
Mitochondrial Dysfunction and Permeability Transition in Neonatal Brain and Lung Injuries

{kind=link}
Abstract
:1. Introduction
1.1. Mitochondria and Perinatal Hypoxia-Ischemia Brain Injury
1.2. Primary Energy Failure in Perinatal HI Insult
1.3. Reperfusion and Secondary Energy Failure in Perinatal HI Insult
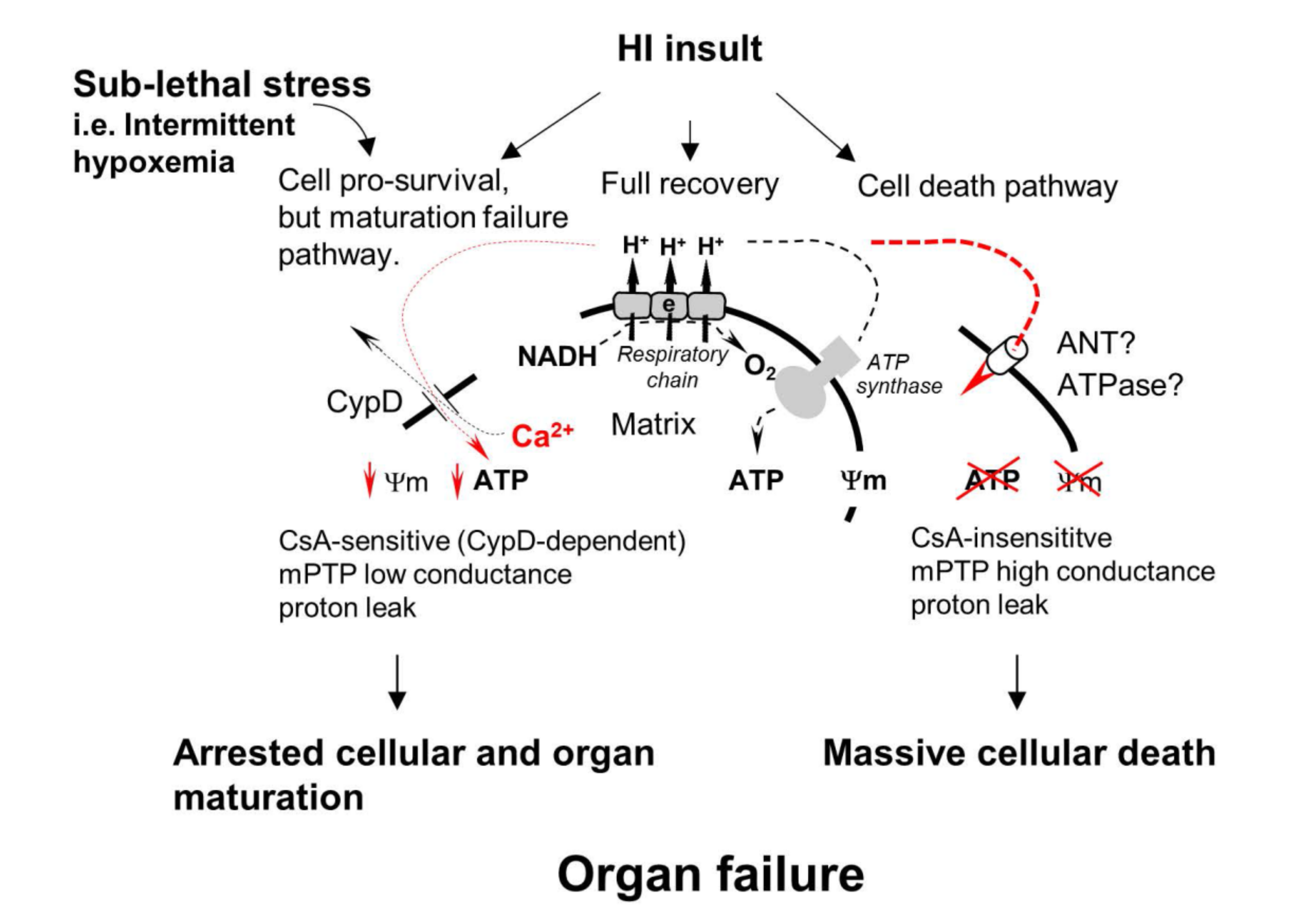
1.4. Mitochondrial Permeability Transition Pore(s) (mPTP) and Secondary Energy Failure
1.5. Mitochondrial Dysfunction in Diseases Driven by Developmental Failure of Brain and Lungs in Premature Infants
1.6. mPTP in the Pathogenesis of Diffuse WMI of Prematurity
1.7. Mitochondrial Dysfunction and mPTP in Neonatal BPD
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katz, V.L.; Dotters, D.J.; Droegemueller, W. Perimortem Cesarean Delivery. Obstet. Gynecol. 1986, 68, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.E.; Gunn, A.J.; Mallard, C.; Gluckman, P.D. Outcome after Ischemia in the Developing Sheep Brain: An Electroencephalographic and Histological Study. Ann. Neurol. 1992, 31, 14–21. [Google Scholar] [CrossRef]
- Murakami, K.; Kondo, T.; Kawase, M.; Chan, P.H. The Development of a New Mouse Model of Global Ischemia: Focus on the Relationships between Ischemia Duration, Anesthesia, Cerebral Vasculature, and Neuronal Injury Following Global Ischemia in Mice. Brain Res. 1998, 780, 304–310. [Google Scholar] [CrossRef]
- Juul, S.E.; Aylward, E.; Richards, T.; McPherson, R.J.; Kuratani, J.; Burbacher, T.M. Prenatal Cord Clamping in Newborn Macaca Nemestrina: A Model of Perinatal Asphyxia. Dev. Neurosci. 2007, 29, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Siesjö, B.K.; Wieloch, T. Cerebral Metabolism in Ischaemia: Neurochemical Basis for Therapy. Br. J. Anaesth. 1985, 57, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Windle, W.F. Brain Damage by Asphyxia at Birth. Sci. Am. 1969, 221, 76–84. [Google Scholar] [CrossRef]
- Myers, R.E. Two Patterns of Perinatal Brain Damage and Their Conditions of Occurrence. Am. J. Obstet. Gynecol. 1972, 112, 246–276. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Brucklacher, R.M.; Vannucci, S.J. Glycolysis and Perinatal Hypoxic-Ischemic Brain Damage. Dev. Neurosci. 2005, 27, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Yager, J.Y.; Brucklacher, R.M.; Vannucci, R.C. Cerebral Energy Metabolism during Hypoxia-Ischemia and Early Recovery in Immature Rats. Am. J. Physiol. Heart Circ. Physiol. 1992, 262, H672–H677. [Google Scholar] [CrossRef]
- Moorcraft, J.; Bolas, N.M.; Ives, N.K.; Ouwerkerk, R.; Smyth, J.; Rajagopalan, B.; Hope, P.L.; Radda, G.K. Global and Depth Resolved Phosphorus Magnetic Resonance Spectroscopy to Predict Outcome after Birth Asphyxia. Arch. Dis. Child. 1991, 66, 1119–1123. [Google Scholar] [CrossRef] [Green Version]
- Cady, E.B.; Iwata, O.; Bainbridge, A.; Wyatt, J.S.; Robertson, N.J. Phosphorus Magnetic Resonance Spectroscopy 2 h after Perinatal Cerebral Hypoxia-Ischemia Prognosticates Outcome in the Newborn Piglet. J. Neurochem. 2008, 107, 1027–1035. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial Metabolism of Reactive Oxygen Species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Fukuchi, T.; Katayama, Y.; Kamiya, T.; McKee, A.; Kashiwagi, F.; Terashi, A. The Effect of Duration of Cerebral Ischemia on Brain Pyruvate Dehydrogenase Activity, Energy Metabolites, and Blood Flow during Reperfusion in Gerbil Brain. Brain Res. 1998, 792, 59–65. [Google Scholar] [CrossRef]
- Ljunggren, B.; Schutz, H.; Siesjö, B.K. Changes in Energy State and Acid-Base Parameters of the Rat Brain during Complete Compression Ischemia. Brain Res. 1974, 73, 277–289. [Google Scholar] [CrossRef]
- Hillered, L.; Ernster, L.; Siesjo, B.K. Influence of in Vitro Lactic Acidosis and Hypercapnia on Respiratory Activity of Isolated Rat Brain Mitochondria. J. Cereb. Blood Flow Metab. 1984, 4, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folbergrová, J.; Ljunggren, B.; Norberg, K.; Siesjö, B.K. Influence of Complete Ischemia on Glycolytic Metabolites, Citric Acid Cycle Intermediates, and Associated Amino Acids in the Rat Cerebral Cortex. Brain Res. 1974, 80, 265–279. [Google Scholar] [CrossRef]
- Sahni, P.V.; Zhang, J.; Sosunov, S.; Galkin, A.; Niatsetskaya, Z.; Starkov, A.; Brookes, P.S.; Ten, V.S. Krebs Cycle Metabolites and Preferential Succinate Oxidation Following Neonatal Hypoxic-Ischemic Brain Injury in Mice. Pediatr. Res. 2018, 83, 491–497. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Stepanova, A.; Niatsetskaya, Z.; Sosunov, S.; Arndt, S.; Murphy, M.P.; Galkin, A.; Ten, V.S. Attenuation of Oxidative Damage by Targeting Mitochondrial Complex I in Neonatal Hypoxic-Ischemic Brain Injury. Free Radic. Biol. Med. 2018, 124, 517–524. [Google Scholar] [CrossRef]
- Johnston, M.V. Excitotoxicity in Perinatal Brain Injury. Brain Pathol. 2006, 15, 234–240. [Google Scholar] [CrossRef]
- Brorson, J.R.; Manzolillo, P.A.; Miller, R.J. Ca2+ Entry via AMPA/KA Receptors and Excitotoxicity in Cultured Cerebellar Purkinje Cells. J. Neurosci. 1994, 14, 187–197. [Google Scholar] [CrossRef]
- Schinder, A.F.; Olson, E.C.; Spitzer, N.C.; Montal, M. Mitochondrial Dysfunction Is a Primary Event in Glutamate Neurotoxicity. J. Neurosci. 1996, 16, 6125–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utkina-Sosunova, I.V.; Niatsetskaya, Z.V.; Sosunov, S.A.; Ratner, V.I.; Matsiukevich, D.; Ten, V.S. Nelfinavir Inhibits Intra-Mitochondrial Calcium Influx and Protects Brain against Hypoxic-Ischemic Injury in Neonatal Mice. PLoS ONE 2013, 8, e62448. [Google Scholar] [CrossRef]
- Palmer, C.; Brucklacher, R.M.; Christensen, M.A.; Vannucci, R.C. Carbohydrate and Energy Metabolism during the Evolution of Hypoxic-Ischemic Brain Damage in the Immature Rat. J. Cereb. Blood Flow Metab. 1990, 10, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorek, A.; Takei, Y.; Cady, E.B.; Wyatt, J.S.; Penrice, J.; Edwards, A.D.; Peebles, D.; Wylezinska, M.; Owen-Reece, H.; Kirkbride, V.; et al. Delayed (“Secondary”) Cerebral Energy Failure after Acute Hypoxia-Ischemia in the Newborn Piglet: Continuous 48-Hour Studies by Phosphorus Magnetic Resonance Spectroscopy. Pediatr. Res. 1994, 36, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, R.C.; Towfighi, J.; Vannucci, S.J. Secondary Energy Failure after Cerebral Hypoxia–Ischemia in the Immature Rat. J. Cereb. Blood Flow Metab. 2004, 24, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Groenendaal, F.; de Vries, L.S. Fifty Years of Brain Imaging in Neonatal Encephalopathy Following Perinatal Asphyxia. Pediatr. Res. 2017, 81, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Puka-Sundvall, M.; Wallin, C.; Gilland, E.; Hallin, U.; Wang, X.; Sandberg, M.; Karlsson, J.-O.; Blomgren, K.; Hagberg, H. Impairment of Mitochondrial Respiration after Cerebral Hypoxia–Ischemia in Immature Rats: Relationship to Activation of Caspase-3 and Neuronal Injury. Dev. Brain Res. 2000, 125, 43–50. [Google Scholar] [CrossRef]
- Niatsetskaya, Z.V.; Sosunov, S.A.; Matsiukevich, D.; Utkina-Sosunova, I.V.; Ratner, V.I.; Starkov, A.A.; Ten, V.S. The Oxygen Free Radicals Originating from Mitochondrial Complex I Contribute to Oxidative Brain Injury Following Hypoxia-Ischemia in Neonatal Mice. J. Neurosci. 2012, 32, 3235–3244. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Allen, K.L.; Bates, T.E.; Clark, J.B. Effect of Reperfusion Following Cerebral Ischaemia on the Activity of the Mitochondrial Respiratory Chain in the Gerbil Brain. J. Neurochem. 1995, 65, 1698–1703. [Google Scholar] [CrossRef] [PubMed]
- Ten, V.S.; Yao, J.; Ratner, V.; Sosunov, S.; Fraser, D.A.; Botto, M.; Sivasankar, B.; Morgan, B.P.; Silverstein, S.; Stark, R.; et al. Complement Component C1q Mediates Mitochondria-Driven Oxidative Stress in Neonatal Hypoxic-Ischemic Brain Injury. J. Neurosci. 2010, 30, 2077–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ma, X.; Yu, W.; Lou, Z.; Mu, D.; Wang, Y.; Shen, B.; Qi, S. Reperfusion Promotes Mitochondrial Dysfunction Following Focal Cerebral Ischemia in Rats. PLoS ONE 2012, 7, e46498. [Google Scholar] [CrossRef]
- Lust, W.D.; Taylor, C.; Pundik, S.; Selman, W.R.; Ratcheson, R.A. Ischemic Cell Death: Dynamics of Delayed Secondary Energy Failure during Reperfusion Following Focal Ischemia. Metab. Brain Dis. 2002, 17, 113–121. [Google Scholar] [CrossRef]
- Wisnowski, J.L.; Wu, T.-W.; Reitman, A.J.; McLean, C.; Friedlich, P.; Vanderbilt, D.; Ho, E.; Nelson, M.D.; Panigrahy, A.; Blüml, S. The Effects of Therapeutic Hypothermia on Cerebral Metabolism in Neonates with Hypoxic-Ischemic Encephalopathy: An in Vivo 1 H-MR Spectroscopy Study. J. Cereb. Blood Flow Metab. 2016, 36, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Hodge, T.; Colombini, M. Regulation of Metabolite Flux through Voltage-Gating of VDAC Channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- O’Rourke, B. Mitochondrial Ion Channels. Annu. Rev. Physiol. 2007, 69, 19–49. [Google Scholar] [CrossRef] [Green Version]
- Zoratti, M.; Szabò, I. The Mitochondrial Permeability Transition. Biochim. Biophys. Acta Rev. Biomembr. 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Niatsetskaya, Z.; Sosunov, S.; Stepanova, A.; Goldman, J.; Galkin, A.; Neginskaya, M.; Pavlov, E.; Ten, V. Cyclophilin D–Dependent Oligodendrocyte Mitochondrial Ion Leak Contributes to Neonatal White Matter Injury. J. Clin. Invest. 2020, 130, 5536–5550. [Google Scholar] [CrossRef]
- Szabó, I.; Zoratti, M. The Mitochondrial Megachannel Is the Permeability Transition Pore. J. Bioenerg. Biomembr. 1992, 24, 111–117. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Dmitry, Z.; Antonenko, Y.; Perini, S. Calcium Modulation of Mitochondrial Inner Membrane Channel Activity. Biochem. Biophys. Res. Commun. 1991, 176, 1183–1188. [Google Scholar] [CrossRef]
- Urbani, A.; Giorgio, V.; Carrer, A.; Franchin, C.; Arrigoni, G.; Jiko, C.; Abe, K.; Maeda, S.; Shinzawa-Itoh, K.; Bogers, J.F.M.; et al. Purified F-ATP Synthase Forms a Ca2+-Dependent High-Conductance Channel Matching the Mitochondrial Permeability Transition Pore. Nat. Commun. 2019, 10, 4341. [Google Scholar] [CrossRef] [Green Version]
- Mnatsakanyan, N.; Llaguno, M.C.; Yang, Y.; Yan, Y.; Weber, J.; Sigworth, F.J.; Jonas, E.A. A Mitochondrial Megachannel Resides in Monomeric F1FO ATP Synthase. Nat. Commun. 2019, 10, 5823. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Tropschug, M.; Heimpel, S.; Heidkämper, D.; Klingenberg, M. A Large Ca 2+ -Dependent Channel Formed by Recombinant ADP/ATP Carrier from Neurospora Crassa Resembles the Mitochondrial Permeability Transition Pore. Biochemistry 2002, 41, 11804–11811. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Klingenberg, M. Mitochondrial ADP/ATP Carrier Can Be Reversibly Converted into a Large Channel by Ca 2+. Biochemistry 1996, 35, 8483–8488. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental Shift of Cyclophilin D Contribution to Hypoxic-Ischemic Brain Injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef]
- Hilton, G.D.; Nunez, J.L.; Bambrick, L.; Thompson, S.M.; McCarthy, M.M. Glutamate-Mediated Excitotoxicity in Neonatal Hippocampal Neurons Is Mediated by MGluR-Induced Release of Ca ++ from Intracellular Stores and Is Prevented by Estradiol. Eur. J. Neurosci. 2006, 24, 3008–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A Forty-Kilodalton Protein of the Inner Membrane Is the Mitochondrial Calcium Uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX Is an Essential Component of Mitochondrial Na+/Ca2+ Exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Ichas, F.; Mazat, J.-P. From Calcium Signaling to Cell Death: Two Conformations for the Mitochondrial Permeability Transition Pore. Switching from Low- to High-Conductance State. Biochim. Biophys. Acta Bioenerg. 1998, 1366, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Molkentin, J.D. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-Dependent Mitochondrial Permeability Transition Regulates Some Necrotic but Not Apoptotic Cell Death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Kantrow, S.P.; Piantadosi, C.A. Release of Cytochrome c from Liver Mitochondria during Permeability Transition. Biochem. Biophys. Res. Commun. 1997, 232, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.; Tamrakar, P.; Rosenthal, R.E.; Fiskum, G. Calcium Uptake and Cytochrome c Release from Normal and Ischemic Brain Mitochondria. Neurochem. Int. 2018, 117, 15–22. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.-L. Mitochondrial Calcium and the Permeability Transition in Cell Death. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D Is a Component of Mitochondrial Permeability Transition and Mediates Neuronal Cell Death after Focal Cerebral Ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, R.S.; Lee, T.-F.; Liu, J.-Q.; Chaudhary, H.; Brocks, D.R.; Bigam, D.L.; Cheung, P.-Y. Cyclosporine Treatment Reduces Oxygen Free Radical Generation and Oxidative Stress in the Brain of Hypoxia-Reoxygenated Newborn Piglets. PLoS ONE 2012, 7, e40471. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.H.; Lee, J.H.; Lee, K.-H.; Bae, E.J.; Sung, D.K.; Chang, Y.S.; Park, W.S. Cyclosporine A Attenuates Hypoxic–Ischemic Brain Injury in Newborn Rats. Brain Res. 2010, 1359, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Leger, P.-L.; De Paulis, D.; Branco, S.; Bonnin, P.; Couture-Lepetit, E.; Baud, O.; Renolleau, S.; Ovize, M.; Gharib, A.; Charriaut-Marlangue, C. Evaluation of Cyclosporine A in a Stroke Model in the Immature Rat Brain. Exp. Neurol. 2011, 230, 58–66. [Google Scholar] [CrossRef]
- Fang, J.; Chavez-Valdez, R.; Flock, D.L.; Avaritt, O.; Saraswati, M.; Robertson, C.; Martin, L.J.; Northington, F.J. An Inhibitor of the Mitochondrial Permeability Transition Pore Lacks Therapeutic Efficacy Following Neonatal Hypoxia Ischemia in Mice. Neuroscience 2019, 406, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Starkov, A.A.; Fiskum, G. Cyclosporin A-Insensitive Permeability Transition in Brain Mitochondria. J. Biol. Chem. 2003, 278, 27382–27389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustovetsky, N.; Dubinsky, J.M. Limitations of Cyclosporin A Inhibition of the Permeability Transition in CNS Mitochondria. J. Neurosci. 2000, 20, 8229–8237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A. White Matter Injury in the Preterm Infant: Pathology and Mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Kinney, H.C.; Jensen, F.E.; Rosenberg, P.A. The Developing Oligodendrocyte: Key Cellular Target in Brain Injury in the Premature Infant. Int. J. Dev. Neurosci. 2011, 29, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested Preoligodendrocyte Maturation Contributes to Myelination Failure in Premature Infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Attwell, D. The Energetics of CNS White Matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative Aspects of the Brain Growth Spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Erecinska, M.; Cherian, S.; Silver, I.A. Energy Metabolism in Mammalian Brain during Development. Prog. Neurobiol. 2004, 73, 397–445. [Google Scholar] [CrossRef] [PubMed]
- Bates, T.E.; Almeida, A.; Heales, S.J.R.; Clark, J. Postnatal Development of the Complexes of the Electron Transport Chain in Isolated Rat Brain Mitochondria. Dev. Neurosci. 1994, 16, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.I.; Hadera, M.G.; Tavares, J.M.; Kotter, M.R.N.; Sonnewald, U. Characterization of Glucose-Related Metabolic Pathways in Differentiated Rat Oligodendrocyte Lineage Cells. Glia 2016, 64, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.J.; Wang, K.; Köroğlu, Ö.; Di Fiore, J.; Kc, P. Intermittent Hypoxic Episodes in Preterm Infants: Do They Matter? Neonatology 2011, 100, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Poets, C.F.; Roberts, R.S.; Schmidt, B.; Whyte, R.K.; Asztalos, E.V.; Bader, D.; Bairam, A.; Moddemann, D.; Peliowski, A.; Rabi, Y.; et al. Association Between Intermittent Hypoxemia or Bradycardia and Late Death or Disability in Extremely Preterm Infants. JAMA 2015, 314, 595. [Google Scholar] [CrossRef] [PubMed]
- Juliano, C.; Sosunov, S.; Niatsetskaya, Z.; Isler, J.A.; Utkina-Sosunova, I.; Jang, I.; Ratner, V.; Ten, V. Mild Intermittent Hypoxemia in Neonatal Mice Causes Permanent Neurofunctional Deficit and White Matter Hypomyelination. Exp. Neurol. 2015, 264, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Jouaville, L.S.; Mazat, J.-P. Mitochondria Are Excitable Organelles Capable of Generating and Conveying Electrical and Calcium Signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Korge, P.; Yang, L.; Yang, J.H.; Wang, Y.; Qu, Z.; Weiss, J.N. Protective Role of Transient Pore Openings in Calcium Handling by Cardiac Mitochondria. J. Biol. Chem. 2011, 286, 34851–34857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzozero, O.A.; Sanchez, P.; Tetzloff, S.U. Effect of ATP Depletion on the Palmitoylation of Myelin Proteolipid Protein in Young and Adult Rats. J. Neurochem. 1999, 72, 2610–2616. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, R.; Wong, A.; Silva, J.; Li, M.; Itoh, A.; Horiuchi, M.; Itoh, T.; Pleasure, D.; Cortopassi, G. Oligodendroglial Differentiation Induces Mitochondrial Genes and Inhibition of Mitochondrial Function Represses Oligodendroglial Differentiation. Mitochondrion 2010, 10, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Y.; He, J.; Zhang, H.; Xiao, L.; Nazarali, A.; Zhang, Z.; Zhang, D.; Tan, Q.; Kong, J.; et al. Hyperforin Promotes Mitochondrial Function and Development of Oligodendrocytes. J. Neurochem. 2011, 119, 555–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziabreva, I.; Campbell, G.; Rist, J.; Zambonin, J.; Rorbach, J.; Wydro, M.M.; Lassmann, H.; Franklin, R.J.M.; Mahad, D. Injury and Differentiation Following Inhibition of Mitochondrial Respiratory Chain Complex IV in Rat Oligodendrocytes. Glia 2010, 58, 1827–1837. [Google Scholar] [CrossRef] [Green Version]
- McEvoy, C.T.; Jain, L.; Schmidt, B.; Abman, S.; Bancalari, E.; Aschner, J.L. Bronchopulmonary Dysplasia: NHLBI Workshop on the Primary Prevention of Chronic Lung Diseases. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. 3), S146–S153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treciokas, L.J. The Effect of “Oxygen Poisoning” on the Alveolar Cell Mitochondria as Revealed by Electron Microscopy. Aeromed. Acta 1959, 30, 674–677. [Google Scholar]
- Rosenbaum, R.M.; Wittner, M.; Lenger, M. Mitochondrial and Other Ultrastructural Changes in Great Alveolar Cells of Oxygen-Adapted and Poisoned Rats. Lab. Investig. 1969, 20, 516–528. [Google Scholar] [PubMed]
- Huang, K.; Rabold, R.; Abston, E.; Schofield, B.; Misra, V.; Galdzicka, E.; Lee, H.; Biswal, S.; Mitzner, W.; Tankersley, C.G. Effects of Leptin Deficiency on Postnatal Lung Development in Mice. J. Appl. Physiol. 2008, 105, 249–259. [Google Scholar] [CrossRef]
- Vohwinkel, C.U.; Lecuona, E.; Sun, H.; Sommer, N.; Vadász, I.; Chandel, N.S.; Sznajder, J.I. Elevated CO 2 Levels Cause Mitochondrial Dysfunction and Impair Cell Proliferation. J. Biol. Chem. 2011, 286, 37067–37076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, V.; Starkov, A.; Matsiukevich, D.; Polin, R.A.; Ten, V.S. Mitochondrial Dysfunction Contributes to Alveolar Developmental Arrest in Hyperoxia-Exposed Mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Ratner, V.; Sosunov, S.A.; Niatsetskaya, Z.V.; Utkina-Sosunova, I.V.; Ten, V.S. Mechanical Ventilation Causes Pulmonary Mitochondrial Dysfunction and Delayed Alveolarization in Neonatal Mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Ten, V.S. Mitochondrial Dysfunction in Alveolar and White Matter Developmental Failure in Premature Infants. Pediatr. Res. 2017, 81, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Porzionato, A.; Zaramella, P.; Macchi, V.; Sarasin, G.; Di Giulio, C.; Rigon, A.; Grisafi, D.; Dedja, A.; Chiandetti, L.; De Caro, R. Cyclosporine and Hyperoxia-Induced Lung Damage in Neonatal Rats. Respir. Physiol. Neurobiol. 2013, 187, 41–46. [Google Scholar] [CrossRef]
- Matthew, E.; Pun, R.; Simonich, M.; Iwamoto, H.; Dedman, J. Cyclosporin A Protects Lung Function from Hyperoxic Damage. Am. J. Physiol. Lung Cell. Mol. Physiol. 1999, 276, 786–795. [Google Scholar] [CrossRef]
- Matthew, E.; Kutcher, L.; Dedman, J. Protection of Lungs from Hyperoxic Injury: Gene Expression Analysis of Cyclosporin: A Therapy. Physiol. Genom. 2003, 14, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Pagano, A.; Donati, Y.; Métrailler, I.; Barazzone Argiroffo, C. Mitochondrial Cytochrome c Release Is a Key Event in Hyperoxia-Induced Lung Injury: Protection by Cyclosporin A. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L275–L283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budinger, G.R.S.; Mutlu, G.M.; Urich, D.; Soberanes, S.; Buccellato, L.J.; Hawkins, K.; Chiarella, S.E.; Radigan, K.A.; Eisenbart, J.; Agrawal, H.; et al. Epithelial Cell Death Is an Important Contributor to Oxidant-Mediated Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2011, 183, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Steer, J.H.; Mann, T.S.; Lo, S.Z.Y.; Inglis, J.J.; Yap, H.S.; Henry, P.J.; Joyce, D.A. Early Induction of Uncoupling Protein-2 in Pulmonary Macrophages in Hyperoxia-Associated Lung Injury. Inhal. Toxicol. 2013, 25, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wang, J.; Hu, M.; Yang, Y.; Guo, L.; Xu, J.; Lei, C.; Jiao, Y.; Xu, J. Uncoupling Protein 2 Increases Susceptibility to Lipopolysaccharide-Induced Acute Lung Injury in Mice. Mediat. Inflamm. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Jung, Y.W. Protective Effect of Right Ventricular Mitochondrial Damage by Cyclosporine A in Monocrotaline-Induced Pulmonary Hypertension. Korean Circ. J. 2018, 48, 1135. [Google Scholar] [CrossRef] [PubMed]
- Ten, V.S.; Ratner, V. Mitochondrial Bioenergetics and Pulmonary Dysfunction: Current Progress and Future Directions. Paediatr. Respir. Rev. 2020, 34, 37–45. [Google Scholar] [CrossRef]
- Matsiukevich, D.; Randis, T.M.; Utkina-Sosunova, I.; Polin, R.A.; Ten, V.S. The State of Systemic Circulation, Collapsed or Preserved Defines the Need for Hyperoxic or Normoxic Resuscitation in Neonatal Mice with Hypoxia–Ischemia. Resuscitation 2010, 81, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S. Ten, V.; Stepanova, A.A.; Ratner, V.; Neginskaya, M.; Niatsetskaya, Z.; Sosunov, S.; Starkov, A. Mitochondrial Dysfunction and Permeability Transition in Neonatal Brain and Lung Injuries. Cells 2021, 10, 569. https://doi.org/10.3390/cells10030569
S. Ten V, Stepanova AA, Ratner V, Neginskaya M, Niatsetskaya Z, Sosunov S, Starkov A. Mitochondrial Dysfunction and Permeability Transition in Neonatal Brain and Lung Injuries. Cells. 2021; 10(3):569. https://doi.org/10.3390/cells10030569
Chicago/Turabian StyleS. Ten, Vadim, Anna A. Stepanova, Veniamin Ratner, Maria Neginskaya, Zoya Niatsetskaya, Sergey Sosunov, and Anatoly Starkov. 2021. "Mitochondrial Dysfunction and Permeability Transition in Neonatal Brain and Lung Injuries" Cells 10, no. 3: 569. https://doi.org/10.3390/cells10030569