
Hepatic Wnt1 Inducible Signaling Pathway Protein 1 (WISP-1/CCN4) Associates with Markers of Liver Fibrosis in Severe Obesity

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.2. Blood Sampling and Tissue Collection
2.3. Histology and Immunohistochemistry
2.4. Analytical Procedures
2.5. Cell Culture
2.6. Gene Expression Analyses
2.7. Analysis of WISP1 Expression in Human Tissue Panel
2.8. Statistical Analyses
3. Results
3.1. Circulating CCN4 and Anthropometric and Biochemical Parameters
3.2. Association of Hepatic CCN4 Expression with Fibrosis Markers
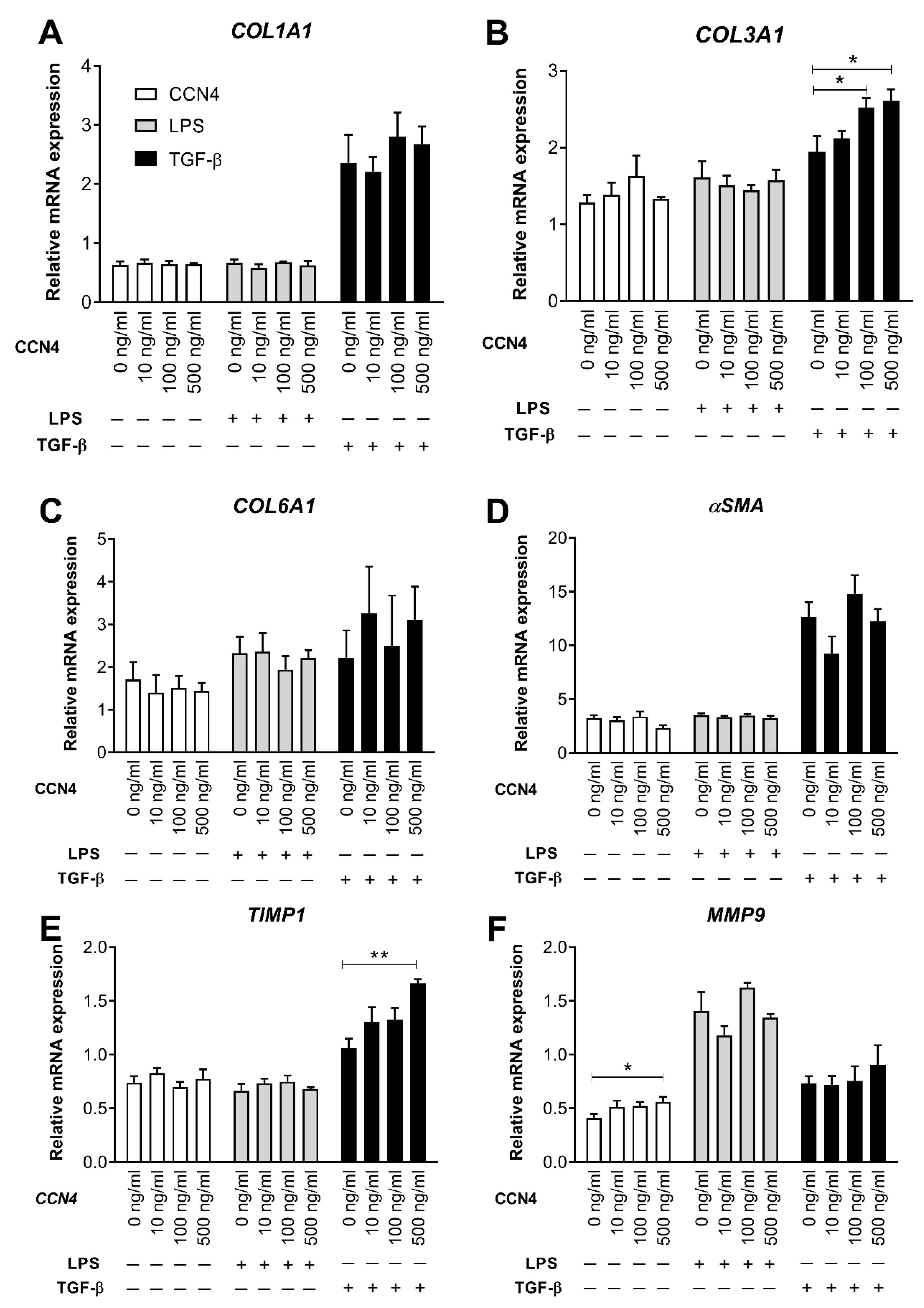
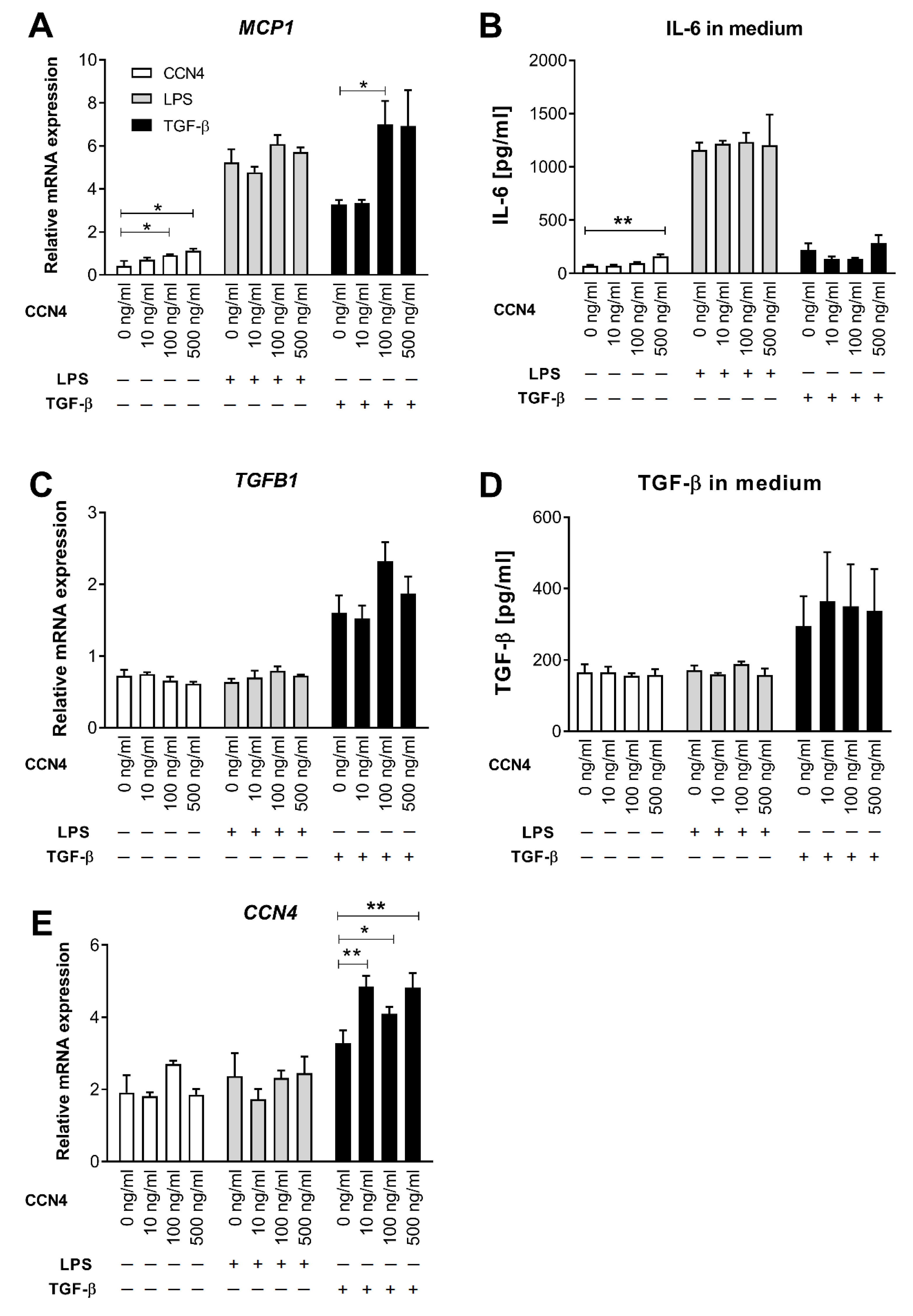
3.3. Effects of CCN4 Treatment in Human Hepatic Stellate Cells
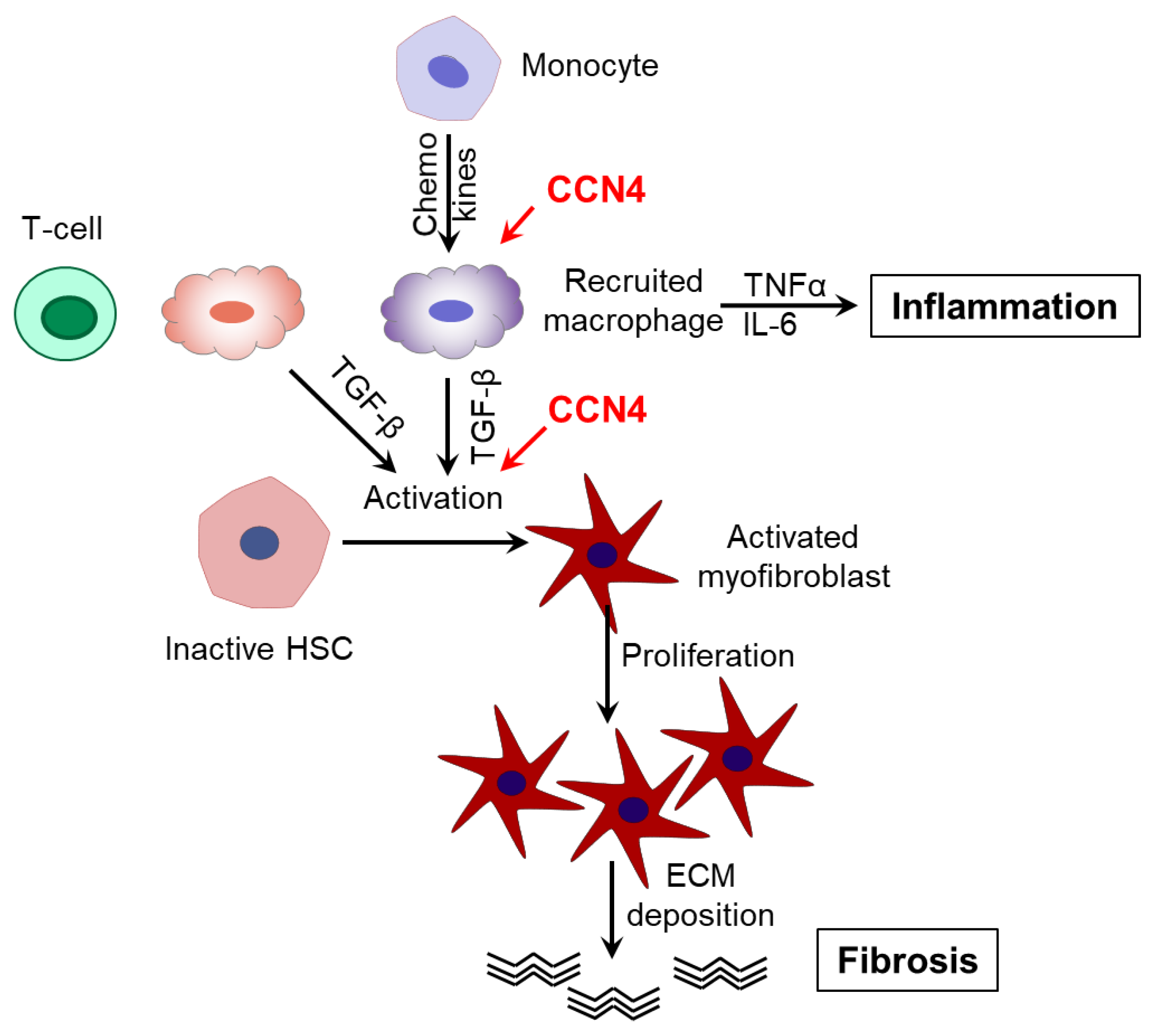
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metab. Clin. Exp. 2020, 111S, 154299. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Eng. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Manon-Jensen, T.; Genovese, F.; Kristensen, J.H.; Nielsen, M.J.; Sand, J.M.; Hansen, N.U.; Bay-Jensen, A.C.; Bager, C.L.; Krag, A.; et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G807–G830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, C.; Luedde, T.; Sauerbruch, T.; Scholten, D.; Streetz, K.; Tacke, F.; Tolba, R.; Trautwein, C.; Trebicka, J.; Weiskirchen, R. Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects. Fibrogenes. Tissue Repair 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Sanchez, N.; Valencia-Rodriguez, A.; Coronel-Castillo, C.; Vera-Barajas, A.; Contreras-Carmona, J.; Ponciano-Rodriguez, G.; Zamora-Valdes, D. The cellular pathways of liver fibrosis in non-alcoholic steatohepatitis. Ann. Transl. Med. 2020, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.; Osawa, Y.; Kimura, K. Wnt/beta-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 2018, 19, 3103. [Google Scholar] [CrossRef] [Green Version]
- Brigstock, D.R. The CCN family: A new stimulus package. J. Endocrinol. 2003, 178, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berschneider, B.; Konigshoff, M. WNT1 inducible signaling pathway protein 1 (WISP1): A novel mediator linking development and disease. Int. J. Biochem. Cell Biol. 2011, 43, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, I.; Chiquet-Ehrismann, R. CCN4/WISP1 (WNT1 inducible signaling pathway protein 1): A focus on its role in cancer. Int. J. Biochem. Cell Biol. 2015, 62, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Blom, A.B.; Brockbank, S.M.; van Lent, P.L.; van Beuningen, H.M.; Geurts, J.; Takahashi, N.; van der Kraan, P.M.; van de Loo, F.A.; Schreurs, B.W.; Clements, K.; et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: Prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009, 60, 501–512. [Google Scholar] [CrossRef]
- Zhong, X.; Tu, Y.J.; Li, Y.; Zhang, P.; Wang, W.; Chen, S.S.; Li, L.; Chung, A.C.; Lan, H.Y.; Chen, H.Y.; et al. Serum levels of WNT1-inducible signaling pathway protein-1 (WISP-1): A noninvasive biomarker of renal fibrosis in subjects with chronic kidney disease. Am. J. Transl Res. 2017, 9, 2920–2932. [Google Scholar] [PubMed]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, Y.; Ye, W.; Tao, X.; Zhu, J.; Wu, S.; Lou, L. Blockade of CCN4 attenuates CCl4-induced liver fibrosis. Arch. Med. Sci. 2015, 11, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Murahovschi, V.; Pivovarova, O.; Ilkavets, I.; Dmitrieva, R.M.; Docke, S.; Keyhani-Nejad, F.; Gogebakan, O.; Osterhoff, M.; Kemper, M.; Hornemann, S.; et al. WISP1 Is a Novel Adipokine Linked to Inflammation in Obesity. Diabetes 2015, 64, 856–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horbelt, T.; Tacke, C.; Markova, M.; Herzfeld de Wiza, D.; Van de Velde, F.; Bekaert, M.; Van Nieuwenhove, Y.; Hornemann, S.; Rodiger, M.; Seebeck, N.; et al. The novel adipokine WISP1 associates with insulin resistance and impairs insulin action in human myotubes and mouse hepatocytes. Diabetologia 2018, 61, 2054–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, C.; Aleksandrova, K.; Rehfeldt, M.; Murahovschi, V.; Markova, M.; Kemper, M.; Hornemann, S.; Kaiser, U.; Honig, C.; Gerbracht, C.; et al. Assessment of circulating Wnt1 inducible signalling pathway protein 1 (WISP-1)/CCN4 as a novel biomarker of obesity. J. Cell Commun. Signal. 2017. [Google Scholar] [CrossRef]
- Sahin Ersoy, G.; Altun Ensari, T.; Subas, S.; Giray, B.; Simsek, E.E.; Cevik, O. WISP1 is a novel adipokine linked to metabolic parameters in gestational diabetes mellitus. J. Matern. Fetal Neonatal Med. 2016, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Capoccia, D.; De Gioannis, R.; Porzia, A.; Mainiero, F.; Di Martino, M.; Bertoccini, L.; De Bernardinis, M.; Leonetti, F.; et al. WISP1 Is a Marker of Systemic and Adipose Tissue Inflammation in Dysmetabolic Subjects with or without Type 2 Diabetes. J. Endocr. Soc. 2017, 1, 660–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Xu, C.; Markova, M.; Seebeck, N.; Loft, A.; Hornemann, S.; Gantert, T.; Kabisch, S.; Herz, K.; Loske, J.; Ost, M.; et al. High-protein diet more effectively reduces hepatic fat than low-protein diet despite lower autophagy and FGF21 levels. Liver Int. 2020. [Google Scholar] [CrossRef]
- Fernando, R.; Wardelmann, K.; Deubel, S.; Kehm, R.; Jung, T.; Mariotti, M.; Vasilaki, A.; Gladyshev, V.N.; Kleinridders, A.; Grune, T.; et al. Low steady-state oxidative stress inhibits adipogenesis by altering mitochondrial dynamics and decreasing cellular respiration. Redox Biol. 2020, 32, 101507. [Google Scholar] [CrossRef]
- Kotronen, A.; Peltonen, M.; Hakkarainen, A.; Sevastianova, K.; Bergholm, R.; Johansson, L.M.; Lundbom, N.; Rissanen, A.; Ridderstrale, M.; Groop, L.; et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology 2009, 137, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. WJG 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Klimontov, V.V.; Bulumbaeva, D.M.; Fazullina, O.N.; Lykov, A.P.; Bgatova, N.P.; Orlov, N.B.; Konenkov, V.I.; Pfeiffer, A.F.H.; Pivovarova-Ramich, O.; Rudovich, N. Circulating Wnt1-inducible signaling pathway protein-1 (WISP-1/CCN4) is a novel biomarker of adiposity in subjects with type 2 diabetes. J. Cell Commun. Signal. 2020, 14, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.W.; Kang, C.; Goh, J.; Chae, S.I.; Kim, H.C.; Lee, T.J.; Abd El-Aty, A.M.; Jeong, J.H. WISP1 promotes non-alcoholic fatty liver disease and skeletal muscle insulin resistance via TLR4/JNK signaling. J. Cell. Physiol. 2018, 233, 6077–6087. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Li, Q.; Liu, P.; Liu, Y.; Li, J.; Wasserloos, K.; Chao, W.; You, M.; Oury, T.D.; Chhinder, S.; et al. WNT1-inducible signaling pathway protein 1 contributes to ventilator-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, V.; Dargere, D.; Vidaud, M.; De Gouville, A.C.; Huet, S.; Martinez, V.; Gauthier, J.M.; Ba, N.; Sobesky, R.; Ratziu, V.; et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 1999, 30, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Brigstock, D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012, 17, 2495–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Chen, C.C.; Monzon, R.I.; Lau, L.F. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell. Biol. 2013, 33, 2078–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Y.C.; Wang, J.J.; Dong, S.; Hu, J.W.; Hu, L.J.; Yang, G.M.; Zheng, Y.X.; Xiong, W.J. Wnt-induced secreted protein 1/CCN4 in liver fibrosis both in vitro and in vivo. Clin. Lab. 2014, 60, 29–35. [Google Scholar] [CrossRef] [PubMed]
- van den Bosch, M.H.; Gleissl, T.A.; Blom, A.B.; van den Berg, W.B.; van Lent, P.L.; van der Kraan, P.M. Wnts talking with the TGF-beta superfamily: WISPers about modulation of osteoarthritis. Rheumatology 2016, 55, 1536–1547. [Google Scholar] [CrossRef] [Green Version]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis—a systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Iredale, J.P. Macrophages: Central regulators of hepatic fibrogenesis and fibrosis resolution. J. Hepatol. 2012, 56, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Values |
|---|---|
| n | 35 |
| Gender (n, men/women) | 9/26 |
| Age (years) | 46.7 ± 1.8 |
| Weight (kg) | 124.7 ± 3.9 |
| BMI (kg/m2) | 42.5 ± 0.7 |
| Fat mass (%) | 53.3 ± 1.6 |
| IHL (%) a | 13.2 ± 2.4 |
| AST (U/L) | 25.5 ± 2.4 |
| ALT (U/L) | 31.9 ± 2.2 |
| GGT (U/L) | 33.7 ± 5.3 |
| Creatinine (μmol/L) | 74.5 ± 4.2 |
| Urea (mmol/L) | 5.4 ± 0.9 |
| Uric acid (μmol/L) | 315.0 ± 15.5 |
| Cholesterol (mmol/L) | 4.15 ± 0.18 |
| HDL-c (mmol/L) | 0.96 ± 0.03 |
| LDL-c (mmol/L) | 1.90 ± 0.16 |
| Triglyceride (mmol/L) | 3.01 ± 0.23 |
| NEFA (mmol/L) | 1.85 ± 0.56 |
| Fasting glucose (mmol/L) | 7.24 ± 0.43 |
| Fasting insulin (mU/L) | 15.3 ± 1.5 |
| HOMA-IR | 4.94 ± 0.58 |
| HbA1c (%) | 5.9 ± 0.2 |
| Diabetes type 1 (n) | 3 |
| Diabetes type 2 (n) | 8 |
| Hepatic steatosis (n) | 26 |
| Ballooning (n) | 10 |
| Lobular inflammation (n) | 28 |
| Hepatic fibrosis (n) | 24 |
| NASH (n) | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pivovarova-Ramich, O.; Loske, J.; Hornemann, S.; Markova, M.; Seebeck, N.; Rosenthal, A.; Klauschen, F.; Castro, J.P.; Buschow, R.; Grune, T.; et al. Hepatic Wnt1 Inducible Signaling Pathway Protein 1 (WISP-1/CCN4) Associates with Markers of Liver Fibrosis in Severe Obesity. Cells 2021, 10, 1048. https://doi.org/10.3390/cells10051048
Pivovarova-Ramich O, Loske J, Hornemann S, Markova M, Seebeck N, Rosenthal A, Klauschen F, Castro JP, Buschow R, Grune T, et al. Hepatic Wnt1 Inducible Signaling Pathway Protein 1 (WISP-1/CCN4) Associates with Markers of Liver Fibrosis in Severe Obesity. Cells. 2021; 10(5):1048. https://doi.org/10.3390/cells10051048
Chicago/Turabian StylePivovarova-Ramich, Olga, Jennifer Loske, Silke Hornemann, Mariya Markova, Nicole Seebeck, Anke Rosenthal, Frederick Klauschen, José Pedro Castro, René Buschow, Tilman Grune, and et al. 2021. "Hepatic Wnt1 Inducible Signaling Pathway Protein 1 (WISP-1/CCN4) Associates with Markers of Liver Fibrosis in Severe Obesity" Cells 10, no. 5: 1048. https://doi.org/10.3390/cells10051048