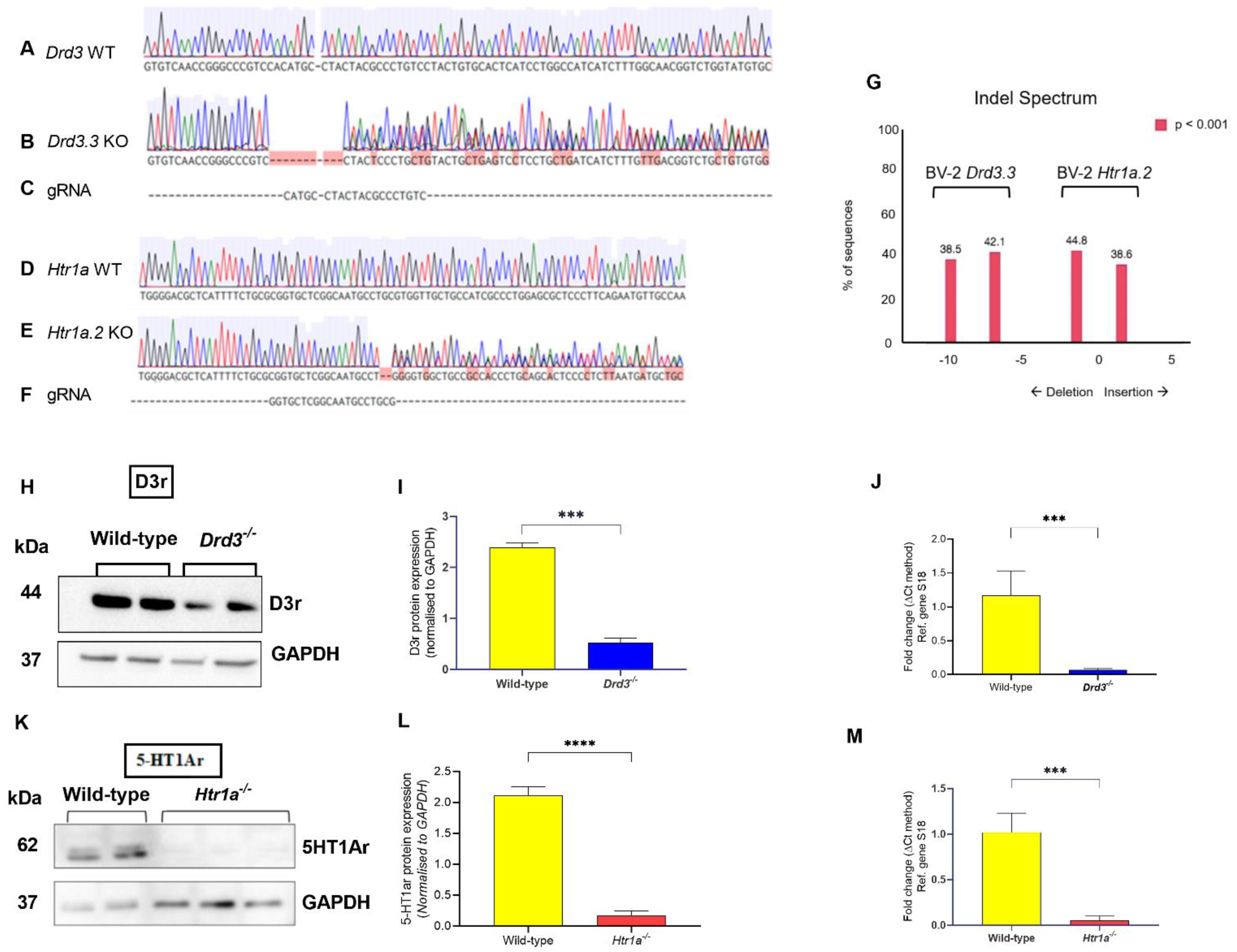
3.1. Generation of Stable Drd3−/− and Htr1a−/− Cell Lines
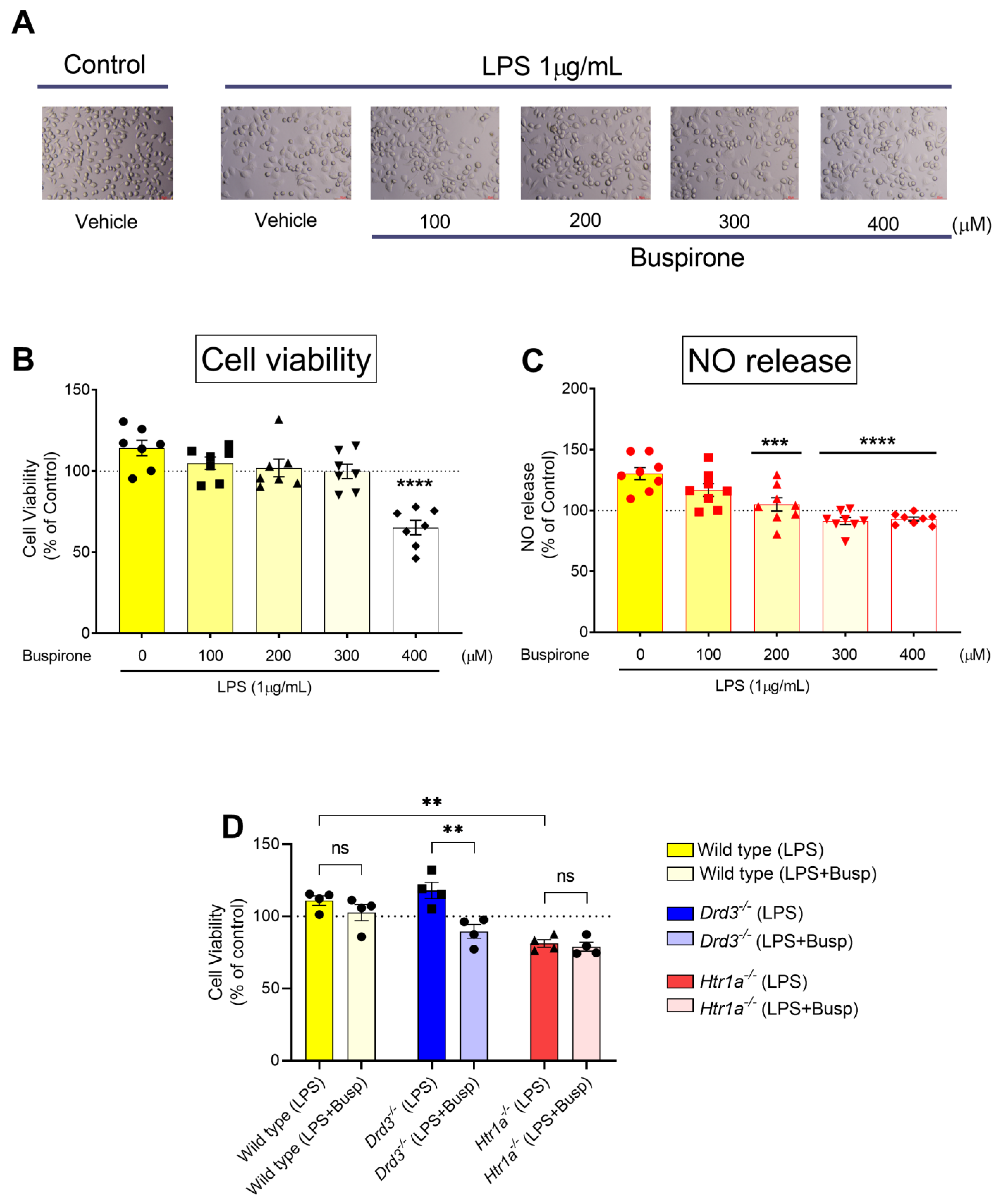
3.2. Establishing the Optimal Buspirone Concentration and Assessing the Effects of Drd3 and Htr1a Gene Deletions and Buspirone Treatment on Cell Viability Following Exposure to LPS
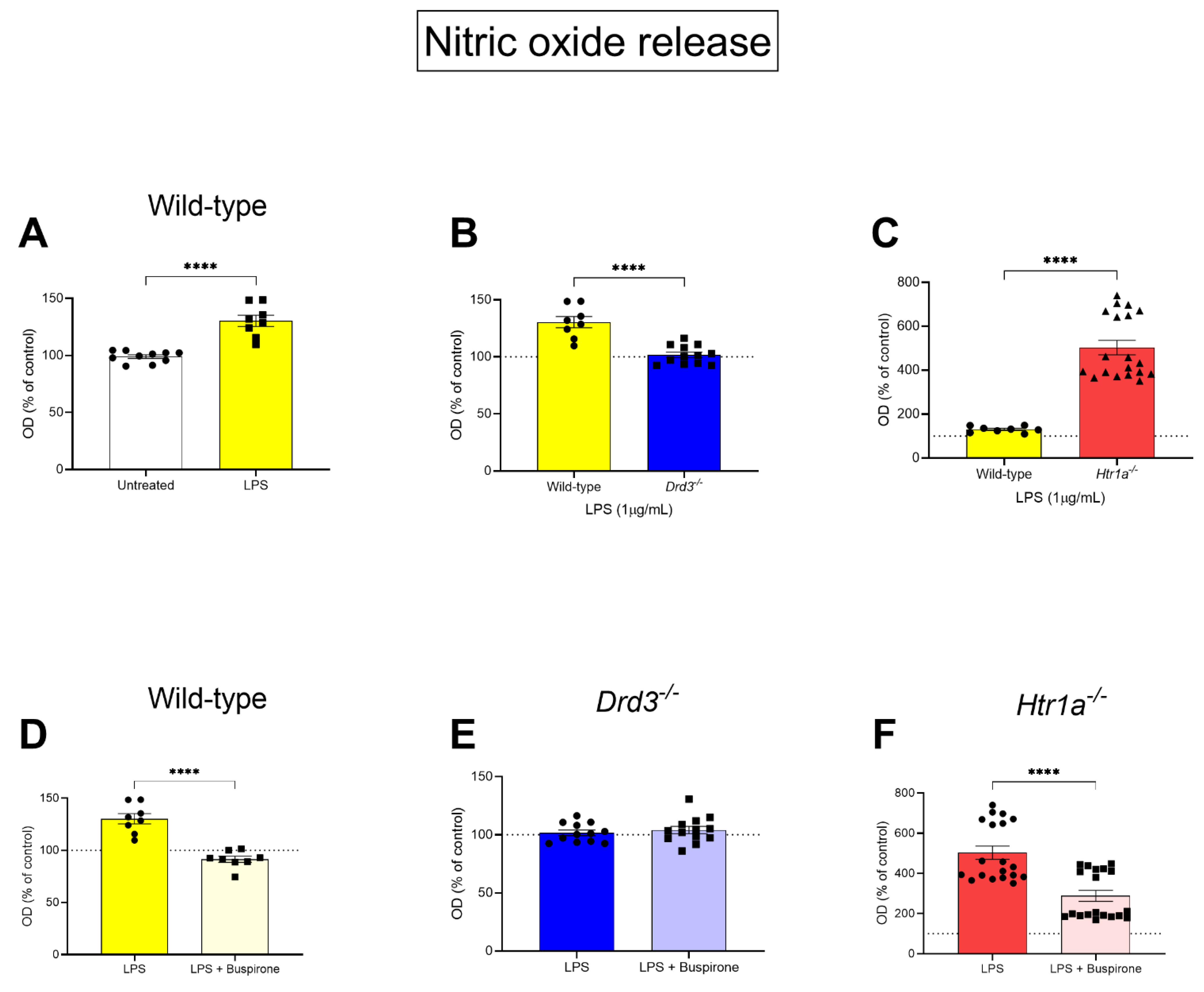
3.3. Nitric Oxide Release in Wild-Type, Drd3−/− and Htr1a−/− Microglial Cells Following LPS Challenge and Buspirone Treatment
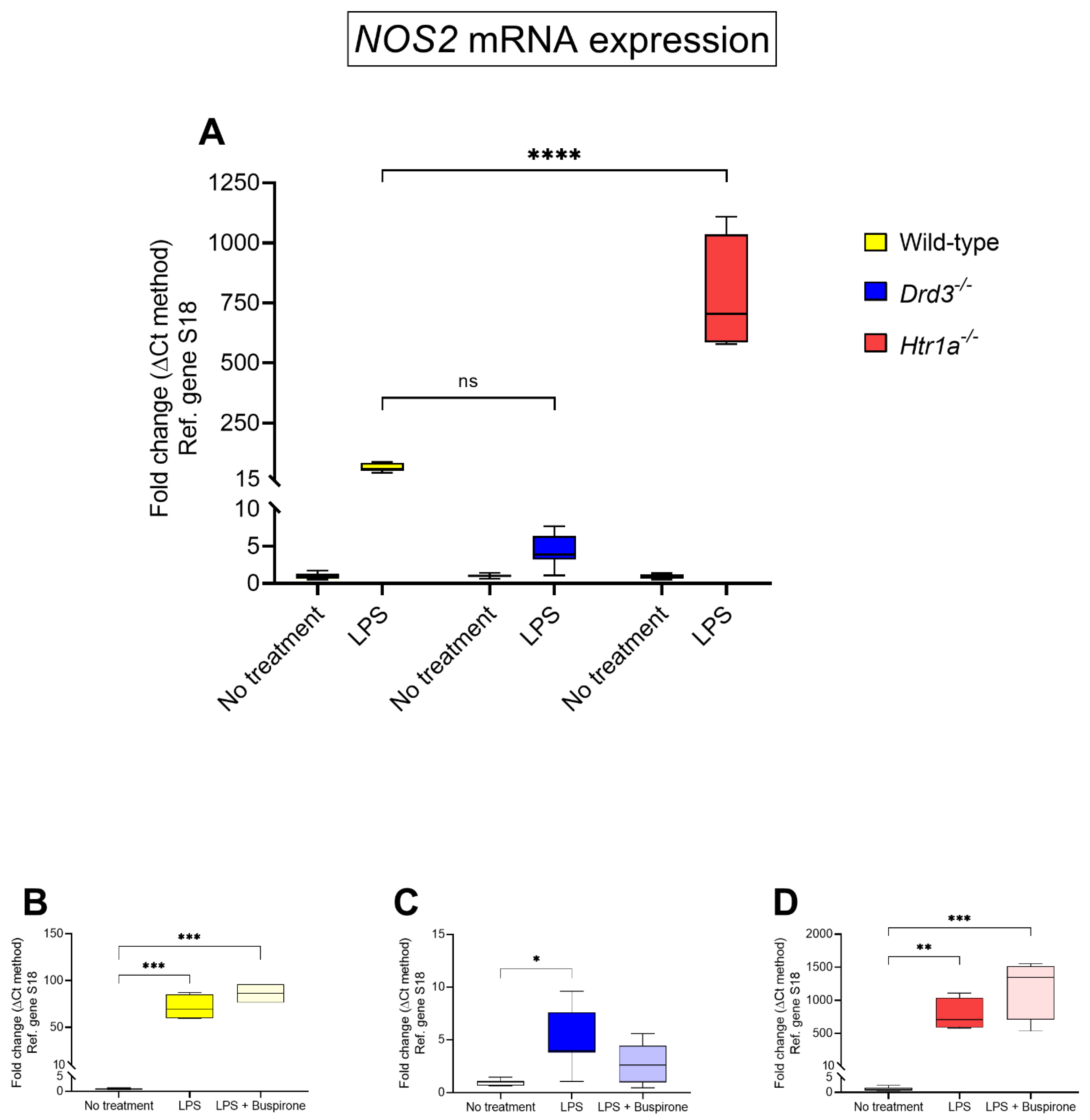
3.4. Effects of Buspirone Treatment on Inducible Nitric Oxide Synthetase (NOS2) Gene Expression in Wild Type, Drd3−/− and Htr1a−/− Knockout Microglial Cells after Exposure to LPS
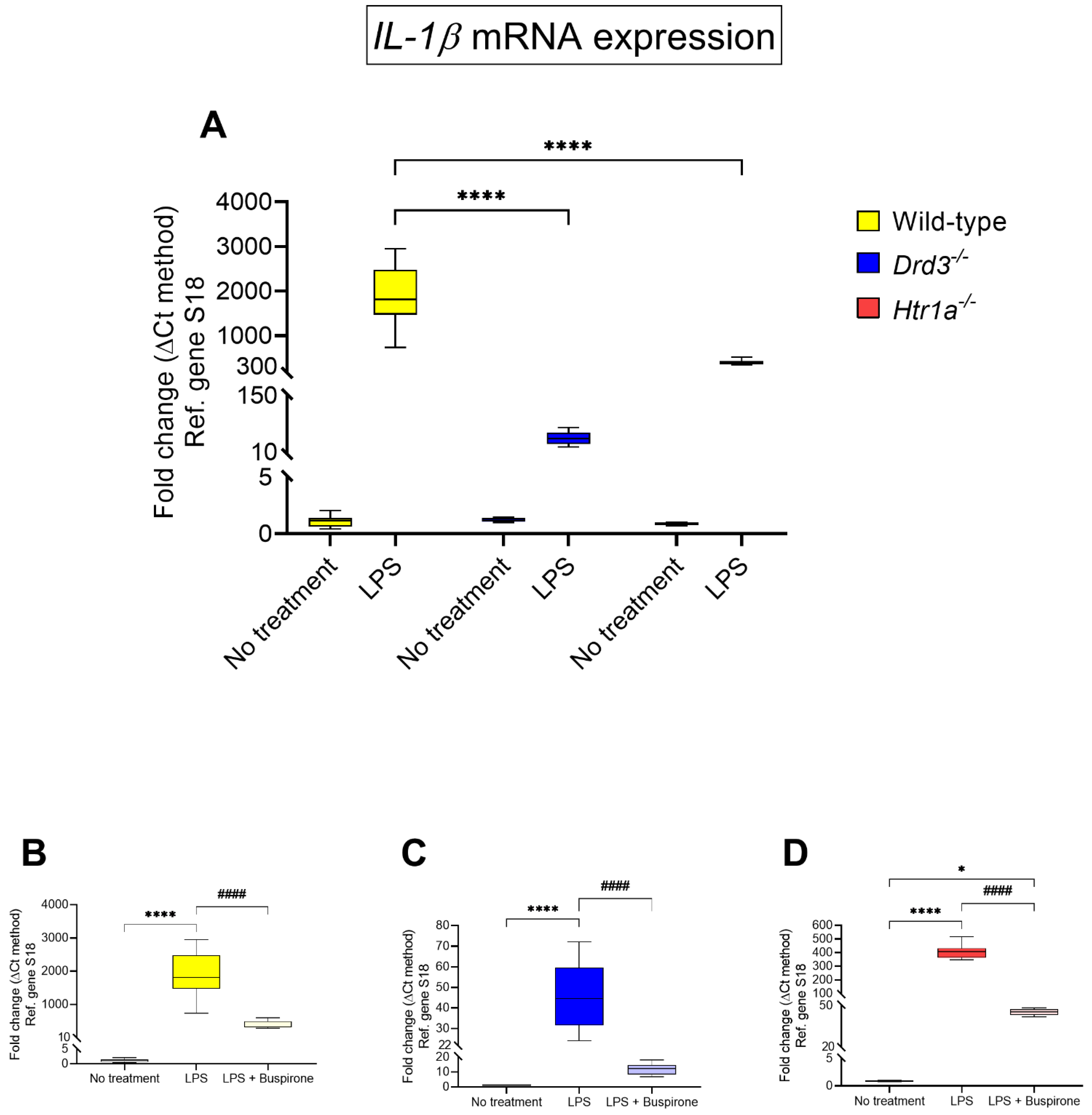
3.5. Effects of Buspirone Treatment on Interleukin-1β (IL-1β) Gene Expression in Wild Type, Drd3−/− and Htr1a−/− Knockout Microglial Cells after Exposure to LPS
3.6. Effects of Buspirone Treatment on Tumor Necrosis Factor-α (TNF-α) Gene Expression in Wild Type, Drd3−/− and Htr1a−/− Knockout Microglial Cells after Exposure to LPS
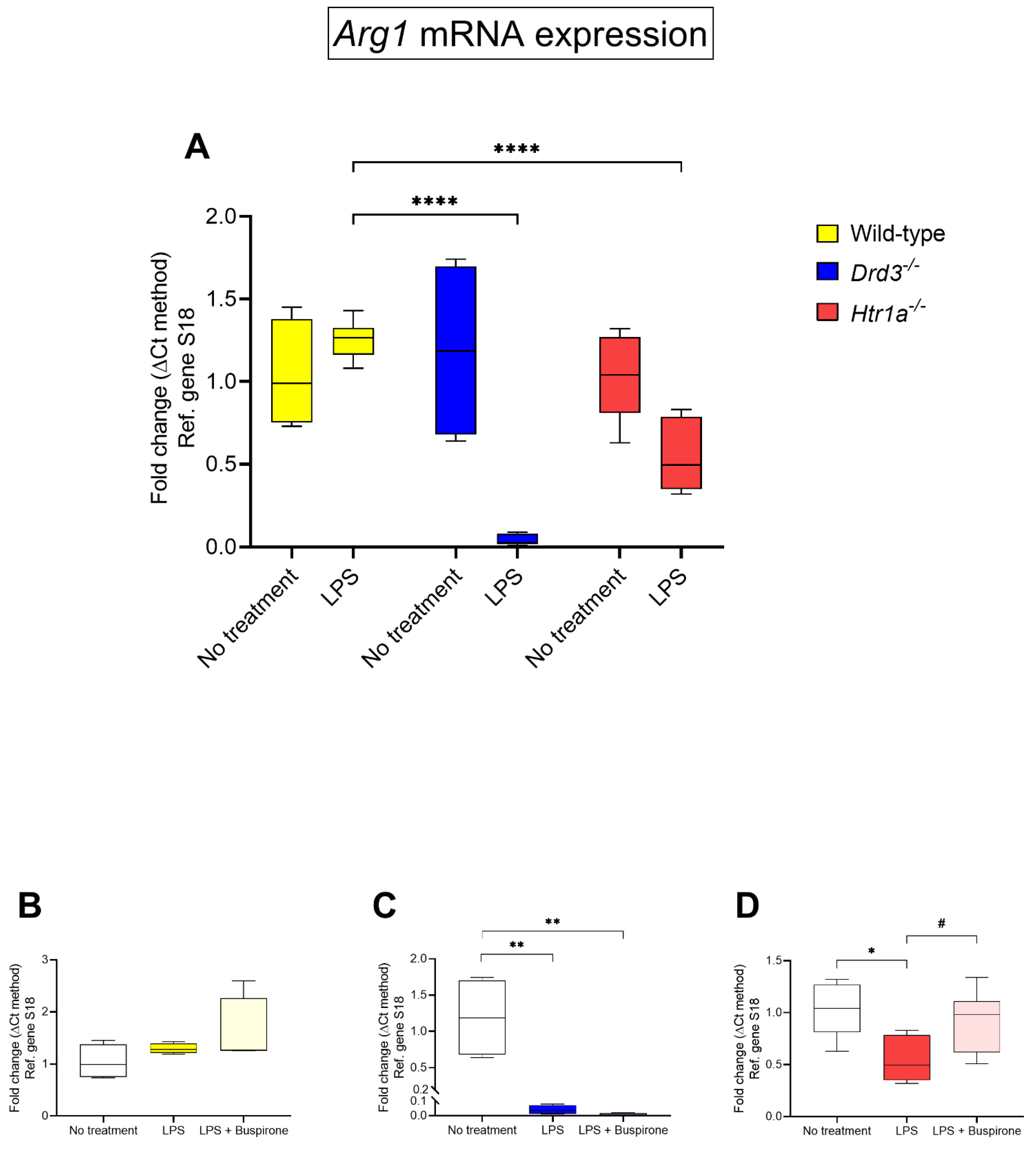
3.7. Effects of Buspirone Treatment on Arginase 1 (Arg1) Gene Expression in Wild Type, Drd3−/− and Htr1a−/− Knockout Microglial Cells after Exposure to LPS
3.8. Effects of Buspirone Treatment on Found in Inflammatory Zone 1 (FIZZ1) Gene Expression in Wild Type, Drd3−/− and Htr1a−/− Knockout Microglial Cells after Exposure to LPS
4. Discussion
4.1. Targeting Drd3 and Htr1a Genes to Study the Effects of Buspirone in Microglial Cells
4.2. Genotype-Specific and Drug-Related Changes in BV-2 Cell Viability and Nitric Oxide Release after LPS Challenge
4.3. Genotype-Specific and Drug-Related Changes of Pro-Inflammatory Cytokines in LPS-Stimulated BV-2 Microglia
4.4. Genotype-Specific and Drug-Related Changes of Anti-Inflammatory Cytokines in LPS-Stimulated BV-2 Microglia
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Honorato, J.; Catalan, M. La buspirona: Un nuevo fármaco ansiolítico no benzodiacepínico [buspirone: A new non-benzodiazepine anxiolytic drug]. Rev. Clin. Esp. 1990, 186, 286–291. [Google Scholar]
- Caliendo, G.; Santagada, V.; Perissutti, E.; Fiorino, F. Derivatives as 5HT1A receptor ligands-past and present. Curr. Med. Chem. 2005, 12, 1721–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Garcia, A.L.; Newman-Tancredi, A.; Leonardo, E.D. 5-ht(1a) receptors in mood and anxiety: Recent insights into autoreceptor versus heteroreceptor function. Psychopharmacology 2014, 231, 623–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raber, J.; Wienclaw, R.A.; Cataldo, L.J. The Gale Encyclopedia of Mental Health 2012; Gale: Detroit, MI, USA, 2012; pp. 264–267. [Google Scholar]
- Kim, S.W.; Fowler, J.S.; Skolnick, P.; Muench, L.; Kang, Y.; Shea, C.; Logan, J.; Kim, H.; Carter, P.; King, P.; et al. Therapeutic doses of buspirone block D3 receptors in the living primate brain CORRIGENDUM. Int. J. Neuropsychopharmacol. 2014, 17, 1354. [Google Scholar] [CrossRef] [Green Version]
- Leggio, G.M.; Camillieri, G.; Platania, C.B.M.; Castorina, A.; Marrazzo, G.; Torrisi, S.A.; Nona, C.N.; D’Agata, V.; Nobrega, J.; Stark, H.; et al. Dopamine D3 receptor is necessary for ethanol consumption: An approach with buspirone. Neuropsychopharmacology 2014, 39, 2017–2028. [Google Scholar] [CrossRef]
- Bhatia, A.; Lencher, J.; Saadabadi, A. Biochemistry, Dopamine Receptors; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Leggio, G.M.; Torrisi, S.A.; Castorina, A.; Platania, C.B.M.; Impellizzeri, A.A.R.; Fidilio, A.; Caraci, F.; Bucolo, C.; Drago, F.; Salomone, S. Dopamine D3 receptor-dependent changes in alpha6 GABAA subunit expression in striatum modulate anxiety-like behaviour: Responsiveness and tolerance to diazepam. Eur. Neuropsychopharmacol. 2015, 25, 1427–1436. [Google Scholar] [CrossRef]
- Marzagalli, R.; Leggio, G.; Bucolo, C.; Pricoco, E.; Keay, K.; Cardile, V.; Castorina, S.; Salomone, S.; Drago, F.; Castorina, A. Genetic blockade of the dopamine D3 receptor enhances hippocampal expression of PACAP and receptors and alters their cortical distribution. Neuroscience 2016, 316, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; D’Amico, A.; Scuderi, S.; Leggio, G.; Drago, F.; D’Agata, V. Dopamine D3 receptor deletion increases tissue plasminogen activator (tPA) activity in prefrontal cortex and hippocampus. Neuroscience 2013, 250, 546–556. [Google Scholar] [CrossRef]
- Castorina, A.; Broome, S.T.; Louangaphay, K.; Keay, K.A.; Leggio, G.M.; Musumeci, G. Dopamine: An immune transmitter. Neural Regen. Res. 2020, 15, 2173–2185. [Google Scholar] [CrossRef]
- McKenna, F.; McLaughlin, P.; Lewis, B.; Sibbring, G.; Cummerson, J.; Bowen-Jones, D.; Moots, R. Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: A flow cytometric study. J. Neuroimmunol. 2002, 132, 34–40. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Coronel, I.; Florán, B. Dopamine receptors and neurodegeneration. Aging Dis. 2015, 6, 349–368. [Google Scholar] [CrossRef] [Green Version]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcón, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J. Neuroinflamm. 2019, 16, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgueta, D.; Aymerich, M.S.; Contreras, F.; Montoya, A.; Celorrio, M.; Rojo-Bustamante, E.; Riquelme, E.; Gonzalez, H.; Vasquez, M.; Franco, R.; et al. Pharmacologic antagonism of dopamine receptor D3 attenuates neurodegeneration and motor impairment in a mouse model of Parkinson’s disease. Neuropharmacology 2017, 113, 110–123. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van Der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 2018, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.-C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef]
- Yang, B.; Li, R.; Greenlief, C.M.; Fritsche, K.L.; Gu, Z.; Cui, J.; Lee, J.C.; Beversdorf, D.Q.; Sun, G.Y. Unveiling anti-oxidative and anti-inflammatory effects of docosahexaenoic acid and its lipid peroxidation product on lipopolysaccharide-stimulated BV-2 microglial cells. J. Neuroinflamm. 2018, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, J.; You, Z. Switching of the microglial activation phenotype is a possible treatment for depression disorder. Front. Cell. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef]
- Sun, G.Y.; Li, R.; Yang, B.; Fritsche, K.L.; Beversdorf, D.Q.; Lubahn, D.B.; Geng, X.; Lee, J.C.; Greenlief, C.M. Quercetin potentiates Docosahexaenoic Acid to suppress Lipopolysaccharide-induced Oxidative/inflammatory responses, alter lipid peroxidation products, and enhance the adaptive stress pathways in BV-2 Microglial cells. Int. J. Mol. Sci. 2019, 20, 932. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.-P.; Cheng, Z.-Y.; He, L. The modulatory role of dopamine receptors in brain neuroinflammation. Int. Immunopharmacol. 2019, 76, 105908. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Weinreb, O.; Mandel, S.A.; Youdim, M.B.H.; Frenkel, D. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J. Neurochem. 2013, 129, 434–447. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouwels, S.D.; Wiersma, V.R.; Fokkema, I.E.; Berg, M.; Hacken, N.H.T.; Berge, M.V.D.; Heijink, I.; Faiz, A. Acute cigarette smoke-induced eQTL affects formyl peptide receptor expression and lung function. Respirology 2021, 26, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Henn, A.; Lund, S.; Hedtjarn, M.; Schrattenholz, A.; Porzgen, P.; Leist, M. The suitability of bv2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009, 26, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Bucolo, C.; Leggio, G.M.; Maltese, A.; Castorina, A.; D’Agata, V.; Drago, F. Dopamine-3 receptor modulates intraocular pressure: Implications for glaucoma. Biochem. Pharm. 2012, 83, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Castorina, A.; Adorno, A.; Mazzone, V.; Carnazza, M.L.; D’Agata, V. PACAP and VIP affect NF1 expression in rat malignant peripheral nerve sheath tumor (MPNST) cells. Neuropeptides 2010, 44, 45–51. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time pcr data by the comparative c(t) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Castorina, A.; Leggio, G.M.; Giunta, S.; Magro, G.G.; Scapagnini, G.; Drago, F.; D’Agata, V. Neurofibromin and Amyloid precursor protein expression in Dopamine D3 receptor knock-out mice brains. Neurochem. Res. 2010, 36, 426–434. [Google Scholar] [CrossRef]
- Castorina, A.; Vogiatzis, M.; Kang, J.W.; Keay, K.A. PACAP and VIP expression in the periaqueductal grey of the rat following sciatic nerve constriction injury. Neuropeptides 2019, 74, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, G.D.; Lancaster, S.P.; Montague-Clouse, J. The association of buspirone and its metabolite 1-pyrimidinylpiperazine in the remission of comorbid anxiety with depressive features and alcohol dependency. Psychopharmacol. Bull. 1991, 27, 163–170. [Google Scholar]
- Le Foll, B.; Payer, R.; Di Ciano, P.; Guranda, M.; Nakajima, S.; Tong, J.; Mansouri, E.; Wilson, A.A.; Houle, S.; Meyer, J.H.; et al. Occupancy of Dopamine D3 and D2 Receptors by Buspirone: A [11C]-(+)-PHNO PET Study in Humans. Neuropsychopharmacology 2015, 41, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.M.; Rutter, H.; Killick, R.; Hodges, A. [P1-208]: CRISPR/CAS9-mediated gene editing of trem2 in monocytic and microglial cell lines. Alzheimer’s Dement. 2017, 13, P322. [Google Scholar] [CrossRef]
- Hanger, B.; Couch, A.; Rajendran, L.; Srivastava, D.P.; Vernon, A.C. Emerging developments in human induced pluripotent stem cell-derived microglia: Implications for modelling psychiatric disorders with a neurodevelopmental origin. Front. Psychiatry 2020, 11, 789. [Google Scholar] [CrossRef]
- Prante, O.; Dörfler, M.; Gmeiner, P. Dopamine Receptor Subtype-Selective Drugs: D2-Like Receptors. In The Dopamine Receptors; Humana Press: Totowa, NJ, USA, 2010; pp. 101–135. [Google Scholar]
- Twayana, K.S.; Chaudhari, N.; Ravanan, P. Prolonged lipopolysaccharide exposure induces transient immunosuppression in BV2 microglia. J. Cell. Physiol. 2019, 234, 1889–1903. [Google Scholar] [CrossRef]
- McGuiness, B.; Gibney, S.M.; Beumer, W.; Versnel, M.A.; Sillaber, I.; Harkin, A.; Drexhage, H.A. Exaggerated increases in microglia proliferation, brain inflammatory response and sickness behaviour upon lipopolysaccharide stimulation in non-obese diabetic mice. Neuroimmunomodulation 2016, 23, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, S.; Furube, E.; Itoh, M.; Nakashima, T.; Miyata, S. Robust increase of microglia proliferation in the fornix of hippocampal axonal pathway after a single LPS stimulation. J. Neuroimmunol. 2015, 285, 31–40. [Google Scholar] [CrossRef]
- Krabbe, G.; Matyash, V.; Pannasch, U.; Mamer, L.; Boddeke, H.W.; Kettenmann, H. Activation of serotonin receptors promotes microglial injury-induced motility but attenuates phagocytic activity. Brain Behav. Immun. 2012, 26, 419–428. [Google Scholar] [CrossRef]
- Stojakovic, A.; Paz-Filho, G.; Arcos-Burgos, M.; Licinio, J.; Wong, M.-L.; Mastronardi, C.A. Role of the IL-1 pathway in dopaminergic neurodegeneration and decreased voluntary movement. Mol. Neurobiol. 2016, 54, 4486–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenkgrub, J.; Joniec-Maciejak, I.; Sznejder-Pacholek, A.; Wawer, A.; Ciesielska, A.; Bankiewicz, K.; Czlonkowska, A.; Czlonkowski, A. Effect of human interleukin-10 on the expression of nitric oxide synthases in the mptp-based model of parkinson’s disease. Pharmacol. Rep. 2013, 65, 44–49. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, X.-Y.; Ye, M.-L.; Luo, C.-X.; Wu, H.-Y.; Hu, Y.; Zhou, Q.-G.; Wu, D.-L.; Zhu, L.-J.; Zhu, N.-Y. Neuronal nitric oxide synthase alteration accounts for the role of 5-HT1A receptor in modulating anxiety-related behaviors. J. Neurosci. 2010, 30, 2433–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgueta, D.; Contreras, F.; Prado, C.; Montoya, A.; Ugalde, V.; Chovar, O.; Villagra, R.; Henriquez, C.; Abellanas, M.A.; Aymerich, M.S.; et al. Dopamine receptor d3 expression is altered in cd4(+) t-cells from parkinson’s disease patients and its pharmacologic inhibition attenuates the motor impairment in a mouse model. Front. Immunol. 2019, 10, 981. [Google Scholar] [CrossRef] [Green Version]
- Leal, M.C.; Casabona, J.C.; Puntel, M.; Pitossi, F.J. Interleukin-1beta and tumor necrosis factor-alpha: Reliable targets for protective therapies in parkinson’s disease? Front. Cell. Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Lewin, A.S.; Ahmed, C.M.; Biswal, M.R.; Li, H.; Han, P. The 5ht1a agonist xaliproden exhibits anti-oxidant and anti-inflammatory properties and protects the retina in a mouse model of geographic atrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4428. [Google Scholar]
- Besser, M.J.; Ganor, Y.; Levite, M. Dopamine by itself activates either D2, D3 or D1/D5 dopaminergic receptors in normal human T-cells and triggers the selective secretion of either IL-10, TNFα or both. J. Neuroimmunol. 2005, 169, 161–171. [Google Scholar] [CrossRef]
- Sharifi, H.; Nayebi, A.M.; Farajnia, S. 8-OH-DPAT (5-HT1A agonist) Attenuates 6-Hydroxy- dopamine-induced catalepsy and modulates inflammatory cytokines in rats. Iran J. Basic Med. Sci. 2013, 16, 1270–1275. [Google Scholar]
- Cunha, C.; Gomes, C.; Vaz, A.R.; Brites, D. Exploring new inflammatory biomarkers and pathways during LPS-induced M1 polarization. Mediat. Inflamm. 2016, 2016, 1–17. [Google Scholar] [CrossRef]
- Menzies, F.M.; Henriquez, F.L.; Alexander, J.; Roberts, C.W. Sequential expression of macrophage anti-microbial/inflammatory and wound healing markers following innate, alternative and classical activation. Clin. Exp. Immunol. 2010, 160, 369–379. [Google Scholar] [CrossRef]
- Zanin, R.F.; Braganhol, E.; Bergamin, L.S.; Campesato, L.F.I.; Zanotto-Filho, A.; Moreira, J.C.F.; Morrone, F.B.; Sévigny, J.; Schetinger, M.R.C.; Wyse, A.T.D.S.; et al. Differential macrophage activation alters the expression profile of NTPDase and ecto-5′-Nucleotidase. PLoS ONE 2012, 7, e31205. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, F.; Xue, L.; Wang, B.; Li, J.; Chen, Y.; Chen, T. Methamphetamine causes neurotoxicity by promoting polarization of macrophages and inflammatory response. Hum. Exp. Toxicol. 2017, 37, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Chourbaji, S.; Brandwein, C.; Vogt, M.A.; Dormann, C.; Mueller, R.; Drescher, K.; Gross, G.; Gass, P. Dopamine receptor 3 (D3) knockout mice show regular emotional behaviour. Pharm. Res. 2008, 58, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Afzelius, P.; Nielsen, S.D.; Hofmann, B.; Nielsen, J.O. The serotonin analogue buspirone increases the function of PBMC from HIV-infected Individuals In Vitro. Scand. J. Infect. Dis. 1997, 29, 117–120. [Google Scholar] [CrossRef]
- Freire-Garabal, M.; Núñez-Iglesias, M.J.; Balboa, J.; Fernández-Rial, J.; Rey-Méndez, M. Effects of buspirone on the immune response to stress in mice. Pharm. Biochem. Behav. 1995, 51, 821–825. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Ref. Seq.) | Forward Sequence 5′-3′ Reverse Sequence 3′-5′ | Location | Tm (°C) |
|---|---|---|---|
| Mouse Drd3 (NC_0.000082.6) | (AAAC) CATGCCTACTACGCCCTGTC (CACC) GACAGGGCGTAGTAGGCATG | 472 491 | 69.7 73.9 |
| Mouse Htr1a (NC_0.000079.6) | (CACC) GGTGCTCGGCAATGCCTGCG (AAAC) CGCAGGCATTGCCGAGCACC | 501 520 | 84.2 80.0 |
| Gene (Ref. Seq.) | Forward Sequence 5′-3′ Reverse Sequence 3′-5′ | Location | Tm (°C) | Length (BP) |
|---|---|---|---|---|
| Mouse Drd3 (NC_0.000082.6) | TTGTTGTCTGTGTTCGCCCA AGGGTCCCATCATTCATGCC | 23 883 | 67.8 68.2 | 861 |
| Mouse Htr1a (NC_0.000079.6) | AGTGAAATGGACAGCGCGA AATGAGCCAAGTGAGCGAGA | 27 840 | 67 65.1 | 814 |
| Gene (Ref. Seq.) | Forward Sequence 5′-3′ Reverse Sequence 3′-5′ | Location | Tm (°C) | Length (BP) |
|---|---|---|---|---|
| Mouse Drd3 (NC_0.000082.6) | CCAGTTCTTACAGCACTGCCT CCGGAGCAGCATGTACCATAA | 209 688 | 63.3 66.9 | 480 |
| Mouse Htr1a (NC_0.000079.6) | CCCTTCGAAACTCCCCAGAAA GAGCCGATGAGATAGTTGGCA | 239 581 | 68.2 66.4 | 343 |
| Gene | GeneBank Accession Number | Primer Sequence | Length (bp) |
|---|---|---|---|
| S18 | NM_011296.2 | Fwd 5′ CCCTGAGAAGTTCCAGCACA 3′ Rev 5′ GGTGAGGTCGATGTCTGCTT 3′ | 145 |
| Drd3 | NM_007877 | Fwd 5′ GGGGTGACTGTCCTGGTCTA 3′ Rev 5′ AAGCCAGGTCTGATGCTGAT 3′ | 100 |
| IL-1β | NM_008361.4 | Fwd 5′ GCTACCTGTGTCTTTCCCGT 3′ Rev 5′ CATCTCGGAGCCTGTAGTGC 3′ | 164 |
| TNF-α | NM_013693.3 | Fwd 5′ ATGGCCTCCCTCTCATCAGT 3′ Rev 5′ TTTGCTACGACGTGGGCTAC 3′ | 97 |
| FIZZ1 | NM_020509.3 | Fwd 5′ AGCTGATGGTCCCAGTGAAT 3′ Rev 5′ AGTGGAGGGATAGTTAGCTGG 3′ | 98 |
| Arg1 | NM_007482.3 | Fwd 5′ ACAAGACAGGGCTCCTTTCAG 3′ Rev 5′ TTAAAGCCACTGCCGTGTTC 3′ | 105 |
| NOS2 | NM_010927.4 | Fwd 5′ TACCAAAGTGACCTGAAAGAGG 3′ Rev 5′ TCATCTTGTATTGTTGGGCTGA 3′ | 89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas Broome, S.; Fisher, T.; Faiz, A.; Keay, K.A.; Musumeci, G.; Al-Badri, G.; Castorina, A. Assessing the Anti-Inflammatory Activity of the Anxiolytic Drug Buspirone Using CRISPR-Cas9 Gene Editing in LPS-Stimulated BV-2 Microglial Cells. Cells 2021, 10, 1312. https://doi.org/10.3390/cells10061312
Thomas Broome S, Fisher T, Faiz A, Keay KA, Musumeci G, Al-Badri G, Castorina A. Assessing the Anti-Inflammatory Activity of the Anxiolytic Drug Buspirone Using CRISPR-Cas9 Gene Editing in LPS-Stimulated BV-2 Microglial Cells. Cells. 2021; 10(6):1312. https://doi.org/10.3390/cells10061312
Chicago/Turabian StyleThomas Broome, Sarah, Teagan Fisher, Alen Faiz, Kevin A. Keay, Giuseppe Musumeci, Ghaith Al-Badri, and Alessandro Castorina. 2021. "Assessing the Anti-Inflammatory Activity of the Anxiolytic Drug Buspirone Using CRISPR-Cas9 Gene Editing in LPS-Stimulated BV-2 Microglial Cells" Cells 10, no. 6: 1312. https://doi.org/10.3390/cells10061312