
Dysregulation of Multiple Signaling Neurodevelopmental Pathways during Embryogenesis: A Possible Cause of Autism Spectrum Disorder



 and
and
Abstract
:1. Introduction
2. Types of Autism Spectrum Disorder
3. Etiology and Pathophysiology of ASD
3.1. Genetic Factors
3.2. Neuropathological and Neurotransmitter Level Abnormalities in the ASD Brain
3.2.1. Olivocerebellar Impairment
Cerebellum Impairment in ASD and Decrease in Purkinje Cells
Cholinergic Receptor Changes in the Cerebellum
Inferior Olivary Complex (IOC) Pathology
Deep Cerebellar Nuclei (DCN) Pathology
Brain Stem Impairment Pathology
3.2.2. Limbic System Impairment
Hippocampus Pathology
Entorhinal Cortex Pathology
Amygdala Pathology
Anterior Cingulate Cortex (ACC) Pathology
Posterior Cingulate Cortex (PCC) Pathology
Default Network Defects Pathology
3.2.3. Neocortical Pathology
Cortical Dendritic Impairment
Abnormal Organization of Cortical Minicolumns Pathology
Abnormality in Frontal Lobe Growth Pathology
3.3. Non-Genetic Factors Implicated in ASD
3.3.1. Gastrointestinal Implications
3.3.2. Immune System Imbalance in ASD
3.3.3. Neuropeptides in ASD (VIP, BDNF, CGRP)
4. Impairment of Developmental Pathways
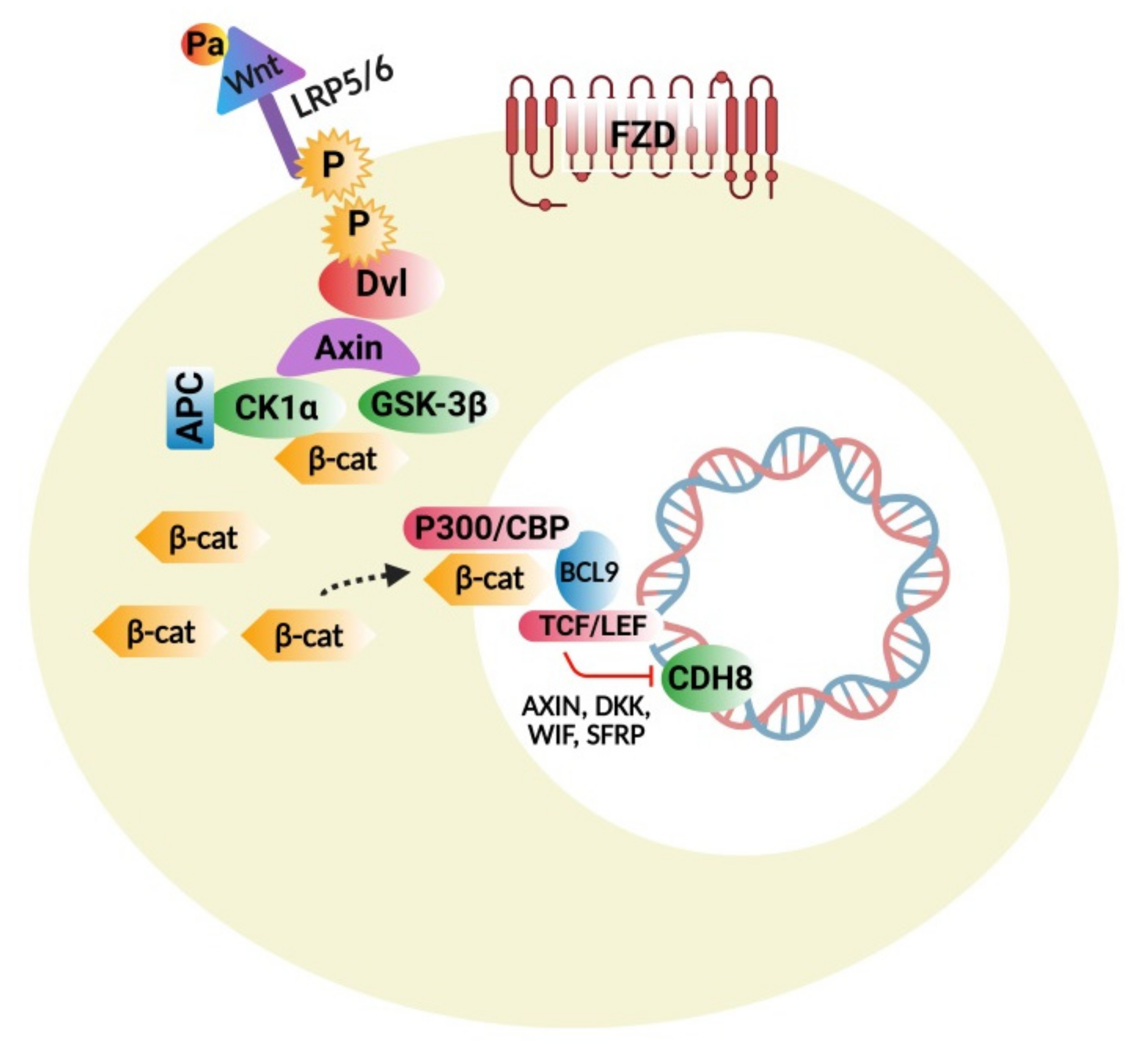
4.1. Wnt Protein and β-Catenin Signaling Pathways
4.1.1. Wnt/β-Catenin Signaling Disturbances
4.1.2. Wnt Pathway Modulators
4.1.3. Interconnections of Wnt Signaling with Developmental and Inflammatory Pathways
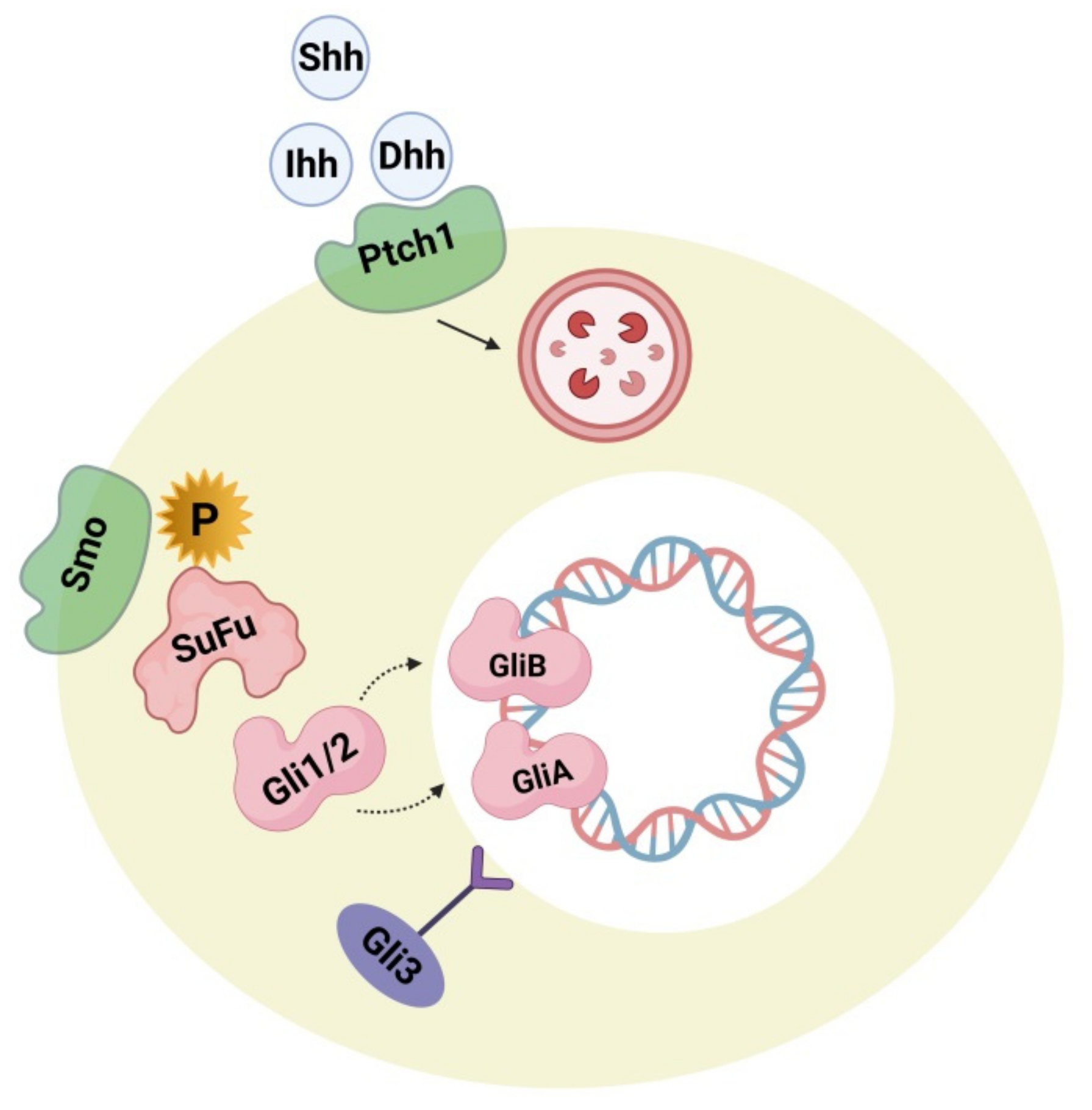
4.2. Hedgehog Signaling Pathway
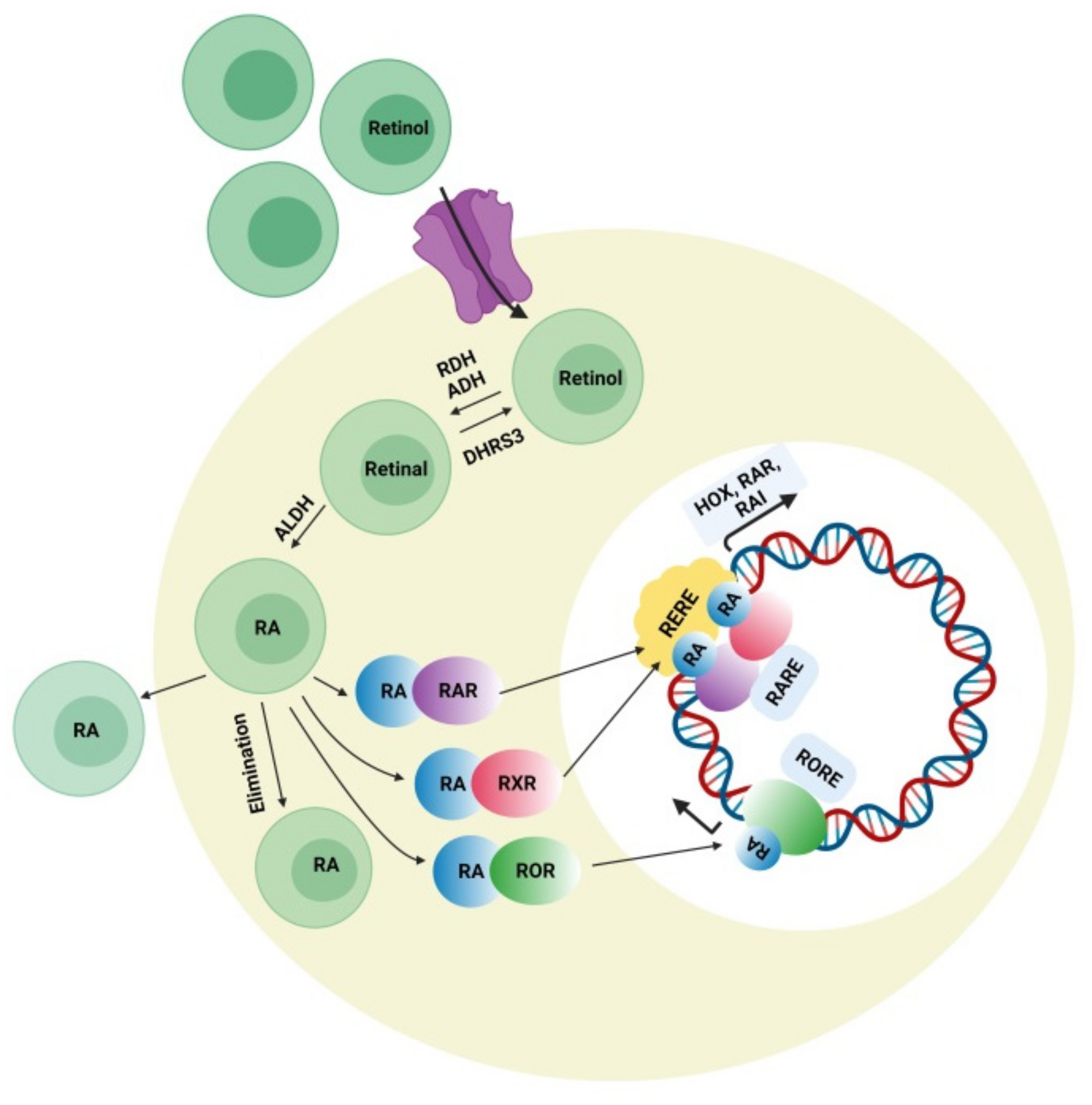
4.3. Retinoic Acid (RA) Signaling Pathway
5. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, X.; Lv, C.-C.; Tian, J.; Miao, R.-J.; Xi, W.; Hertz-Picciotto, I.; Qi, L. Prenatal and Perinatal Risk Factors for Autism in China. J. Autism Dev. Disord. 2010, 40, 1311–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, A.J.; Brugha, T.S.; Erskine, H.E.; Scheurer, R.W.; Vos, T.; Scott, J.G. The epidemiology and global burden of autism spectrum disorders. Psychol. Med. 2015, 45, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Daley, T.C. From symptom recognition to diagnosis: Children with autism in urban India. Soc. Sci. Med. 2004, 58, 1323–1335. [Google Scholar] [CrossRef]
- Fusar-Poli, L.; Brondino, N.; Politi, P.; Aguglia, E. Missed diagnoses and misdiagnoses of adults with autism spectrum disorder. Eur. Arch. Psychiatry Clin. Neurosci. 2020, 1–12. [Google Scholar] [CrossRef]
- Vismara, L.A.; Rogers, S.J. Behavioral Treatments in Autism Spectrum Disorder: What Do We Know? Annu. Rev. Clin. Psychol. 2010, 6, 447–468. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.P.; Myers, S.M.; American Academy of Pediatrics, the Council on Children with Disabilities. Identification and Evaluation of Children with Autism Spectrum Disorders. Pediatrics 2007, 120, 1183–1215. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Fombonne, E. Pervasive developmental disorders in preschool children. JAMA J. Am. Med Assoc. 2001, 285, 3093–3099. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorders—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar]
- Karande, S. Autism: A review for family physicians. Indian J. Med Sci. 2006, 60, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, V. A Clinical Experience of Autism in India. J. Dev. Behav. Pediatr. 2008, 29, 331–333. [Google Scholar] [CrossRef]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Zeldovich, L. The Evolution of ‘Autism’ as a Diagnosis, Explained. 2018. Available online: https://www.spectrumnews.org/news/evolution-autism-diagnosisexplained (accessed on 31 January 2021).
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; (DSM-5); American Psychiatric Association: Washington, DC, USA, 2013; p. 5. [Google Scholar]
- Asperger’s Syndrome. Available online: https://www.autism-society.org/what-is/aspergers-syndrome/ (accessed on 28 January 2021).
- Centre for Disease Control and Prevention (CDC). Autism Spectrum Disorder. Available online: https://www.cdc.gov/ncbddd/autism/hcp-dsm.html (accessed on 13 April 2021).
- Laurvick, C.L.; de Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett syndrome in Australia: A review of the epidemiology. J. Pediatr. 2006, 148, 347–352. [Google Scholar] [CrossRef]
- Volkmar, F.R.; Reichow, B.; McPartland, J. Classification of autism and related conditions: Progress, challenges, and opportunities. Dialog. Clin. Neurosci. 2012, 14, 229–237. [Google Scholar]
- Walker, D.R.; Thompson, A.; Zwaigenbaum, L.; Goldberg, J.; Bryson, S.E.; Mahoney, W.J.; Strawbridge, C.P.; Szatmari, P. Specifying PDD-NOS: A Comparison of PDD-NOS, Asperger Syndrome, and Autism. J. Am. Acad. Child Adolesc. Psychiatry 2004, 43, 172–180. [Google Scholar] [CrossRef]
- Karabekiroglu, K. Pervasive Developmental Disorder- not Otherwise Specified: Specifying and Differentiating. In Autism Spectrum Disorders: The Role of Genetics in Diagnosis and Treatment; IntechOpen: London, UK, 2011. [Google Scholar]
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Zwaigenbaum, L.; Szatmari, P.; Bryson, S.E.; MacLean, J.E.; Tuff, L.P.; Bartolucci, G.; Tuff, L. Pregnancy and birth com-plications in autism and liability to the broader autism phenotype. J. Am. Acad. Child. Adolesc. Psychiatry 2002, 41, 572–579. [Google Scholar] [CrossRef]
- Folstein, S.; Rutter, M. Infantile autism: A genetic study of 21 twin pairs. J. Child Psychol. Psychiatry 1977, 18, 297–321. [Google Scholar] [CrossRef]
- Bailey, A.; Le Couteur, A.; Gottesman, I.; Bolton, P.; Simonoff, E.; Yuzda, E.; Rutter, M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol. Med. 1995, 25, 63–77. [Google Scholar] [CrossRef]
- Casanova, M.F.; Buxhoeveden, D.P.; Switala, A.E.; Roy, E. Minicolumnar pathology in autism. Neurology 2002, 58, 428–432. [Google Scholar] [CrossRef]
- Critchley, H.D.; Daly, E.M.; Bullmore, E.T.; Williams, S.C.; Van Amelesvoort, T.; Robertson, D.M.; Rowe, A.; Phillips, M.; McAlonan, G.; Howlin, P.; et al. The functional neuroanatomy of social behaviour: Changes in cerebral blood flow in people with autistic disorder process facial expressions. Brain 2000, 123, 2203–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.F. Intestinal Pathophysiology in Autism. Exp. Biol. Med. 2003, 228, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Bruining, H.; De Sonneville, L.; Swaab, H.; De Jonge, M.; Kas, M.; Van Engeland, H.; Vorstman, J. Dissecting the Clinical Heterogeneity of Autism Spectrum Disorders through Defined Genotypes. PLoS ONE 2010, 5, e10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoda, Y.; Sadakata, T.; Furuichi, T. Animal models of autism spectrum disorder (ASD): A synaptic-level approach to autistic-like behavior in mice. Exp. Anim. 2013, 62, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong Association of De Novo Copy Number Mutations with Autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Mikhail, F.M.; Lose, E.J.; Robin, N.H.; Descartes, M.D.; Rutledge, K.D.; Rutledge, S.L.; Korf, B.R.; Carroll, A.J. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am. J. Med Genet. Part A 2011, 155, 2386–2396. [Google Scholar] [CrossRef]
- Ronemus, M.; Iossifov, I.; Levy, D.; Wigler, M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 2014, 15, 133–141. [Google Scholar] [CrossRef]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic Dysfunction in Neurodevelopmental Disorders Associated with Autism and Intellectual Disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Riordan, M.; Bhat, M.A. Genetic aspects of autism spectrum disorders: Insights from animal models. Front. Cell. Neurosci. 2014, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The Genetics of Autism. Pediatrics 2004, 113, e472–e486. [Google Scholar] [CrossRef] [Green Version]
- Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380, 42–77. [Google Scholar] [CrossRef] [Green Version]
- Miles, J.H. Autism spectrum disorders—A genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Randolph-Gips, M.M.; Srinivasan, P. Modeling autism: A systems biology approach. J. Clin. Bioinform. 2012, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sabo, A.; Neale, B.M.; Nagaswamy, U.; Stevens, C.; Lim, E.; Bodea, C.A.; Muzny, D.; Reid, J.G.; Banks, E.; et al. Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013, 9, e1003443. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Oblak, A.; Gibbs, T.T.; Blatt, G.J. Reduced Serotonin Receptor Subtypes in a Limbic and a Neocortical Region in Autism. Autism Res. 2013, 6, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Berkel, S.; Marshall, C.R.; Weiss, B.; Howe, J.L.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef]
- Sato, D.; Lionel, A.C.; Leblond, C.S.; Prasad, A.; Pinto, D.; Walker, S.; O’Connor, I.; Russell, C.; Drmic, I.E.; Hamdan, F.F.; et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 2012, 90, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-G.; Kishikawa, S.; Higgins, A.W.; Seong, I.-S.; Donovan, D.J.; Shen, Y.; Lally, E.; Weiss, L.A.; Najm, J.; Kutsche, K.; et al. Disruption of Neurexin 1 Associated with Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.S.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.P.; Hamel, B.; Chelly, J.; et al. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 2009, 15, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Zweier, C.; de Jong, E.K.; Zweier, M.; Orrico, A.; Ousager, L.B.; Collins, A.L.; Bijlsma, E.K.; Oortveld, M.A.; Ekici, A.B.; Reis, A.; et al. CNTNAP2 and NRXN1 are mutated in autosomal recessive Pitt-Hopkins-like men-tal retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 2009, 85, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Ba, W.; Van Der Raadt, J.; Kasri, N.N. Rho GTPase signaling at the synapse: Implications for intellectual disability. Exp. Cell Res. 2013, 319, 2368–2374. [Google Scholar] [CrossRef]
- Govek, E.E.; Newey, S.E.; Akerman, C.J.; Cross, J.R.; Van der Veken, L.; Van Aelst, L. The X-linked mental retarda-tion protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat. Neurosci. 2004, 7, 364–372. [Google Scholar] [CrossRef]
- Boda, B.; Alberi, S.; Nikonenko, I.; Node-Langlois, R.; Jourdain, P.; Moosmayer, M.; Parisi-Jourdain, L.; Muller, D. The Mental Retardation Protein PAK3 Contributes to Synapse Formation and Plasticity in Hippocampus. J. Neurosci. 2004, 24, 10816–10825. [Google Scholar] [CrossRef] [Green Version]
- Dubos, A.; Combeau, G.; Bernardinelli, Y.; Barnier, J.-V.; Hartley, O.; Gaertner, H.; Boda, B.; Muller, D. Alteration of Synaptic Network Dynamics by the Intellectual Disability Protein PAK3. J. Neurosci. 2012, 32, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Meng, Y.; Hanna, A.; Janus, C.; Jia, Z. Abnormal Long-Lasting Synaptic Plasticity and Cognition in Mice Lacking the Mental Retardation Gene Pak3. J. Neurosci. 2005, 25, 6641–6650. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, R.J.; Bear, M.F. The Autistic Neuron: Troubled Translation? Cell 2008, 135, 401–406. [Google Scholar] [CrossRef] [Green Version]
- Yenkoyan, K.; Grigoryan, A.; Fereshetyan, K.; Yepremyan, D. Advances in understanding the pathophysiology of autism spec-trum disorders. Behav. Brain Res. 2017, 331, 92–101. [Google Scholar] [CrossRef]
- Sanchez-Ortiz, E.; Cho, W.; Nazarenko, I.; Mo, W.; Chen, J.; Parada, L.F. NF1 regulation of RAS/ERK signaling is required for appropriate granule neuron progenitor expansion and migration in cerebellar development. Genes Dev. 2014, 28, 2407–2420. [Google Scholar] [CrossRef] [Green Version]
- Matthes, J.; Herzig, S. Voltage-gated Calcium Channels and Autism Spectrum Disorders. Curr. Mol. Pharmacol. 2015, 8, 123–132. [Google Scholar] [CrossRef]
- Blatt, G.J. The Neuropathology of Autism. Science 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, M.F.; El-Baz, A.S.; Kamat, S.S.; Dombroski, B.A.; Khalifa, F.; Elnakib, A.; Soliman, A.; Allison-McNutt, A.; Switala, A.E. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol. Commun. 2013, 1, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggio, M.G.; Chiricozzi, F.R.; Clausi, S.; Tedesco, A.M.; Molinari, M. The neuropsychological profile of cerebellar damage: The sequencing hypothesis. Cortex 2011, 47, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Ramnani, N. The primate cortico-cerebellar system: Anatomy and function. Nat. Rev. Neurosci. 2006, 7, 511–522. [Google Scholar] [CrossRef]
- Whitney, E.R.; Kemper, T.L.; Bauman, M.L.; Rosene, D.L.; Blatt, G.J. Cerebellar Purkinje Cells are Reduced in a Subpopulation of Autistic Brains: A Stereological Experiment Using Calbindin-D28k. Cerebellum 2008, 7, 406–416. [Google Scholar] [CrossRef]
- Martin-Ruiz, C.; Lee, M.; Perry, R.; Baumann, M.; Court, J.; Perry, E. Molecular analysis of nicotinic receptor expression in autism. Mol. Brain Res. 2004, 123, 81–90. [Google Scholar] [CrossRef]
- Lippiello, P. Nicotinic cholinergic antagonists: A novel approach for the treatment of autism. Med. Hypotheses 2006, 66, 985–990. [Google Scholar] [CrossRef]
- Brondino, N.; Fusar-Poli, L.; Panisi, C.; Damiani, S.; Barale, F.; Politi, P. Pharmacological Modulation of GABA Function in Autism Spectrum Disorders: A Systematic Review of Human Studies. J. Autism Dev. Disord. 2016, 46, 825–839. [Google Scholar] [CrossRef]
- Scheibel, M.E.; Scheibel, A.B. Observations on the intracortical relations of the climbing fibers of the cerebellum. A Golgi study. J. Comp. Neurol. 1954, 101, 733–763. [Google Scholar] [CrossRef]
- Bauman, M.; Kemper, T.L. Histoanatomic observations of the brain in early infantile autism. Neurology 1985, 35, 866. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic Observations of the Brain in Autism in the Neurobiology of Autism; Bauman, M.L., Kemper, T.L., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 1994; pp. 119–145. [Google Scholar]
- Yip, J.; Soghomonian, J.J.; Blatt, G.J. Decreased GAD65 mRNA levels in select subpopulations of neurons in the cerebellar dentate nuclei in autism: An in situ hybridization study. Autism Res. 2009, 2, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A clinicopathological study of autism. Brain 1998, 121, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Chan-Palay, V. Cerebellar Dentate Nucleus: Organization, Cytology and Transmitters; Springer: Berlin, Germany, 1977. [Google Scholar]
- Jou, R.J.; Minshew, N.J.; Melhem, N.M.; Keshavan, M.S.; Hardan, A.Y. Brainstem volumetric alterations in children with autism. Psychol. Med. 2008, 39, 1347–1354. [Google Scholar] [CrossRef] [Green Version]
- Rodier, P.M. The Early Origins of Autism. Sci. Am. 2000, 282, 56–63. [Google Scholar] [CrossRef]
- Raymond, G.V.; Bauman, M.L.; Kemper, T.L. Hippocampus in autism: A Golgi analysis. Acta Neuropathol. 1995, 91, 117–119. [Google Scholar] [CrossRef]
- Sparks, B.F.; Friedman, S.D.; Shaw, D.W.; Aylward, E.H.; Echelard, D.; Artru, A.A.; Maravilla, K.R.; Giedd, N.; Munson, J.; Dawson, G.; et al. Brain structural abnormalities in young children with autism spectrum disorder. Neurology 2002, 59, 184–192. [Google Scholar] [CrossRef]
- Schumann, C.M.; Hamstra, J.; Goodlin-Jones, B.L.; Lotspeich, L.J.; Kwon, H.; Buonocore, M.H.; Lammers, C.R.; Reiss, A.L.; Amaral, D.G. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J. Neurosci. 2004, 24, 6392–6401. [Google Scholar] [CrossRef]
- Allman, J.M.; Hakeem, A.; Erwin, J.M.; Nimchinsky, E.; Hof, P. The anterior cingulate cortex. The evolution of an interface between emotion and cognition. Ann. N. Y. Acad. Sci. 2001, 935, 107–117. [Google Scholar] [CrossRef]
- Robertson, R.T.; Kaitz, S.S. Thalamic connections with limbic cortex. I. Thalamocortical projections. J. Comp. Neurol. 1981, 195, 501–525. [Google Scholar] [CrossRef]
- Bush, G.; Luu, P.; Posner, M.I. Cognitive and emotional influences in anterior cingulate cortex. Trends Cogn. Sci. 2000, 4, 215–222. [Google Scholar] [CrossRef]
- Nieuwenhuis, S.; Yeung, N.; van den Wildenberg, W.; Ridderinkhof, K.R. Electrophysiological correlates of an-terior cingulate function in a go/no-go task: Effects of response conflict and trial type frequency. Cogn. Affect. Behav. Neurosci. 2003, 3, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, U.; Firth, C. The biological basis of social interaction. Curr. Dir. Psychol. Sci. 2001, 10, 151–155. [Google Scholar] [CrossRef]
- Mundy, P. Annotation: The neural basis of social impairments in autism: The role of the dorsal medial-frontal cortex and anterior cingulate system. J. Child Psychol. Psychiatry 2003, 44, 793–809. [Google Scholar] [CrossRef]
- Sidman, R.L.; Rakic, P. Development of the Human Central Nervous System in Histology and Histopathology of the Nervous System; Haymaker, W., Adams, S., Eds.; CC Thomas: Springfield, MA, USA, 1982. [Google Scholar]
- Assaf, M.; Jagannathan, K.; Calhoun, V.D.; Miller, L.; Stevens, M.C.; Sahl, R.; O’Boyle, J.G.; Schultz, R.T.; Pearlson, G.D. Abnormal functional connectivity of default mode sub-networks in autism spectrum disorder patients. NeuroImage 2010, 53, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Monk, C.S.; Peltier, S.J.; Wiggins, J.L.; Weng, S.-J.; Carrasco, M.; Risi, S.; Lord, C. Abnormalities of intrinsic functional connectivity in autism spectrum disorders. NeuroImage 2009, 47, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courchesne, E.; Pierce, K. Brain overgrowth in autism during a critical time in development: Implications for frontal pyramidal neuron and interneuron development and connectivity. Int. J. Dev. Neurosci. 2005, 23, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y. Gastrointestinal Issues in Autism Spectrum Disorder. Harv. Rev. Psychiatry 2014, 22, 104–111. [Google Scholar] [CrossRef]
- Rabinowitz, K.; Mayer, L. Working out mechanisms of controlled/physiologic inflammation in the GI tract. Immunol. Res. 2012, 54, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Long, T.M.; Nisa, S.; Donnenberg, M.S.; Hassel, B.A. Enteropathogenic Escherichia coli Inhibits Type I Interferon- and RNase L-Mediated Host Defense To Disrupt Intestinal Epithelial Cell Barrier Function. Infect. Immun. 2014, 82, 2802–2814. [Google Scholar] [CrossRef] [Green Version]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Noriega, D.B.; Savelkoul, H.F.J. Immune dysregulation in autism spectrum disorder. Eur. J. Nucl. Med. Mol. Imaging 2014, 173, 33–43. [Google Scholar] [CrossRef]
- Nelson, K.B.; Grether, J.K.; Croen, L.A.; Dambrosia, J.M.; Dickens, B.F.; Jelliffe, L.L.; Hansen, R.L.; Phillips, T.M. Neuropeptides and neurotrophins in neonatal blood of children with autism or mental retardation. Ann. Neurol. 2001, 49, 597–606. [Google Scholar] [CrossRef]
- Edvinsson, L. Sensory nerves in man and their role in primary headaches. Cephalalgia 2001, 21, 761–764. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Varela, N.; Chorny, A.; Delgado, M. Therapeutical Approaches of Vasoactive Intestinal Peptide as a Pleiotropic Immunomodulator. Curr. Pharm. Des. 2007, 13, 1113–1139. [Google Scholar] [CrossRef]
- Samsam, M.; Covenas, R.; Ahangari, R.; Yajeya, J.; Narvaez, J.A. Role of neuropeptides in migraine: Where do they stand in the latest expert recommendations in migraine treatment? Drug Dev. Res. 2007, 68, 294–314. [Google Scholar] [CrossRef]
- Kafitz, K.W.; Rose, C.R.; Thoenen, H.; Konnerth, A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nat. Cell Biol. 1999, 401, 918–921. [Google Scholar] [CrossRef]
- Ray, M.; Weickert, C.S.; Webster, M.J. Decreased BDNF and TrkB mRNA expression in multiple cortical areas of patients with schizophrenia and mood disorders. Transl. Psychiatry 2014, 4, e389. [Google Scholar] [CrossRef]
- Samsam, M.; Coveñas, R.; Ahangari, R.; Yajeya, J.; Narváez, J.A.; Tramu, G. Simultaneous depletion of neurokinin A, substance P and calcitonin gene-related peptide from the caudal trigeminal nucleus of the rat during electrical stimulation of the trigeminal ganglion. Pain 2000, 84, 389–395. [Google Scholar] [CrossRef]
- Brondino, N.; Fusar-Poli, L.; Rocchetti, M.; Bertoglio, F.; Bloise, N.; Visai, L.; Politi, P. BDNF levels are associated with autistic traits in the general population. Psychoneuroendocrinology 2018, 89, 131–133. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012. [Google Scholar] [CrossRef] [Green Version]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Hormozdiari, F.; Penn, O.; Borenstein, E.; Eichler, E.E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015. [Google Scholar] [CrossRef]
- Kwan, V.; Unda, B.K.; Singh, K.K. Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Platt, R.J.; Zhou, Y.; Slaymaker, I.M.; Shetty, A.S.; Weisbach, N.R.; Kim, J.-A.; Sharma, J.; Desai, M.; Sood, S.; Kempton, H.R.; et al. Chd8 Mutation Leads to Autistic-like Behaviors and Impaired Striatal Circuits. Cell Rep. 2017, 19, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Kalkman, H.O. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol. Autism 2012, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Sousa, K.M.; Villaescusa, J.C.; Cajanek, L.; Ondr, J.K.; Castelo-Branco, G.; Hofstra, W.; Bryja, V.; Palmberg, C.; Bergman, T.; Wainwright, B.; et al. Wnt2 Regulates Progenitor Proliferation in the Developing Ventral Midbrain. J. Biol. Chem. 2010, 285, 7246–7253. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.-Q.; Chen, W.-W.; Jiang, L.; Liu, K.; Yung, W.-H.; Fu, A.K.; Ip, N.Y. Overproduction of Upper-Layer Neurons in the Neocortex Leads to Autism-like Features in Mice. Cell Rep. 2014, 9, 1635–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Sil, P.C. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol. Res. 2019, 142, 251–261. [Google Scholar] [CrossRef]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Katoh, M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, Z.-Y.; Liu, W.-P.; Zhao, M.-R. Crosstalk between Wnt/β-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol. Ther. 2014, 16, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hayward, P.; Kalmar, T.; Arias, A.M. Wnt/Notch signalling and information processing during development. Development 2008, 135, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Boyle, S.C.; Kim, M.; Valerius, M.T.; McMahon, A.P.; Kopan, R. Notch pathway activation can replace the requirement for Wnt4 and Wnt9b in mesenchymal-to-epithelial transition of nephron stem cells. Development 2011, 138, 4245–4254. [Google Scholar] [CrossRef] [Green Version]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-β Pathways Impinge on Hedgehog Signaling Complexity: An Open Window on Cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef] [Green Version]
- Koopmans, T.; Eilers, R.; Menzen, M.; Halayko, A.; Gosens, R. β-catenin directs nuclear factor-κB p65 output via CREB-binding pro-tein/p300 in human airway smooth muscle. Front. Immunol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; An, N.; Haydon, R.C.; Zhou, Q.; Cheng, H.; Peng, Y.; Jiang, W.; Luu, H.H.; Vanichakarn, P.; Szatkowski, J.P.; et al. Tyrosine kinase inhibitor STI-571/Gleevec down-regulates the β-catenin signaling activity. Cancer Lett. 2003, 193, 161–170. [Google Scholar] [CrossRef]
- Fuccillo, M.V.; Joyner, A.L.; Fishell, G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 772–783. [Google Scholar] [CrossRef]
- Álvarez-Buylla, A.; Ihrie, R.A. Sonic hedgehog signaling in the postnatal brain. Semin. Cell Dev. Biol. 2014, 33, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.A.; Blaeser, A.S.; Coley, A.A.; Xie, Y.; Shepard, K.A.; Harwell, C.C.; Gao, W.-J.; Garcia, A.D.R. Sonic hedgehog signaling in astrocytes mediates cell type-specific synaptic organization. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reynolds, K.; Ji, Y. Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Al-Ayadhi, L.Y. Relationship between Sonic Hedgehog Protein, Brain-Derived Neurotrophic Factor and Oxidative Stress in Autism Spectrum Disorders. Neurochem. Res. 2011, 37, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanizadeh, A. Malondialdehyde, Bcl-2, Superoxide Dismutase and Glutathione Peroxidase may Mediate the Association of Sonic Hedgehog Protein and Oxidative Stress in Autism. Neurochem. Res. 2012, 37, 899–901. [Google Scholar] [CrossRef]
- Bashir, S.; Halepoto, D.M.; Al-Ayadhi, L. Serum level of desert hedgehog protein in autism spectrum disorder: Preliminary results. Med. Princ. Pr. 2014, 23, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Halepoto, D.M.; Bashir, S.; Zeina, R.; Al-Ayadhi, L.Y. Correlation between Hedgehog (Hh) Protein Family and Brain-Derived Neurotrophic Factor (BDNF) in Autism Spectrum Disorder (ASD). J. Coll. Physicians Surg. Pak. 2015, 25, 882–885. [Google Scholar]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The Crosstalk of mTOR/S6K1 and Hedgehog Pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2016, 8, 833–845. [Google Scholar] [CrossRef] [Green Version]
- Goldani, A.A.S.; Downs, S.R.; Widjaja, F.; Lawton, B.; Hendren, R.L. Biomarkers in Autism. Front. Psychiatry 2014, 5, 100. [Google Scholar] [CrossRef] [PubMed]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-Derived Hedgehog Proteins Modulate Th Differentiation and Disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohja, K.; Gozal, E.; Fahnestock, M.; Cai, L.; Cai, J.; Freedman, J.H.; Switala, A.; El-Baz, A.; Barnes, G.N. Neuroimmunologic and Neurotrophic Interactions in Autism Spectrum Disorders: Relationship to Neuroinflammation. NeuroMol. Med. 2018, 20, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Faravelli, I.; Bucchia, M.; Rinchetti, P.; Nizzardo, M.; Simone, C.; Frattini, E.; Corti, S. Motor neuron derivation from human embryonic and induced pluripotent stem cells: Experimental approaches and clinical perspectives. Stem Cell Res. Ther. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [Green Version]
- Lai, X.; Wu, X.; Hou, N.; Liu, S.; Li, Q.; Yang, T.; Miao, J.; Dong, Z.; Chen, J.; Li, T. Vitamin A Deficiency Induces Autistic-Like Behaviors in Rats by Regulating the RARβ-CD38-Oxytocin Axis in the Hypothalamus. Mol. Nutr. Food Res. 2018, 62, 1700754. [Google Scholar] [CrossRef]
- Cheng, B.; Zhu, J.; Yang, T.; Guo, M.; Lai, X.; Li, Q.; Chen, J.; Li, T. Vitamin A deficiency increases the risk of gastrointestinal comorbidity and exacerbates core symptoms in children with autism spectrum disorder. Pediatr. Res. 2021, 89, 211–216. [Google Scholar] [CrossRef]
- Xu, X.; Li, C.; Gao, X.; Xia, K.; Guo, H.; Li, Y.; Hao, Z.; Zhang, L.; Gao, D.; Xu, C.; et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2017, 28, 48–68. [Google Scholar] [CrossRef]
- Khatri, N.; Man, H.-Y. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front. Mol. Neurosci. 2019, 12, 109. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3036–3051. [Google Scholar] [CrossRef]
- Ryu, J.; Bar-Shalita, T.; Granovsky, Y.; Weissman-Fogel, I.; Torres, E. Personalized Biometrics of Physical Pain Agree with Psychophysics by Participants with Sensory over Responsivity. J. Pers. Med. 2021, 11, 93. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Levels | Clinical Symptoms | ||
|---|---|---|---|
| Social Communication | Repetitive Behavior | ||
| Level 1 | Requires extensive medical support | Severe impairment in verbal and non-verbal communication; a deficit in social interactions; less response to social overtures (e.g., rarely starts an interaction if they have some words of intelligible speech and respond only to direct social overtures. | Rigid behavior; extreme problems coping with change; repetitive or restricted behavior marked by interferences in body functioning in all spheres; great difficulty changing action or focus. |
| Level 2 | Requires medical support | Marked impairment in verbal and non-verbal communication; deficit in social interaction even with support; minimum responses to social overtures like simple spoken sentences; less interest in interaction, and odd behavior in non-verbal communication | Rigid behavior; difficulty coping with change, repetitive or restricted behavior affecting various functions in different contexts; trouble changing action or focus. |
| Level 3 | Requires support | Noticeable impairments in social interaction without support; problems initiating interactions with people and appears to have less interest in social communication (e.g., affected person speaks full sentences with others but to-and-fro conversation fails and attempts to make friends typically not successful and odd), | Rigid behavior causes difficulty with functioning in several contexts; problems switching from one activity to another; deficit in behavior while organizing and planning inhibits independence. |
| S. No. | Genes implicated in ASD and ID | Functions and Effects | References |
|---|---|---|---|
| 1. | Copy number variants (CNV) Variation in genes like NRXNI (neurexin 1 α), CNTNAP2 (contactin-associated protein-like), NLGN4 (neuroligin), SHANK1, SHANK3 (SH3 and multiple ankyrin repeat domains 3) | Neuronal and synaptic functions: -NRXNI, NLGN4, and CNTNAP2 causes synaptic adhesion -SHANK performs scaffoldings of postsynaptic density protein and the formation and maturation of dendritic spines. | [42,43,44,45,46] |
| 2. | OPHN1 (oligophrenin-1), MEGAP and SRGAP3 (SLIT-ROBO Rho-GTPase activating gene), OCRL1 (oculocerebrorenal syndrome of Lowe), ARHGEF6 (Rac/Cdc42 Guanine Nucleotide Exchange Factor 6), ARHGEF9 (Rac/Cdc42 Guanine Nucleotide Exchange Factor 9), FGD1 (encodes guanine nucleotide exchange factor which activates Rho GTPase Cdc42), LIMK1 (LIM domain kinase 1 regulator of actin dynamics), PAK3 (p21-activated kinase), and IQSEC2 (IQ (aa 347–376) and the SEC7 domains) | -Functions as effectors or regulators of Rho GTPases or Rac and Cdc42 and code for proteins linked with GTPase signaling and -Perform an important role in synaptic transmission, neurite outgrowth, and differentiation, dendrites branching, spine maintenance, and formation. | [47,48] |
| 3. | PAK3 | PAK3, protein, is associated with the p21-activating kinases (PAK) family, they are downstream effectors for Rac and Cdc42. Its downregulation causes spine abnormalities and defects in synaptic plasticity. | [49,50,51] |
| 4. | FMR1 FMRP: Fragile X mental retardation protein | FMR1 acts as a translational repressor show 15–30% rate of autism | [52] |
| 5. | TSC1/2 (Tuberous sclerosis protein 1 and 2) | Gene suppressor and inhibitor of mTOR and shows 25–60% rate of autism. | [53,54] |
| 6. | PTEN (Phosphatase and tensin homolog) | Gene suppressor and inhibitor of P13K and mTOR. | [53,54] |
| 7. | NF1 (Neurofibromin) | Gene suppressor—an inhibitor of PI3K/mTOR signaling | [53,54] |
| 8. | MECP2 (Methyl-CpG-binding protein-2) | Global transcriptional repressor and 100% rate of autism causes Retts syndrome | [53] |
| 9. | UBE3A (E6AP ubiquitin-protein ligase) | Ubiquitination, 40% rate of autism causes Angelman’s syndrome | [53] |
| 10. | CACNA1C (Alpha-1 subunit of a voltage-dependent calcium channel) | L-type voltage-gated calcium channels, 60% rate of autism causes Timothy syndrome | [53,55] |
| 11. | CTNND2 (Adhesive junction-associated δ-catenin protein) | Dendritic morphogenesis, histone modification, and participation in WNT signaling | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhyay, J.; Patra, J.; Tiwari, N.; Salankar, N.; Ansari, M.N.; Ahmad, W. Dysregulation of Multiple Signaling Neurodevelopmental Pathways during Embryogenesis: A Possible Cause of Autism Spectrum Disorder. Cells 2021, 10, 958. https://doi.org/10.3390/cells10040958
Upadhyay J, Patra J, Tiwari N, Salankar N, Ansari MN, Ahmad W. Dysregulation of Multiple Signaling Neurodevelopmental Pathways during Embryogenesis: A Possible Cause of Autism Spectrum Disorder. Cells. 2021; 10(4):958. https://doi.org/10.3390/cells10040958
Chicago/Turabian StyleUpadhyay, Jyoti, Jeevan Patra, Nidhi Tiwari, Nilima Salankar, Mohd Nazam Ansari, and Wasim Ahmad. 2021. "Dysregulation of Multiple Signaling Neurodevelopmental Pathways during Embryogenesis: A Possible Cause of Autism Spectrum Disorder" Cells 10, no. 4: 958. https://doi.org/10.3390/cells10040958