
Extrinsic Regulators of mRNA Translation in Developing Brain: Story of WNTs


Department of Neuroscience and Cell Biology, Robert Wood Johnson Medical School, Rutgers University, Piscataway, NJ 08854, USA
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(2), 253; https://doi.org/10.3390/cells10020253
Submission received: 7 December 2020
/
Revised: 16 January 2021
/
Accepted: 21 January 2021
/
Published: 28 January 2021
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Neocortical Circuit Formation)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Extrinsic molecules such as morphogens can regulate timed mRNA translation events in developing neurons. In particular, Wingless-type MMTV integration site family, member 3 (Wnt3), was shown to regulate the translation of Foxp2 mRNA encoding a Forkhead transcription factor P2 in the neocortex. However, the Wnt receptor that possibly mediates these translation events remains unknown. Here, we report Frizzled member 7 (Fzd7) as the Wnt3 receptor that lays downstream in Wnt3-regulated mRNA translation. Fzd7 proteins co-localize with Wnt3 ligands in developing neocortices. In addition, the Fzd7 proteins overlap in layer-specific neuronal subpopulations expressing different transcription factors, Foxp1 and Foxp2. When Fzd7 was silenced, we found decreased Foxp2 protein expression and increased Foxp1 protein expression, respectively. The Fzd7 silencing also disrupted the migration of neocortical glutamatergic neurons. In contrast, Fzd7 overexpression reversed the pattern of migratory defects and Foxp protein expression that we found in the Fzd7 silencing. We further discovered that Fzd7 is required for Wnt3-induced Foxp2 mRNA translation. Surprisingly, we also determined that the Fzd7 suppression of Foxp1 protein expression is not Wnt3 dependent. In conclusion, it is exhibited that the interaction between Wnt3 and Fzd7 regulates neuronal identity and the Fzd7 receptor functions as a downstream factor in ligand Wnt3 signaling for mRNA translation. In particular, the Wnt3-Fzd7 signaling axis determines the deep layer Foxp2-expressing neurons of developing neocortices. Our findings also suggest that Fzd7 controls the balance of the expression for Foxp transcription factors in developing neocortical neurons. These discoveries are presented in our manuscript within a larger framework of this review on the role of extrinsic factors in regulating mRNA translation.
1. Introduction
1.1. Neocortical Development
In evolutionarily advanced species, the neocortex develops into the central neuronal circuit of higher cognitive function and voluntary motor behavior [1,2,3,4]. The development of this complex laminar structure requires an intricate progression of spatially and temporally controlled molecular events starting from neural stem cells [2,4,5,6,7,8,9,10]. The disruption of these events can lead to abnormal neocortical development associated with neurological and psychiatric disorders, such as epileptic encephalopathy (EE), autism spectrum disorder (ASD), and intellectual disability (ID), which are often expressed simultaneously [11,12]. The neocortical structure gives rise to six horizontal layers (I to VI), segregated principally by cell type and the specific site of neuronal connections. The neocortex, like all other central nervous structures, contains two main types of cells, neurons and glia, along with endothelial cells and microglia. Within the neuronal subtypes, the highly organized positioning of distinct subtypes of glutamatergic projection neurons (also known as pyramidal neurons) defines each layer during neocortical development. Each layer’s pyramidal neurons form specific axonal projections, with different targets determining the canonical circuitry within the cortex [1,2,7,13,14]. Pyramidal neurons in the upper layers II and III solely project their axons to other areas within the neocortex (intracortically within the ipsilateral hemisphere or to contralateral targets via the corpus callosum). In contrast, pyramidal neurons in deeper layers V and VI predominantly project out of the cortex to sites such as the thalamus, brainstem, and spinal cord. Interestingly, glutamatergic neurons in layer IV make short-range, local connections to other cortical layers. In addition to the differentiation in their efferent projections, each layer has distinction in their dendritic structures, incoming afferent axons, and transcription factor identities [4]. For example, upper layer glutamatergic neurons express transcription factors Satb2, Brn1, Brn2, Cdp, Cux2, Svet1, and Foxp1 while lower layer neurons express Fezf2, Tbr1, Otx1, Ctip2, and Foxp2 [4].
The primary stem cell source of neocortical projection neurons is radial glial progenitor cells (RGCs), widely known as scaffolds that guide the migration of new neurons from their birth zones close to the ventricles, towards their final destination of the cortical plate [15]. RGCs are multipotent, as they possess the ability to produce diverse types of glutamatergic neurons and glia as well as other progenitor subtypes; they can also proliferate extensively by self-renewal. These properties mark RGCs as a special type of neural stem cell (NSC) in the developing neocortex [3,16], of which the RGCs reside in the ventricular zone (VZ) adjacent to the lateral ventricles (LV). RGCs can give rise to a post-mitotic neuron either directly or indirectly via an intermediate progenitor (IP) cell. IPs populate the space basal to the VZ, known as the subventricular zone (SVZ). Embryonic cortical neurons are generated in an inside-out fashion to populate the six layers of the cortex [2,4,5,6,7,8,9,10]. Earlier-born neurons populate the deepest of the six cortical layers (V–VI), while later born neurons populate progressively more superficial layers (II–IV). These layers contain distinct neuronal subtypes that differ based on morphology, electrophysiological activity, axonal connectivity, and gene expression [17].
1.2. Post-Transcriptional Regulation in Neocortical Development
To achieve a six-layered laminar neocortex, it must go through genesis, migration, and maturation, regulated by transcriptional, post-transcriptional and epigenetic mechanisms. [2,4,5,6,7,8,9,10,18,19,20,21]. Recent discoveries of transcriptional regulation combined with advances in nucleic acid-predicated technologies have spearheaded a series of studies analyzing the neocortex’s genome and transcriptome across its neocortical layers [22,23]. Investigations on neurological disorders involving the neocortex, such as autism spectrum disorders, highlight the transcriptional framework and concentrate on abnormalities at the genomic and transcriptomic levels [24,25,26,27,28,29,30,31,32]. The precise equilibrium between RGC self-renewal and neuronal differentiation in the developing neocortex is modulated by the intricate network of intrinsic cellular programs and mechanisms. Experiments modulating gain- and loss-of-function show that transcription factors Sox5, Satb2, Fezf2, Tbr1, and Ctip2 comprise a cross-repressive transcriptional circuit that regulates projection neuron specification [17,33,34]. Subsequent studies have identified many additional subtype-specific genes, including transcription factors Brn1/2 and Rorb, which play critical roles in the specification and establishment of the neocortical layers [22,35,36]. Additionally, other studies have recognized the defining epigenetic signatures across differentiating cortical areas during development, evolution, aging, and disease [37,38,39,40,41]. Recent works have shown the importance of de novo somatic mutations, which occur post-zygotically and, thus, are present only in a subset of cells of an affected individual [42]. These somatic mutations contribute to neurodevelopmental diseases, including epileptic encephalopathies, intellectual disability, and autism spectrum disorder. Collectively, these studies support the notion that transcriptional regulation is a critical determinant of mature neuronal subtypes.
Despite the fact that transcription is considered the primary predictor of protein levels under steady state conditions, recent evidence shows that post-transcriptional control plays a dominant role during short-term state transitions, such as differentiation in response to extrinsic stimuli [43,44,45,46]. In order to better comprehend the molecular basis of the vast complexity and diversity encompassing neocortical development, post-transcriptional mechanisms have been characterized, including RNA-binding proteins (RBPs), ribosomal proteins (RPs), micro-RNAs (miRNAs), and long non-coding RNAs (lncRNAs). Understanding the regulation at the post-transcriptional level brings clarity to the transcriptional complexity and spatiotemporal specification of neuronal subtypes generated from RGCs in the developing neocortex [2,4,5,6,7,8,9,10,18]. RBPs play a crucial role in driving neocortical development by controlling the steps of post-transcriptional processing (splicing, transport, stability/decay, localization, and translation) [4]. RBPs and miRNAs regulate groups of functionally related mRNAs along a coordinated pathway of RNA processing, allowing cells to respond with unprecedented efficiency to extrinsic signals [47]. The combination of ribosomal proteins associated with translating polysomes is dynamic in both time and space during cortical development [48]. While the levels of certain mRNAs encoding transcriptional and translational regulators remain constant throughout cortical development, the differentially associated translating polysomes are temporally specific in manner, suggesting that the posttranscriptional regulators themselves could also be subject to regulation [48].
Cis- and trans-encoded translation regulations below significantly influence the fate of a cell. (1). In response to environmental cues, ribosomes are positioned locally in radial glial endfeet to translate mRNAs. (2). Upstream open reading frames (uORFs) repress canonical ORF translation by causing decay, leading to reduced translational efficiency. (3). MicroRNAs incorporated into an Argonaute complex bind to the 3′UTR to repress target mRNA expression. (4). RBPs and RNA helicases either promote or repress mRNA translation. (5). mRNAs and proteins are segregated in dividing cells, driving their asymmetrical inheritance in newborn cells. (6). RNA modifications, such as m6A, may affect mRNA stability and mRNA translation. (7). Microexons can change the reading frame or encode premature stop codons to modulate protein expression. These mechanisms are present and active in progenitors and neurons (reviewed in [9]). It is noted that RBPs play critical roles in the translation of NSC cell fate decisions. The RBP human antigen R (HuR) controls the association between translating polysomes and functionally-related mRNAs encoding transcriptional, translational, and neuronal layer-specific regulators in a developmental stage-specific manner [18,49]. HuR also controls the composition of translational initiation factors, elongation factors, and combinatorial patterns of ribosomal proteins within polysomes [49]. HuR interacts with both common and unique subsets of mRNAs during early and late neurogenesis, suggesting that mRNA targets of RBPs evolve as a function of time [50]. Recent interesting work demonstrates that upstream RBP, Celf1, regulates the 5′ UTR-driven isoform-specific translation of another RBP, Elavl4, which results in the specific development of glutamatergic neurons [12]. These findings reveal a dynamic interplay between distinct RBPs and alternative 5′ UTRs in neuronal development. RBP Fragile X Mental Retardation Protein (FMRP) is another translational regulator that plays a significant role during corticogenesis. At RGC basal endfeet, FMRP integrates mRNAs encoding signals and cytoskeletal factors which are associated with autism and neurogenesis [51]. FMRP represses the RGC to IP transition, maintaining RGCs in an undifferentiated state [52]. Thus, these reports reveal how RBPs are important in regulating multiple aspects of embryonic cortical development including mRNA splicing, stability, localization, and translation.
Multipotent RGCs are transcriptionally primed, allowing the rapid generation of diverse progeny. Post-transcriptional regulation, including translation, refines gene expression to specify cell fates [9]. RGCs express mRNAs that maintain stem cell proliferation and self-renewal in the stages of cortical embryonic development. In addition, they also express mRNAs that help to promote neurogenesis (transcriptional priming or pre-patterning) [53]. During the stem cell state, translational repression of mRNAs interferes with neuronal cell differentiation: elF4E and its binding partner 4E-T compose a repressive complex that sequesters and represses proneurogenic mRNAs expressed in RGCs [54]. The 4E-T protein targets mRNAs via RBPs, such as Smaug2, in an undifferentiated state [55]. In order to generate neurons, the disruption of these relevant translational repression complexes occurs with external proneurogenic signals by mechanisms that remain unknown, enabling the translation of mRNAs mediating neurogenesis. Nanos1, an RBP that promotes neurogenesis presumably by repressing self-renewal genes, suggests that appropriate neuronal differentiation requires both the translational derepression of proneurogenic mRNAs as well as the de novo repression of mRNAs associated in stem cell maintenance [55].
1.3. Neocortical Development Orchestrated with Extracellular Signals
RGCs possess an elongated radial morphology that allows it to access extrinsic signals originating from the meninges, vasculature, newborn neurons, cerebrospinal fluid, and ingrowing axons; many of which regulate RGC cell fate decisions [56,57]. Collective proteomic analyses from the cell-surface of RGCs and newborn neurons have revealed rich growth factors in the developing cortex, including many previously uncharacterized autocrine and paracrine interactions [57]. Extrinsic signals generically control RGC proliferation and differentiation while regulating the specification of particular neuronal subtypes [58]. Namely, epidermal growth factors (EGFs) influence several cells including neural stem cells (NSCs), oligodendrocytes, astrocytes, and neurons with different effects on their proliferation, migration, and differentiation [59]. In multiple neuronal systems, extracellular factors can also regulate mRNA translation specificity [60,61]. For example, nerve growth factor (NGF) stimulates the translation of eukaryotic elongation factor 1A-1 (eEF1A-1) mRNA by specifically recruiting it to polyribosomes in neuronally differentiated PC12 cells [60]. Brain-derived neurotrophic factor (BDNF) also regulates the translation of a select group of mRNAs during neuronal development, especially within neuronal dendrites in the mammalian target of rapamycin (mTOR)-dependent pathway [61].
Timed extracellular signaling events are essential in determining normal neocortical development. One major developmental event is the ingrowth of thalamic axons at mid-neurogenesis [62]. The thalamus, a brain structure in the vertebrate diencephalon, plays a central role in regulating diverse functions of the cerebral cortex with guidance mechanisms for thalamocortical axons (TCAs) [63]. When the mouse thalamus undergoes embryonic neurogenesis, several Wnt ligands, such as WNT3, WNT3A, and WNT7B, are expressed [48,64] and involved in the development and control of TCA projection in thalamic glutamatergic neurons [65]. The TCAs grow through the subpallium and reach the cortex by E14.5 [66]. It was shown that the E14.5 mouse thalamus produces a diffusible factor that promotes the proliferation of cortical precursors over a restricted developmental window [67], suggesting that thalamic afferents control the cortical area size by promoting the division of a specific population of neural progenitor cells. The timed ingrowth of thalamocortical axons is accompanied by the secretion of extracellular factors into the developing neocortex; however, the underlying cellular and molecular mechanisms still remain unknown.
1.4. WNT Signaling in Neuronal Diseases
The WNT signaling pathway is an evolutionarily conserved signal transduction pathway that regulates a wide range of cellular functions including cell proliferation, cell fate determination, apoptosis, cell migration, and cell polarity during development and stem cell maintenance in adults [68,69,70]. WNT proteins are lipid-modified glycoproteins that are about 350–400 amino acids in length [71] and act as ligands that interact with Frizzled (FZD) receptors which are located on the cell surface, to activate intracellular signaling pathways [71,72,73]. FZD receptors are a family of G protein-coupled receptor proteins that have seven-pass transmembrane domains and act as the primary receptors for WNT signaling. Once FZD receptors are activated, a signal is intracellularly transduced causing the activation of protein Disheveled (Dvl or Dsh), which induces the WNT signal branching off into multiple downstream pathways that can be categorized into the canonical WNT/β-catenin pathway and the non-canonical WNT pathway [73,74,75]. During canonical WNT signaling, β-catenin is released from scaffolding proteins, such as Axin, and helps accelerate messaging between the cell membrane and nucleus [76]. β-catenin is then localized to the nucleus where it forms a complex with DNA bound TCF/LEF transcription factor resulting in the activation of WNT-responsive genes [74,77,78,79]. Non-canonical pathways are independent of β-catenin signaling and possess the ability to activate additional signals that regulate several cellular behaviors, including planar cell polarity, cell movements during gastrulation, and the cell migration of neural crest cells [80,81,82,83].
In addition to embryonic development and cancer progression, the WNT pathway has been identified as a key contributor to Alzheimer’s (AD) and metabolic diseases. The WNT pathway is strongly linked to AD pathogenesis because the pathway is fundamental to the development of the central nervous system [84]. Several studies have reported a causative link between WNT signaling and autism. Mice mutants for Dvl1 and Dvl3 showed reduced β-catenin expression causing premature deep layer neurogenesis of neural progenitors in precise brain regions during embryonic development. This expressed an adverse effect on the establishment of neural connections in the future prefrontal cortex, which was observed with serious deficits in brain size and social behavior in adults [85]. The administration of a GSK-3 inhibitor to activate the canonical WNT pathway in utero can reverse these deficits caused by diminished β-catenin expression. WNT signaling is also linked to the pathogenesis of Parkinson’s disease via the dysregulation of the expression of WNT pathway components in brain tissue of Parkinson’s disease patients [86]. Further studies revealed that activation of the WNT/β-catenin pathway in astrocytes could recover neighboring midbrain dopaminergic neurons after post-injury in vivo. Thus, it can serve as a therapeutic approach to promote recovery of midbrain dopaminergic neurons [87,88]. Patients with schizophrenia show altered GSK3 activity [89] as well as increased expression of WNT1, which can lead to synaptic rearrangement and plasticity [90]. Additionally, several SNPs in FZD3 were reported to be associated with susceptibility to schizophrenia [91]. The relevance of WNT signaling in depression, bipolar disorder, epilepsy, seizures has also been reported [92].
1.5. mRNA Translations Regulated by WNTs Signaling
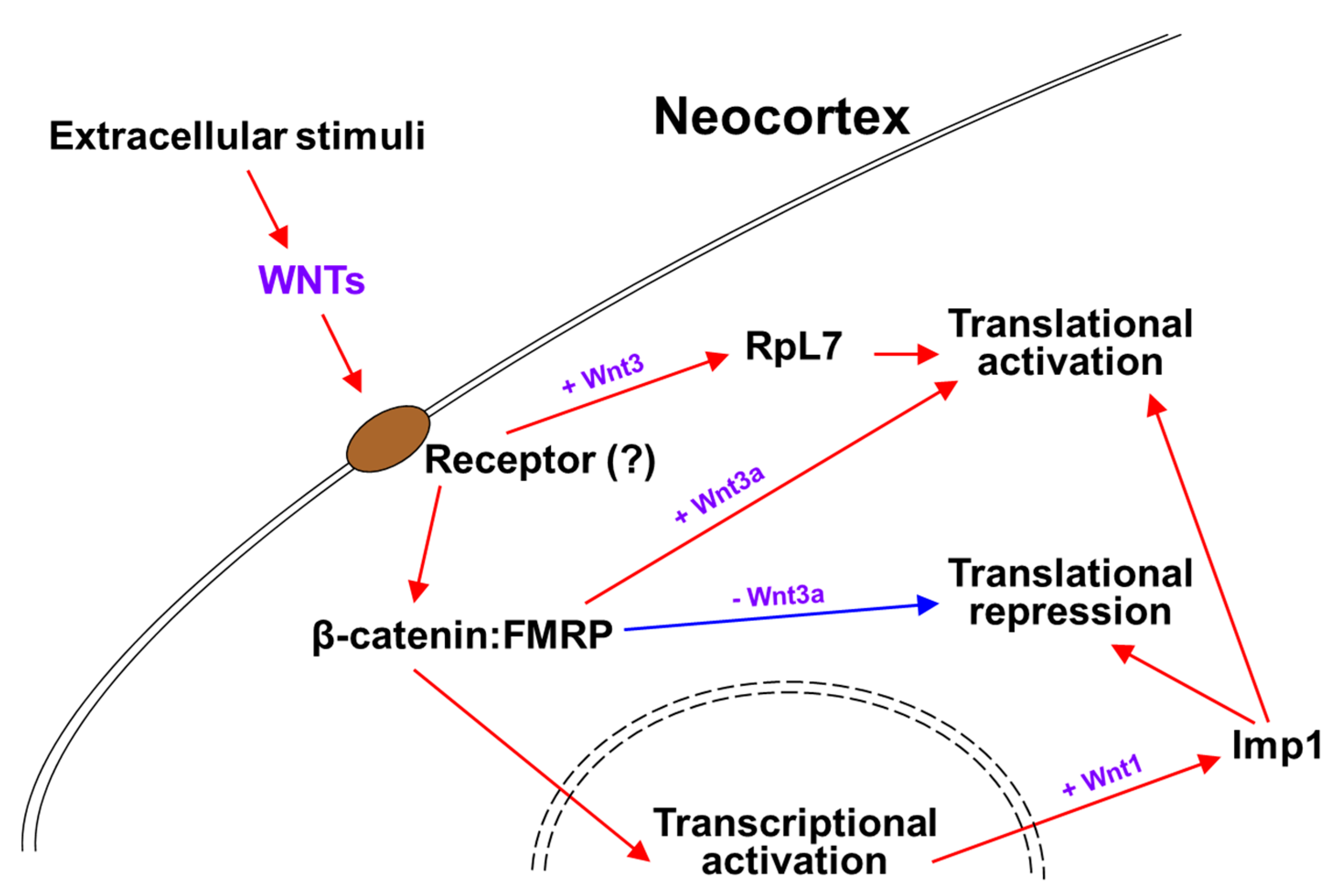
Neural stem cell (NSC) niches are abundant in extrinsic signals that regulate their proliferation, differentiation, and cell fate specification. These environmentally driven signaling pathways make NSC likely to modulate RBP expression and/or its interactions with target mRNAs [57]. WNT signaling is related to Fragile-X syndrome (FXS), a heritable form of autism caused by a deficiency of RBP Fragile-X mental retardation protein (FMRP), resulting in variable degrees of autism-like behavior. FMRP functions as a negative regulator of protein translation at the RGC basal endfeet. FXS patients showed reduced expression of WNT7a and reduced β-catenin-dependent signaling, and in the brain of Fmr1 (FMRP gene) knockout mice the translation of Wnt2 mRNA was found to be reduced [93,94,95,96]. The connection of FMRP to WNT signaling was extended with the discovery of β-catenin and FMRP complex functioning in WNT-dependent translation regulation [97]. The interaction between β-catenin and FMRP leads to a basal recruitment of β-catenin to the messenger ribonucleoprotein and translational pre-initiation complex, fulfilling a translational repressor function (Figure 1). Wnt3a stimulation provokes this function in part by separating β-catenin from the pre-initiation complex (Figure 1) [97]. Another example of how extracellular WNT regulates cytoplasmic mRNA translation is shown in Wnt1 signaling, which enhances embryonic NSC maintenance and proliferation by promoting RBP Imp1 expression during cortical development (Figure 1) [98]. Not only does it silence proneurogenic mRNAs, the Imp1 also translationally increases the stability and expression of pro-self-renewal mRNAs, such as Hmga2, in order to maintain NSCs in an undifferentiated state (Figure 1) [98]. Consistent with these functions of Imp1 from Wnt1 signaling, the cortex specific Imp1 ablation resulted in reduced NSC self-renewal and accelerated neuronal and glial differentiation [98].
Transcription factors, such as Foxp2, that promote layer-specific neuronal differentiation are highly regulated during development [99]. In our previous study, it was found that timed Wnt3 secretion from thalamocortical axons around mid-neurogenesis regulates the composition of ribosomal proteins, including Rpl7, in the subset of mRNAs that are associated with polysomes (Figure 1) [48]. Wnt3 promotes Foxp2 mRNA association and translation in polysomes through the regulation of its 3′UTR (Figure 1). The conditional ablation of thalamic Wnt3 causes damage to the specification of Foxp2-postive deep layer neurons [48]. How do these extrinsic cues promote and support post-transcriptional machinery? Many RBPs contain multiple consensus phosphorylation sites, implying that they can be regulated by phosphorylation [100]. For instance, HuR phosphorylation is important for regulating the translational control of the autism-associated mRNAs, Foxp1 and Foxp2, in the developing cortex [50]. For accurate responses to growth factor stimuli in several other environments, the receptor-mediated phosphorylation of RBPs might provide a mechanism that allows the rapid and reversible alteration of the translation subsets of mRNAs which regulate NSC biology.
However, the WNT receptor that may mediate these mRNA translation events remains unknown. In neurogenesis of neocortical cells, it is critical to discover which receptor transfers the extrinsic Wnt signaling arriving from the thalamus into the neocortex, to regulate the mRNA translation in developing neocortical neurons. Here, we demonstrate that Frizzled member 7 (Fzd7) as a Wnt3 receptor functions downstream in the Wnt3-regulated translation. The Fzd7 co-localizes with Wnt3 in developing neocortices and is co-expressed in neurons expressing Foxp1 or Foxp2 in the distinct layers of neocortices. The silencing and overexpression of Fzd7 resulted in changes in the Foxp1 and Foxp2 protein expressions, respectively. We further found that Fzd7 is required for the Wnt3 induced Foxp2 mRNA translation via 3′UTR, while the novel effect on Foxp1 is not Wnt3 dependent. In conclusion, we show that the interaction between Wnt3 and Fzd7 regulates neuronal identity and that Fzd7 tightly controls the expression of Foxp transcription factors in developing neocortical neurons.
2. Methods
2.1. Animals and In Utero Electroporation
For all embryonic experiments, mice of both genders were used without sex determination. Experiments involving animals were carried out in accordance with Rutgers University Medical School’s Institutional Animal Care and Use Committee. For timed pregnancies, adult pregnant female CD-1 mice were purchased from Charles River Laboratories. In utero electroporation (IUE) was performed at E13 (Fzd7 shRNA) or E16 (Fzd7 OE) and analyzed at P0 as previously described [101]. Co-electroporation was accomplished with 1 µL of mixed plasmids containing 4 µg/µL of control or Fzd7 shRNA/OE vector along with 1 µg/µL of CAG-GFP reporter (4:1 ratio). For each IUE, at least three transfected neocortices were used in experiments and this is indicated in figure legends with the “n” value. Three to five images from different sections of each transfected neocortex were used for the quantifications of immunostaining.
2.2. Primary Neuronal Culture and N2a Cell Transfection/Culture
Alternating E13 neocortices in the same litter were co-electroporated (IUE) with Ctrl shRNA and CAG-GFP or Fzd7 shRNA and CAG-RFP plasmids. Four hours post-IUE, the primary neuronal cells were mixed and in vitro cultured together [12]. The primary neuronal cultures were performed as described [48]. Neuroblastoma N2a cells (ATCC #CCL-131, Manassas, VA, USA) were grown in Dulbecco’s modified eagle medium (Gibco #31053-036, Waltham, MA, USA) containing 10% FBS (Gemini #900-108, West Sacramento, CA, USA), 1% GlutaMAX (Gibco #35050-061, Waltham, MA, USA), 1% Sodium Pyruvate (100mM, Gibco #11360-070, Waltham, MA, USA), and 1% Penicillin/Streptomycin (Corning #30-002-CI, Corning, NY, USA). TrypLE Express (Gibco #12604-021, Waltham, MA, USA) dissociation reagent was used for regular maintenance. Lipofectamine 2000 (Invitrogen #11668-019, Waltham, MA, USA) was used to perform transfections as per the manufacturer’s protocol.
2.3. Plasmids
The shRNA and overexpression plasmids were commercially obtained from OriGene; shRNA: control (#TR30013, Rockville, MD, USA) and Fzd7 (#TG500742B, Rockville, MD, USA); overexpression: control (pCMV6-AC, #PS100020, Rockville, MD, USA) and Fzd7 (NM_008057, #MC202423, Rockville, MD, USA). For the Luciferase-Foxp2_3′UTR construct, the full-length of Foxp2_3′UTR (3922 bp) was subcloned to the downstream of a firefly Luciferase cassette in the pcDNA3.1-Luciferase plasmid as previously described [48].
2.4. Luciferase Reporter Assay
N2a cells were seeded on 12-well plate and co-transfected the next day with 0.4 µg of the Luciferase-Foxp2_3′UTR reporter vector and 2 µg of the Ctrl shRNA or Fzd7 shRNA plasmids per one well. Twenty-four hours after transfection, cells were split into three experimental groups: (1) mock treated (base media only), (2) treated with 100 ng/mL of recombinant Wnt3 (Abnova #H00007473-P01, Taipei, Taiwan), and (3) treated with 100 ng/mL of both Wnt3 and SFRP1 (R&D Systems #5396-SF-025, Minneapolis, MN, USA) for 48 h. After washing with PBS, cells were lysed at room temperature for 15 min with 250 µl of 1X Passive Lysis Buffer (Promega # E1910, Madison, WI, USA), then divided equally into two fractions. One fraction was followed by the Pierce™ 660nm Protein Assay (Thermo Scientific # 22660, Waltham, MA, USA) and Luciferase measurement with the Dual-Luciferase Reporter Assay System (Promega #E1910, Madison, WI, USA). The other fraction of cells was used for total RNA isolation using TRIzol LS (Invitrogen # 10296028, Waltham, MA, USA) for qRT-PCR. Relative Luciferase light units (RLUs) were normalized to the Luciferase mRNA levels measured in duplicate qRT-PCR run in parallel for each condition.
2.5. Quantitative Real-Time PCR
Total RNAs were isolated using TRIzol LS (Invitrogen #10296028, Waltham, MA, USA) following the manufacturer’s instructions, while residual DNAs were removed by incubating the RNAs with the Turbo DNA-free™ kit (Invitrogen #AM1907, Waltham, MA, USA). The Applied Biosystems StepOne real-time system and reagents (TaqMan™ RNA-to-Ct™ 1-step kit, Thermo Fisher #4392938, Waltham, MA, USA) were used to perform qRT-PCR with 50 ng of RNA per reaction and commercially available TaqMan probes of control and target mRNAs below. The results were analyzed using the ∆∆Ct method normalized with control Gapdh/ActB mRNAs [102]. TaqMan FAM probes (Thermo Fisher, Waltham, MA, USA): Gadph, Mm99999915_g1; ActB, Mm01205647_g1; Wnt3, Mm00437336_m1; Fzd7, Mm00433409_s1; Luciferase, Mr03987587_mr.
2.6. Primary Antibodies
The following primary antibodies and dilutions were used for immunocytochemistry: Wnt3 (1:250, rabbit; LifeSpan BioSciences #LS-C774904, Seattle, WA, USA), Fzd7 (1:50, goat, Thermo Fisher #PA5-47232, Waltham, MA, USA), Foxp1 (1:1000, rabbit; Abcam #ab16645, Cambridge, MA, USA), Foxp1 (1:250, mouse; lab stock), Foxp2 (1:250, rabbit; Abnova #PAB12684, Taipei, Taiwan), Foxp2 (1:250, goat; Santa Cruz Biotechnology #sc21069, Dallas, TX, USA), Gapdh (1:300, mouse, Millipore Sigma #MAB374, St. Louis, MO, USA), GFP (1:1000, chicken; Aves Labs #GFP-1020, Tigard, OR, USA), RFP (1:1000, rabbit; Antibodies-Online #ABIN129578, Limerick, ME, USA). Appropriate species-specific Donkey secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA) were used at a 1:250 dilution.
2.7. Immunohistochemistry and Confocal Imaging
Immunohistochemistry was performed as previously described [49]. Briefly, embryonic brains from postnatal day 0 (P0) were dissected and fixed by immersion for 8 h with 4% paraformaldehyde (PFA, Sigma-Aldrich #158127, St. Louis, MO, USA) in 1X PBS, pH 7.4. After fixation, the brains were washed three times in 1× PBS and coronally-sectioned at 70–80 µm using a Leica VT1000S vibratome. Free-floating sections were then incubated in blocking solution (5% Normal donkey serum (Jackson ImmunoResearch #017-000-121, West Grove, PA, USA), 1% Bovine serum albumin (Biomatik #A2134, Cambridge, ON, Canada), 0.1% Glycine (BDH #BDH4156, Radnor, PA, USA) and 0.1% Lysine (Sigma #L5501, St. Louis, MO, USA) in 1X PBS, pH 7.4) and 0.4%Triton (Sigma-Aldrich #X100, St. Louis, MO, USA), gently shaking for 2–4 h at room temperature. Afterwards, the sections were immediately transferred to a solution of primary antibody resuspended in blocking solution +0.4%Triton X-100, and incubated while gently rocking overnight at 4 °C. The following day, sections were washed 3 times in 1X PBS and incubated with secondary antibody diluted in blocking solution without Triton X-100, gently shaking at room temperature for 1–2 h. Next, sections were washed again three times in 1X PBS before being incubated in 1 μg/mL of DAPI (Fisher Scientific #D1306, Waltham, MA, USA) for 10 min. Finally, sections were washed two more times in 1× PBS before being mounted with Vectashield mounting media (Vector Laboratories #H1000, Burlingame, CA, USA). The immunostaining of Wnt3 (ligand protein outside of membrane) was performed without Triton X-100. Primary neuronal and N2a cells were cultured on glass coverslips, with which immunocytochemistry was followed as written above. Then, slides were imaged with an Olympus BX61WI confocal microscope using 10×, 20×, or 60× objective lens and processed using Fluoview FV-1000. All images used in analysis were taken with the same confocal settings per experiment to allow for accurate comparisons of fluorescent intensity.
2.8. Immunohistochemistry Quantification and Statistical Analysis
Ten equal sized bins were framed in each confocal image, of which markers/cells were quantified by separately blinded investigators using Gimp2 software to calculate the percentage of Wnt3/Fzd7/Gapdh or Foxp1/Foxp2-positive neurons among all GFP/RFP cells. The graph bars represent mean and ± SEM, as noted in the figure legends. The number (n) of replicates for each experiment and the appropriate statistical test are noted in figure legends. For example, with three biological replicates (n = 3 animals), we made three to five images from different sections of each transfected neocortex. Then, blinded investigators counted the markers/cells from each image independently. It was not known to them which image was derived from which animal. Statistical significance (Student’s t-test) is reported as: *: p < 0.05, **: p < 0.01, ***: p < 0.005.
All materials available upon request. Contact Dr. Mladen-Roko Rasin ([email protected]).
3. Results
3.1. Wnt3 Morphogen and Fzd7 Receptor Are Co-Localized with Foxp1 and Foxp2 Transcription Factors in Developing Neocortices at E16 and P0
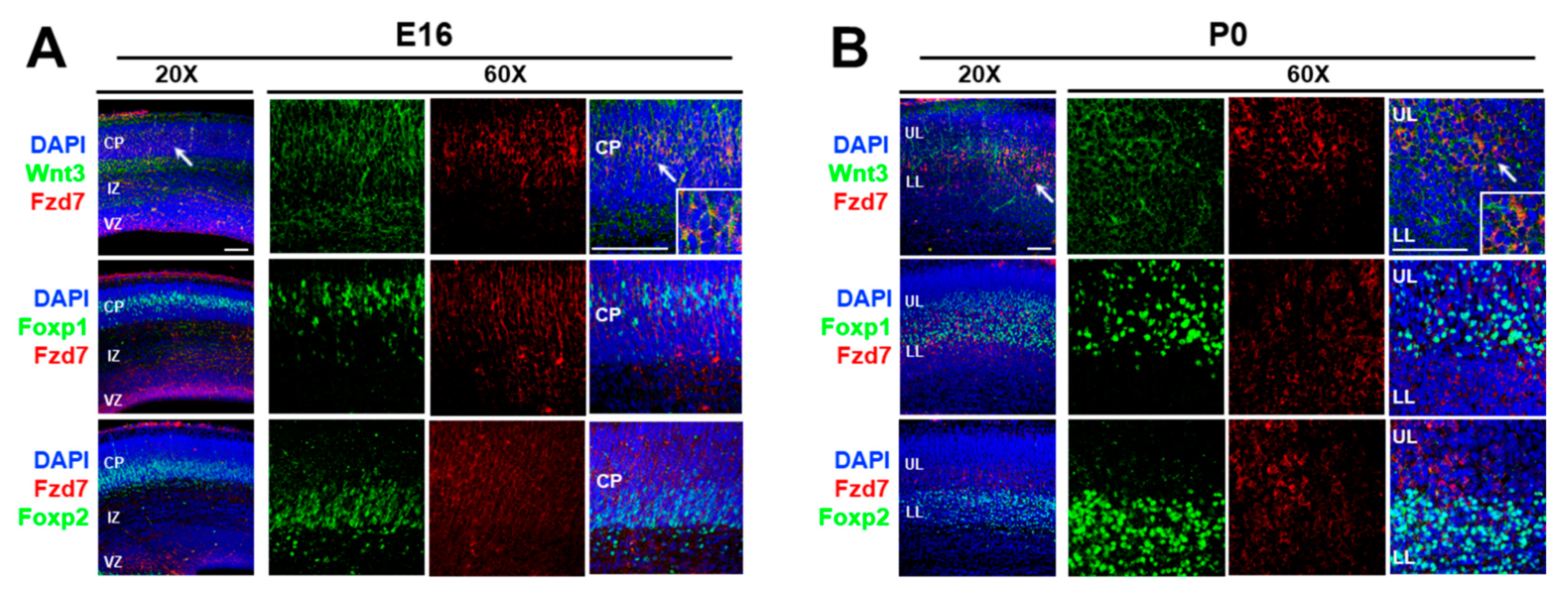
To characterize how thalamic morphogen Wnt3 regulates neocortical mRNA translation, we aimed to identify its receptor. It was reported that Wnt3 binds to the Frizzled 7 (Fzd7) receptor in intestinal stem cells [103]. In addition, the functional interaction between Wnt3 and Fzd7 led to the activation of the Wnt/β-catenin signaling pathway in hepatocellular carcinoma cells [104]. It is known that Fzd7 mRNA is expressed in the developing neocortex. These findings suggested that the action of Wnt3 may be mediated through the Fzd7 receptor in the developing neocortex. Therefore, we first established the expression sites of Fzd7 protein using immunohistochemistry (Figure 2). At E16, the Fzd7 receptors were expressed evenly throughout the cortical plate (CP) (Figure 2A) and colocalized with Wnt3 ligands (arrow in Figure 2A). Furthermore, the Fzd7 protein overlapped in both upper layer and lower layer neurons of the CP expressing Foxp1 and Foxp2, respectively (Figure 2A). The Wnt3/Fzd7 and Fzd7/Foxp1/Foxp2 colocalizations were still present at P0 (Figure 2B). Taken together, these observations suggest that the Wnt3 secreted from the thalamus at E15 [48] can interact with the neocortical Fzd7 receptor in order to timely regulate the protein synthesis of the neocortical Foxp transcription factors.
3.2. Wnt3 Interacts with Fzd7 in Neuroblastoma N2a Cells
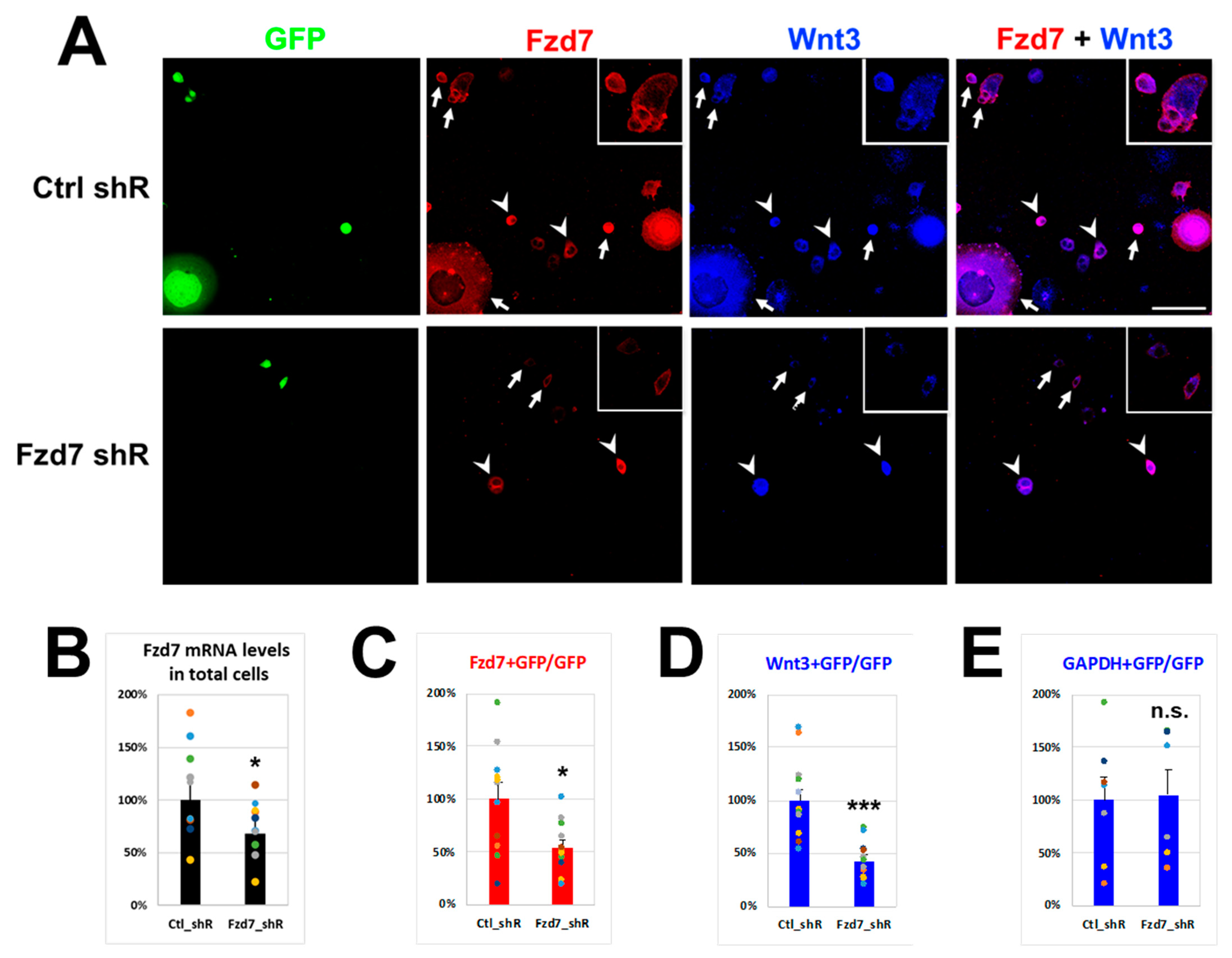
If the extracellular Wnt3 interacts with the Fzd7 receptor in the cell membrane, Fzd7 downregulation should reduce Wnt3 cell membrane immunostaining. To test this, neuroblastoma N2a cells were transfected with either Control (Ctrl) or Fzd7 shRNA plasmid containing Green Fluorescent Protein (GFP) gene (Figure 3). The transfection efficiencies of N2a cells in culture dish with shRNA plasmids (GFP cells) illustrated similarities between Ctrl shRNA (36.0%) and Fzd7 shRNA transfection (34.2%) (Figure S1A). However, Fzd7 mRNA expression was reduced by 68% in total cells with Fzd7 shRNA transfection (Figure 3B), suggesting efficiency of the Fzd7 shRNA in hand. After 3 days of each shRNA transfection, Wnt3 was added into the culture medium at 100 ng/mL that was reported to be sufficient amount to promote Foxp2 translation in neocortical projection neurons [48]. The Fzd7 shRNA transfected GFP cells showed a reduced Fzd7 intensity compared to the Ctrl shRNA transfected GFP cells (arrows in Figure 3A). The difference in Fzd7 intensity was also observed between the Fzd7 shRNA transfected GFP cells (arrows) and non-transfected cells (arrowheads) from the same culture dish (lower panel of Figure 3A). The Fzd7 signal intensity was reduced by 54.2% in the Fzd7 shRNA transfected GFP cells when it was normalized to the signal from the Ctrl shRNA transfected GFP cells (Figure 3C). Thus, these data indicate a successful and significant decrease of Fzd7 protein expression by the shRNA in hand.
We then analyzed the interaction between Wnt3 and Fzd7 at the cell membrane using immunohistochemistry after 4 h of Wnt3 treatment (Figure 3A), with which Wnt3 intensity became clear and strong at the cell membrane compared to no treatment of Wnt3 (Figure S1B). Remarkably, the Wnt3 intensity at the cell membrane also decreased in the Fzd7 shRNA transfected cells when compared to the Ctrl (Figure 3A,D). This finding correlated with the decrease of Fzd7 expression (Figure 3B,D). The intensity of Gapdh protein was used as a negative control and remained unchanged between the Ctrl and Fzd7 shRNA transfected GFP cells (Figure 3E and Figure S1C). We also confirmed that the Fzd7 shRNA did not affect the endogenous Wnt3 mRNA expression (data not shown). These data suggest that the downregulation of Fzd7 receptor reduces the interaction of Wnt3 ligand with the cell membrane.
3.3. Fzd7 Silencing Results in Changes of Foxp Transcription Factors on Primary Neuronal Cells
To characterize the roles of Fzd7 receptor in the neocortical development with the Wnt3 effect on the translation of Foxp transcription factors, we first in utero electroporated (IUE) Ctrl or Fzd7 shRNA plasmid with either CAG-GFP or CAG-RFP reporter, respectively [2,12,50,105]. Ctrl shRNA/CAG-GFP or Fzd7 shRNA/CAG-RFP were transfected in the neocortices of separate embryos of the same litter at E13 and the primary neuronal cell cultures were made together from all neocortices on the same day [12]. After four days in vitro (DIV), the intensities of Foxp proteins were quantified between GFP+ (Ctrl shRNA) and Red Fluorescent Protein positive (RFP+) (Fzd7 shRNA) cells. Intensities of Foxp protein expression were normalized to the intensity (+) observed in non-transfected cell as an intrinsic control of immunostaining quality (Figure 4A). The strong (++) intensity of Foxp2 protein in Fzd7 shRNA cells (arrow: GFP cells; arrowhead: RFP cells in the upper panel of Figure 4A) decreased upon Fzd7 silencing (Figure 4B). In contrast, the strong (++) intensity of Foxp1 staining (lower panel of Figure 4A) increased upon Fzd7 silencing (Figure 4C). These data suggest the Fzd7 receptor regulates both the Foxp2 and Foxp1 expressions in developing neocortical neurons, but in opposing directions. Fzd7 presence promotes Foxp2 expression and suppresses Foxp1 expression.
3.4. Fzd7 Downregulation Disrupts Neuronal Migration and Regulates Foxp2 and Foxp1 Protein Expression In Vivo in Contrasting Ways
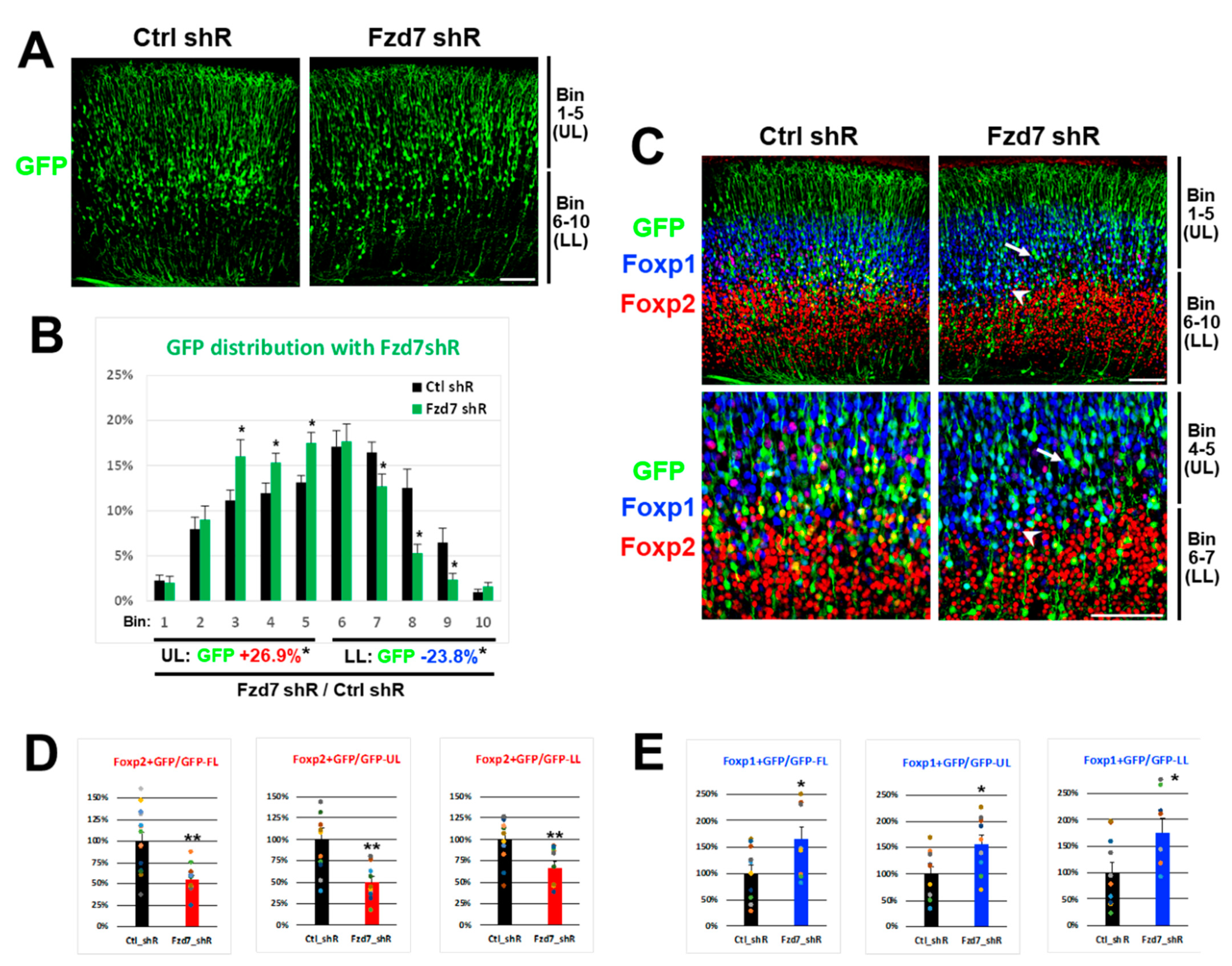
Next, we investigated whether the Fzd7 downregulation in vivo affects the Foxp2 and Foxp1 protein expressions in developing neocortical neurons in a similar fashion as we found in vitro. Either Ctrl or Fzd7 shRNA plasmid was electroporated in utero with a CAG-GFP reporter at E13 and the GFP+ cells in transfected neocortices were analyzed at P0 [12]. When Fzd7 was silenced, the GFP+ axons projected appropriately for the developmental stage and were comparable to the Ctrl (data not shown). However, the distribution of GFP+ cells across 10 bins in the neocortical wall was different between the Ctrl and Fzd7 shRNA (Figure 5A,B). In particular, we found significantly more GFP+ cells in the upper bins (Bin 1–5) than in the lower bins (Bin 6–10) when Fzd7 was silenced (Figure 5A,B).
Consistent with the in vitro data (Figure 4A,B), the in vivo Foxp2 expression was also reduced when Fzd7 was downregulated (Figure 5C,D). The change in the Foxp2 expression affected by Fzd7 silencing was most pronounced in bins 4–5 (arrow in Figure 5C and Figure S2A). In contrast, the Foxp1 expression in vivo increased when Fzd7 was silenced (Figure 5C,E), similar to our in vitro findings (Figure 4A,C). Notably, the increase in Foxp1+/GFP+ cells was observed larger in the lower bins 6–10 (Figure 5E), even though Foxp1 is predominantly expressed in the upper layers of the neocortex (Figure 2B and Figure 5C). In particular, the highest increase in Foxp1 expression was seen in bins 6–7 (arrowhead in Figure 5C and Figure S2B). It is plausible that the disrupted migration of GFP+ cells that were Fzd7 silenced (Figure 5A,B) influences the numbers of Foxp2+ or Foxp1+/GFP+ cells (Figure 5C–E). However, the bins showing a bigger change in either Foxp2 or Foxp1 expression (arrow or arrowhead in Figure 5C and Figure S2) are located to where Fzd7 was found to be normally expressed (Figure 2B). Therefore, taken together with the in vitro findings, the data above further suggest that the Foxp2 and Foxp1 expressions in developing neurons of these regions are regulated by Fzd7 signaling.
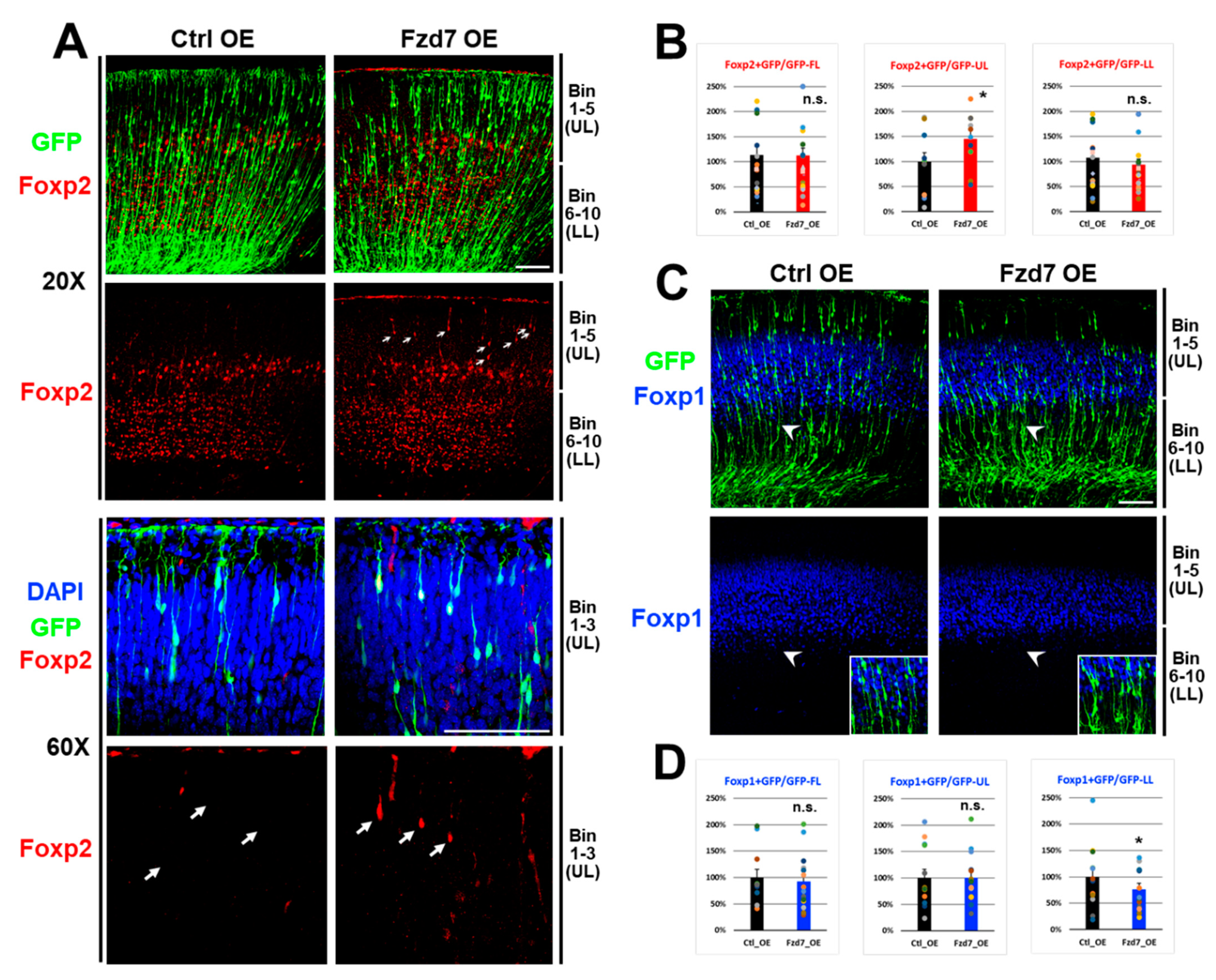
3.5. Fzd7 Overexpression Altered Foxp2 and Foxp1 Protein Expressions in a Layer-Specific Fashion
Since Fzd7 silencing increased the Foxp1 expression present in upper layer neurons, we subsequently hypothesized that Fzd7 overexpression in upper layers will decrease Foxp1 expression. To test this, we in utero electroporated GFP reporter and either Ctrl or Fzd7 overexpression (OE) plasmid at E16 to selectively target upper layer neurons [10], and these transfected neocortices were analyzed at P0 [12]. When Fzd7 was overexpressed, GFP+ cells were reduced in upper bins 1–5 and increased in the lower bins 6–10 (Figure S3A,B). This abnormal distribution of GFP+ cells (Figure S3B) represented that Fzd7 overexpression affects the migration of neuronal GFP cells (Figure S3A), contrasting the migration result when Fzd7 was silenced (Figure 5A,B). Collectively, these data indicate that Fzd7 receptor play an important role in neuronal migration of the neocortical glutamatergic neurons. Considering that the Fzd7 expressions are shown in VZ region (Figure 2A) and its dendrites (Figure S3A), it is possible that the Fzd7 changes may affect neuronal migration from the VZ region containing neocortical stem cells.
When we analyzed the expression of Foxp proteins (Figure 6), we surprisingly found that Fzd7 overexpression resulted in an increase in Foxp2 protein in the upper bins 1–5 (Figure 6A and middle in Figure 6B) while there was no change in Foxp2 expression in the lower bins (right in Figure 6B). Although the Foxp2 protein is normally not expressed in bins 1–3 at P0 neocortex (Figure 2B and Figure 6A), the Fzd7 receptor autonomously induced Foxp2 expression even in neurons of these upper layer bins when Fzd7 was overexpressed (arrows in Figure 6A), which suggests that the change of Foxp2 expression is not due to the defects in migration. These observations additionally imply that Fzd7 regulates the expression of Foxp2. Furthermore, the Fzd7 overexpression decreased the Foxp1 expression in GFP+ neurons (Figure 6C,D). Interestingly, this reduction in Foxp1+/GFP+ cells upon the Fzd7 overexpression was found to be selectively in lower bins 6–7 (arrowhead region in Figure 6C and right in Figure 6D) without the significant change in Foxp1 expression in the upper bins (middle in Figure 6D). Collectively, these findings further support the notion that Fzd7 regulates Foxp2 and Foxp1 expression in developing neocortical neurons.
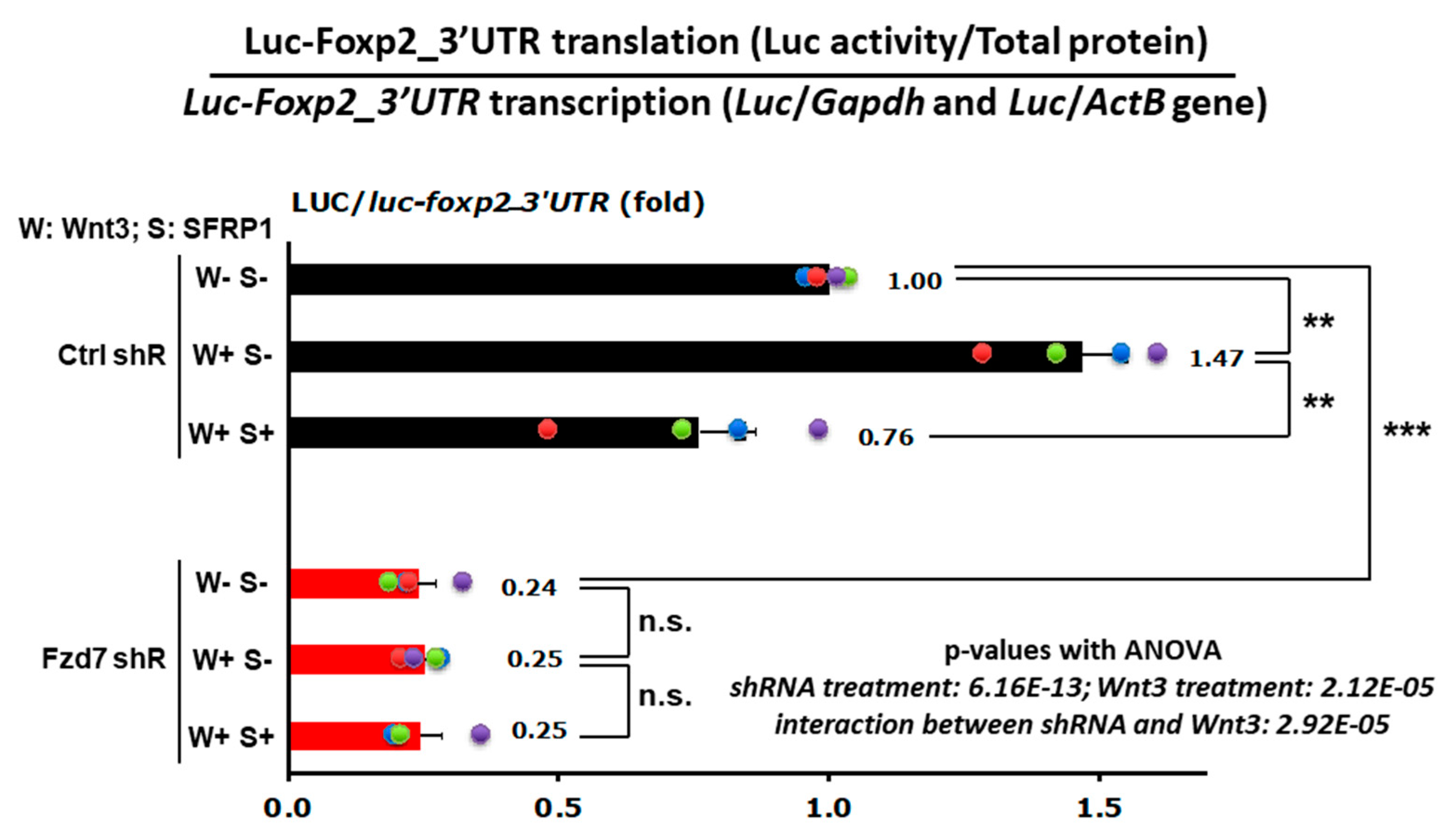
3.6. Wnt3-Fzd7 Signaling Regulates Foxp2 mRNA Translation via Its 3′UTR
The 3′ untranslated region (3′UTR) functions principally on modulating mRNA dissociation with ribosome and translation efficiency [106], including Foxp2 3′UTR [48]. We previously reported that the specific 3′UTR sequence of Foxp2 mRNA regulates translation efficiency in response to Wnt3 signaling in the developing neocortex [48]. First, we repeated these findings in a new set of similar experiments and found that Wnt3 induces mRNA translation via Foxp2_3′UTR also in neuronal N2a cells (Ctrl shRNA, W+S− in Figure 7). To test whether this Wnt3 induced translation of Foxp2 mRNA occurs through the Fzd7 receptor, the control (Ctrl) or Fzd7 shRNA plasmid was transfected into the N2a cells together with the Luciferase-Foxp2_3′UTR reporter construct (Figure 7). Twenty-four hours later, either mock (nothing added), recombinant Wnt3, or recombinant Wnt3 + recombinant SFRP1 (WNT inhibitor) was added to the N2a cell culture medium. After 48 h of the treatment, Luciferase protein activities (RLUs) were measured from total protein extracts of each experimental condition and normalized by the Luciferase mRNA levels (Figure 7) to examine the translational effect (protein/mRNA ratio) [107]. As shown in the primary neuronal cell cultures [48], the neuronal N2a cells also exhibited that treatment with Wnt3 induces translation via the Foxp2–3′UTR (Ctrl shRNA, W+S− in Figure 7) and that this translationally enhanced effect is abolished by the SFRP1 inhibitor (Ctrl shRNA, W+S+ in Figure 7), representing the Wnt3−specific effect to the Foxp2 translation.
Importantly, the Fzd7 silenced cells had reduced Foxp2 translation (Fzd7 shRNA, W–S– in Figure 7) compared to the Ctrl shRNA-transfected cells (Ctrl shRNA, W–S– in Figure 7). In addition, the effect of Wnt3 or inhibitor SFRP1 on Foxp2-3′UTR translation did not occur when Fzd7 was silenced (Fzd7 shRNA in Figure 7). This reduced effect in Wnt3-induced Foxp2 translation is consistent with our observations above which show the decrease in Foxp2 protein expression by Fzd7 shRNA in primary neuronal cells in vitro and developing neocortical neurons in vivo (Figure 4 and Figure 5). Collectively, these data indicate that the Fzd7 receptor functions as a downstream factor for the ligand Wnt3 signaling in timed mRNA translation events of developing neocortices.
4. Discussion
Here we summarize the current state of knowledge on the role of extracellular factors for the intrinsic mRNA translation. In addition, we report that Fzd7 receptor is downstream of the extracellular Wnt3 regulation to Foxp2 mRNA translation. The Fzd7 receptors are expressed broadly throughout the CP layers and colocalized with the Wnt3 ligands (Figure 2) which are secreted from the thalamus at E15 [48]. These Fzd7 proteins also overlapped with distinct Foxp1 and Foxp2 expressions in the upper and lower layers, respectively (Figure 2). In neuroblastoma N2a cells, the Wnt3 intensity at the cell membrane decreases with the Fzd7 downregulation (Figure 3). Our studies above are in agreement with previously reported Wnt3-Fzd7 binding in intestinal stem cells [103] and the functional interaction between Wnt3 and Fzd7 in hepatocellular carcinoma cells [104]. Collectively, these findings indicate that neocortical Fzd7 receptor can transfer the extrinsic WNT signaling arriving from the thalamus to the neocortex in order to regulate the mRNA translation in developing neocortical neurons, which thus may promote neurogenesis of neocortical cells with regulation of developmental transcription factors Foxp1/2 expressions. The Fzd7 is not previously known to regulate translation and neocortical development.
WNT gradient is one of the most important effectors in deciding the developmental stages [108,109]. Our data represent that Wnt3 secreted from the thalamus works with the neocortical Fzd7 receptor, which then regionally regulates the protein levels of neocortical Foxp transcription factors. Therefore, this Wnt3-Fzd7 axis may contribute to the final steps of neuronal maturation, which include circuit formation and synaptogenesis. Forkhead box (Fox) proteins, Foxp1 and Foxp2, are distinct transcription factors which possess different functions in the specific layers of the neocortex [110,111,112,113]. They are strongly linked to human neurodevelopmental disorders (NDDs), including autism spectrum disorder (ASD), intellectual disability (ID), and speech and language disorder [114,115,116]. While FOXP2 mutations predominantly impair speech and language, FOXP1 mutations cause a severe global NDD [112,113]. Therefore, tight control of their expression levels in developing neurons is critical for neurodevelopment. In primary neocortical neuronal cell cultures, it was found that Fzd7 downregulation reduces Foxp2 and induces Foxp1 expression, respectively (Figure 4). These changes in Foxp2 and Foxp1 expressions were also found in the developing neocortex when Fzd7 was downregulated in vivo (Figure 5). In the developing neocortex, this Wnt3-Fzd7-Foxp1/2 signaling can be involved in curing the abnormalities which are associated with NDDs.
The thalamus-specific deletion of Wnt3 reduces the neocortical expression of Foxp2 in lower layers while the Foxp1 protein expression in the upper layers seems to be unaffected in these mice [48]. In addition, the translational effect for Foxp1 protein is absent in Wnt3 treated cells [48]. However, our data showed that the Fzd7 receptor affects both Foxp2 and Foxp1 expression in primary cells and neocortices (Figure 4, Figure 5 and Figure 6), suggesting that the Fzd7 receptor differentially regulates Foxp1 and Foxp2 expression, and that a different ligand possibly acts on the upper layers of Foxp1 than in lower layers of Foxp2. In line with the previous report [48], here we found that Foxp1 expression remains unchanged in primary neuronal cell cultures exposed to Wnt3 ligand (Figure S4). Nonetheless, the Fzd7 shRNA effect resulting in Foxp1 increase still occurred in both Wnt3 treated and Wnt3 non-treated cells (Figure S4). These findings suggest that Foxp1 expression is regulated by the signaling of the Fzd7 receptor, but not by the Wnt3 ligand. A recent report showed that the Fzd7 receptor can physically and functionally interact with the Wnt7b ligand in the hippocampus to modulate dendritic growth and complexity [117]. We observed strong signals of Fzd7 in the dendrites of neocortex when Fzd7 was overexpressed (Figure S3A). It is possible that the Fzd7 receptor in the neocortex may interact with several Wnt ligands to integrate the diverse signals for the complexity in neocortical development.
This study as well as previous findings emphasize the importance of timed extracellular signals in neuronal development, including the neocortex. The exact control of protein expression levels at distinct developmental time points serves the needs of developing cells. The interplay of extrinsic signals and intrinsic protein synthesis rates is critical for normal neurodevelopment. It is easy to envision that slight alterations in these signaling cascades may result in devastating abnormalities within the central nervous system, which would manifest as NDDs.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/10/2/253/s1, Figures S1–S4: Supplementary Figures. This paper has Supplementary Figures in the Supplementary Information that can be found online.
Author Contributions
Conceptualization, M.-R.R., Y.P.; Investigation, Y.P., M.-R.R.; Formal analysis Y.P., M.L., D.L.; Writing, Y.P., M.-R.R.; Funding Acquisition, M.-R.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Institutes of Health (NIH) grants (R01NS075367).
Data Availability Statement
Not applicable.
Acknowledgments
We thank Tatiana Popovitchenko for an initial Fzd7 work and Faizan Rafi for the technical support.
Conflicts of Interest
The authors declare no competing interest.
Materials and Correspondence
Material requests and further information for resources should be directed to the corresponding co-author Mladen-Roko Rasin (e-mail: [email protected]).
References
- Rakic, P. Evolution of the neocortex: A perspective from developmental biology. Nat. Rev. Neurosci. 2009, 10, 724–735. [Google Scholar] [CrossRef] [PubMed]
- DeBoer, E.M.; Kraushar, M.L.; Hart, R.P.; Rasin, M.R. Post-transcriptional regulatory elements and spatiotemporal specification of neocortical stem cells and projection neurons. Neuroscience 2013, 248, 499–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovitchenko, T.; Rasin, M.R. Transcriptional and Post-Transcriptional Mechanisms of the Development of Neocortical Lamination. Front. Neuroanat. 2017, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, D.P.; Srinivasan, K.; Chen, B.; Alcamo, E.; McConnell, S.K. The determination of projection neuron identity in the developing cerebral cortex. Curr. Opin. Neurobiol. 2008, 18, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef]
- Lennox, A.L.; Mao, H.; Silver, D.L. RNA on the brain: Emerging layers of post-transcriptional regulation in cerebral cortex development. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7. [Google Scholar] [CrossRef]
- Hoye, M.L.; Silver, D.L. Decoding mixed messages in the developing cortex: Translational regulation of neural progenitor fate. Curr. Opin. Neurobiol. 2020, 66, 93–102. [Google Scholar] [CrossRef]
- Zahr, S.K.; Kaplan, D.R.; Miller, F.D. Translating neural stem cells to neurons in the mammalian brain. Cell Death Differ. 2019, 26, 2495–2512. [Google Scholar] [CrossRef]
- Pinson, A.; Namba, T.; Huttner, W.B. Malformations of Human Neocortex in Development—Their Progenitor Cell Basis and Experimental Model Systems. Front Cell Neurosci. 2019, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Popovitchenko, T.; Park, Y.; Page, N.F.; Luo, X.; Krsnik, Z.; Liu, Y.; Salamon, I.; Stephenson, J.D.; Kraushar, M.L.; Volk, N.L.; et al. Translational derepression of Elavl4 isoforms at their alternative 5′ UTRs determines neuronal development. Nat. Commun. 2020, 11, 1674. [Google Scholar] [CrossRef] [PubMed]
- Petreanu, L.; Mao, T.; Sternson, S.M.; Svoboda, K. The subcellular organization of neocortical excitatory connections. Nature 2009, 457, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.M.; Lamy, C. Functional maps of neocortical local circuitry. Front. Neurosci. 2007, 1, 19–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric-Sekhar, G.; Hevner, R.F. Malformations of Cerebral Cortex Development: Molecules and Mechanisms. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Hevner, R.F. Growth and folding of the mammalian cerebral cortex: From molecules to malformations. Nat. Rev. Neurosci. 2014, 15, 217–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greig, L.C.; Woodworth, M.B.; Galazo, M.J.; Padmanabhan, H.; Macklis, J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013, 14, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Pilaz, L.J.; Silver, D.L. Post-transcriptional regulation in corticogenesis: How RNA-binding proteins help build the brain. Wiley Interdiscip. Rev. Rna. 2015, 6, 501–515. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.; Kwan, K.Y.; Li, M.; Lefebvre, V.; Sestan, N. Cis-regulatory control of corticospinal system development and evolution. Nature 2012, 486, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Tuoc, T.C.; Narayanan, R.; Stoykova, A. BAF chromatin remodeling complex: Cortical size regulation and beyond. Cell Cycle 2013, 12, 2953–2959. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Sokpor, G.; Pham, L.; Rosenbusch, J.; Stoykova, A.; Staiger, J.F.; Tuoc, T. Epigenetic regulation by BAF (mSWI/SNF) chromatin remodeling complexes is indispensable for embryonic development. Cell Cycle 2016, 15, 1317–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgard, T.G.; Marques, A.C.; Oliver, P.L.; Abaan, H.O.; Sirey, T.M.; Hoerder-Suabedissen, A.; Garcia-Moreno, F.; Molnar, Z.; Margulies, E.H.; Ponting, C.P. A transcriptomic atlas of mouse neocortical layers. Neuron 2011, 71, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoerder-Suabedissen, A.; Oeschger, F.M.; Krishnan, M.L.; Belgard, T.G.; Wang, W.Z.; Lee, S.; Webber, C.; Petretto, E.; Edwards, A.D.; Molnar, Z. Expression profiling of mouse subplate reveals a dynamic gene network and disease association with autism and schizophrenia. Proc. Natl. Acad. Sci.USA 2013, 110, 3555–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, C.W.; Gharani, N.; Millonig, J.H.; Brzustowicz, L.M. Three autism candidate genes: A synthesis of human genetic analysis with other disciplines. Int. J. Dev. Neurosci. 2005, 23, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, J.J.; Shi, Y.; Gujral, M.; Zheng, H.; Malhotra, D.; Jin, X.; Jian, M.; Liu, G.; Greer, D.; Bhandari, A.; et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 2012, 151, 1431–1442. [Google Scholar] [CrossRef] [Green Version]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.A.; Arking, D.E.; Gene Discovery Project of Johns Hopkins & the Autism Consortium; Daly, M.J.; Chakravarti, A. A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009, 461, 802–808. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Jabaudon, D. Fate and freedom in developing neocortical circuits. Nat. Commun. 2017, 8, 16042. [Google Scholar] [CrossRef] [PubMed]
- Toma, K.; Hanashima, C. Switching modes in corticogenesis: Mechanisms of neuronal subtype transitions and integration in the cerebral cortex. Front. Neurosci. 2015, 9, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molyneaux, B.J.; Goff, L.A.; Brettler, A.C.; Chen, H.H.; Hrvatin, S.; Rinn, J.L.; Arlotta, P. DeCoN: Genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron 2015, 85, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, K.; Aramaki, M.; Nakajima, K. Mutually repressive interaction between Brn1/2 and Rorb contributes to the establishment of neocortical layer 2/3 and layer 4. Proc. Natl. Acad. Sci. USA 2016, 113, 3371–3376. [Google Scholar] [CrossRef] [Green Version]
- Hata, K.; Mizukami, H.; Sadakane, O.; Watakabe, A.; Ohtsuka, M.; Takaji, M.; Kinoshita, M.; Isa, T.; Ozawa, K.; Yamamori, T. DNA methylation and methyl-binding proteins control differential gene expression in distinct cortical areas of macaque monkey. J. Neurosci. 2013, 33, 19704–19714. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, Y.; Gotoh, Y. Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci. 2010, 11, 377–388. [Google Scholar] [CrossRef]
- Numata, S.; Ye, T.; Hyde, T.M.; Guitart-Navarro, X.; Tao, R.; Wininger, M.; Colantuoni, C.; Weinberger, D.R.; Kleinman, J.E.; Lipska, B.K. DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 2012, 90, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Petanjek, Z.; Kostovic, I. Epigenetic regulation of fetal brain development and neurocognitive outcome. Proc. Natl Acad. Sci. USA 2012, 109, 11062–11063. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.K.; Yin, J.; Ayoub, A.E.; Emera, D.; Leng, J.; Cotney, J.; Sarro, R.; Rakic, P.; Noonan, J.P. Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 2015, 347, 1155–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Gama, A.M.; Walsh, C.A. Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 2018, 21, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, A.R.; Gsponer, J.; Foster, L.J. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol. 2013, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, F.K.; Lehmann, R. Translational Control during Developmental Transitions. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraushar, M.L.; Popovitchenko, T.; Volk, N.L.; Rasin, M.R. The frontier of RNA metamorphosis and ribosome signature in neocortical development. Int. J. Dev. Neurosci. 2016, 55, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef]
- Kraushar, M.L.; Viljetic, B.; Wijeratne, H.R.; Thompson, K.; Jiao, X.; Pike, J.W.; Medvedeva, V.; Groszer, M.; Kiledjian, M.; Hart, R.P.; et al. Thalamic WNT3 Secretion Spatiotemporally Regulates the Neocortical Ribosome Signature and mRNA Translation to Specify Neocortical Cell Subtypes. J. Neurosci. 2015, 35, 10911–10926. [Google Scholar] [CrossRef] [Green Version]
- Kraushar, M.L.; Thompson, K.; Wijeratne, H.R.; Viljetic, B.; Sakers, K.; Marson, J.W.; Kontoyiannis, D.L.; Buyske, S.; Hart, R.P.; Rasin, M.R. Temporally defined neocortical translation and polysome assembly are determined by the RNA-binding protein Hu antigen R. Proc. Natl. Acad. Sci. USA 2014, 111, E3815–E3824. [Google Scholar] [CrossRef] [Green Version]
- Popovitchenko, T.; Thompson, K.; Viljetic, B.; Jiao, X.; Kontonyiannis, D.L.; Kiledjian, M.; Hart, R.P.; Rasin, M.R. The RNA binding protein HuR determines the differential translation of autism-associated FoxP subfamily members in the developing neocortex. Sci. Rep. 2016, 6, 28998. [Google Scholar] [CrossRef] [Green Version]
- Pilaz, L.-J.; Lennox, A.L.; Rouanet, J.P.; Silver, D.L. Dynamic mRNA Transport and Local Translation in Radial Glial Progenitors of the Developing Brain. Curr. Biol. 2016, 26, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffary, R.; Xie, Z. FMRP Regulates the Transition from Radial Glial Cells to Intermediate Progenitor Cells during Neocortical Development. J. Neurosci. 2011, 31, 1427–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, K.-J.; Ringeling, F.R.; Vissers, C.; Jacob, F.; Pokrass, M.; Jimenez-Cyrus, D.; Su, Y.; Kim, N.-S.; Zhu, Y.; Zheng, L.; et al. Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell 2017, 171, 877–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Smibert, C.A.; Kaplan, D.R.; Miller, F.D. An eIF4E1/4E-T Complex Determines the Genesis of Neurons from Precursors by Translationally Repressing a Proneurogenic Transcription Program. Neuron 2014, 84, 723–739. [Google Scholar] [CrossRef] [Green Version]
- Amadei, G.; Zander, M.A.; Yang, G.; Dumelie, J.G.; Vessey, J.P.; Lipshitz, H.D.; Smibert, C.A.; Kaplan, D.R.; Miller, F.D. A Smaug2-Based Translational Repression Complex Determines the Balance between Precursor Maintenance versus Differentiation during Mammalian Neurogenesis. J. Neurosci. 2015, 35, 15666–15681. [Google Scholar] [CrossRef] [Green Version]
- Taverna, E.; Götz, M.; Huttner, W.B. The Cell Biology of Neurogenesis: Toward an Understanding of the Development and Evolution of the Neocortex. Annu. Rev. Cell Dev. Biol. 2014, 30, 465–502. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Yang, G.; Borrett, M.J.; Clarke, G.; Cancino, G.I.; Zahr, S.K.; Zandstra, P.W.; Kaplan, D.R.; Miller, F.D. Proneurogenic Ligands Defined by Modeling Developing Cortex Growth Factor Communication Networks. Neuron 2016, 91, 988–1004. [Google Scholar] [CrossRef] [Green Version]
- Oishi, K.; Nakajima, K. Subtype Specification of Cerebral Cortical Neurons in Their Immature Stages. Neurochem. Res. 2018, 43, 238–244. [Google Scholar] [CrossRef]
- Scalabrino, G. Epidermal growth factor in the CNS: A beguiling journey from integrated cell biology to multiple sclero-sis. An extensive translational overview. Cell Mol. Neurobiol. 2020, 10. [Google Scholar] [CrossRef]
- Petroulakis, E.; Wang, E. Nerve Growth Factor Specifically Stimulates Translation of Eukaryotic Elongation Factor 1A-1 (eEF1A-1) mRNA by Recruitment to Polyribosomes in PC12 Cells. J. Biol. Chem. 2002, 277, 18718–18727. [Google Scholar] [CrossRef] [Green Version]
- Schratt, G.M.; Nigh, E.A.; Chen, W.G.; Hu, L.; Greenberg, M.E. BDNF Regulates the Translation of a Select Group of mRNAs by a Mammalian Target of Rapamycin-Phosphatidylinositol 3-Kinase-Dependent Pathway during Neuronal Development. J. Neurosci. 2004, 24, 7366–7377. [Google Scholar] [CrossRef] [PubMed]
- López-Bendito, G.; Molnár, Z. Thalamocortical development: How are we going to get there? Nat. Rev. Neurosci. 2003, 4, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y. Development of the thalamus: From early patterning to regulation of cortical functions. Wiley Interdiscip. Rev. Dev. Biol. 2019, 8, e345. [Google Scholar] [CrossRef] [PubMed]
- Bluske, K.K.; Kawakami, Y.; Koyano-Nakagawa, N.; Nakagawa, Y. Differential activity of Wnt/β-catenin signaling in the embryonic mouse thalamus. Dev. Dyn. 2009, 238, 3297–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluske, K.K.; Vue, T.Y.; Kawakami, Y.; Taketo, M.M.; Yoshikawa, K.; Johnson, J.E.; Nakagawa, Y. β-Catenin signaling specifies progenitor cell identity in parallel with Shh signaling in the developing mammalian thalamus. Development 2012, 139, 2692–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicknese, A.R.; Sheppard, A.M.; O’Leary, D.D.; Pearlman, A.L. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J. Neurosci. 1994, 14, 3500–3510. [Google Scholar] [CrossRef] [Green Version]
- Dehay, C.; Savatier, P.; Cortay, V.; Kennedy, H. Cell-Cycle Kinetics of Neocortical Precursors Are Influenced by Embryonic Thalamic Axons. J. Neurosci. 2001, 21, 201–214. [Google Scholar] [CrossRef]
- Eisenmann, D.M. Wnt signaling. WormBook 2005, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gruber, J.; Yee, Z.; Tolwinski, N.S. Developmental Drift and the Role of Wnt Signaling in Aging. Cancers 2016, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Jin, H.J.; Lusk, J.B.; Tolwinski, N.S. Modeling the Role of Wnt Signaling in Human and Drosophila Stem Cells. Genes 2018, 9, 101. [Google Scholar] [CrossRef] [Green Version]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, A.S.; Kuehl, M. Of Wnts and Ribosomes. Prog. Mol. Biol. Transl. Sci. 2018, 153, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Habas, R.; Dawid, I.B. Dishevelled and Wnt signaling: Is the nucleus the final frontier? J. Biol. 2005, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Fagotto, F.; Zhang, T.; Hsu, W.; Vasicek, T.J.; Perry, W.L.; Lee, J.J.; Tilghman, S.M.; Gumbiner, B.M.; Costantini, F. The Mouse Fused Locus Encodes Axin, an Inhibitor of the Wnt Signaling Pathway That Regulates Embryonic Axis Formation. Cell 1997, 90, 181–192. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/ -catenin signaling: Arrows point the way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Tolwinski, N.S.; Wieschaus, E.F. A nuclear escort for β-catenin. Nat. Cell Biol. 2004, 6, 579–580. [Google Scholar] [CrossRef]
- De Calisto, J.; Araya, C.; Marchant, L.; Riaz, C.F.; Mayor, R. Essential role of non-canonical Wnt signalling in neural crest migration. Development 2005, 132, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Kestler, H.A.; Kuehl, M. From individual Wnt pathways towards a Wnt signalling network. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 1333–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Raya, A.; Kawakami, Y.; Callol-Massot, C.; Capdevila, J.; Rodriguez-Esteban, C.; Belmonte, J.C.I. Noncanonical Wnt signaling regulates midline convergence of organ primordia during zebrafish development. Genes Dev. 2005, 19, 164–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A Second Canon. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef] [Green Version]
- De Ferrari, G.V.; E Avila, M.; A Medina, M.; Perez-Palma, E.; I Bustos, B.; A Alarcon, M. Wnt/β-Catenin Signaling in Alzheimer’s Disease. Cns Neurol. Disord. Drug Targets 2014, 13, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Belinson, H.; Nakatani, J.; A Babineau, B.; Birnbaum, R.Y.; Ellegood, J.; Bershteyn, M.; McEvilly, R.J.; Long, J.M.; Willert, K.; Klein, O.D.; et al. Prenatal β-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol. Psychiatry 2016, 21, 1417–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Deng, J.; Pan, Q.; Zhan, Y.; Fan, J.-B.; Zhang, K.; Zhang, Z. Targeted methylation sequencing reveals dysregulated Wnt signaling in Parkinson disease. J. Genet. Genom. 2016, 43, 587–592. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.C.; et al. Reactive astrocytes and Wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef] [Green Version]
- Cervo, P.R.D.V.; A Romanov, R.; Spigolon, G.; Masini, D.; Martín-Montañez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.-H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- McGrath, J.J.; Féron, F.P.; Burne, T.H.J.; Mackay-Sim, A.; Eyles, D.W. The neurodevelopmental hypothesis of schizophrenia: A review of recent developments. Ann. Med. 2003, 35, 86–93. [Google Scholar] [CrossRef]
- Miyaoka, T.; Seno, H.; Ishino, H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999, 38, 1–6. [Google Scholar] [CrossRef]
- Katsu, T.; Ujike, H.; Nakano, T.; Tanaka, Y.; Nomura, A.; Nakata, K.; Takaki, M.; Sakai, A.; Uchida, N.; Imamura, T.; et al. The human frizzled-3 (FZD3) gene on chromosome 8p21, a receptor gene for Wnt ligands, is associated with the susceptibility to schizophrenia. Neurosci. Lett. 2003, 353, 53–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.A.; Montecinos-Oliva, C.; Inestrosa, N.C. Wnt Signaling in the Central Nervous System: New Insights in Health and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 153, 81–130. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Reynoso, M.A.; Ochoa-Hernández, A.B.; Aguilar-Lemarroy, A.; Jave-Suárez, L.F.; Troyo-Sanroman, R.; Barros-Núñez, P. Gene Expression Profiling Identifies WNT7A As a Possible Candidate Gene for Decreased Cancer Risk in Fragile X Syndrome Patients. Arch. Med Res. 2010, 41, 110–118.e2. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular Mechanisms of Fragile X Syndrome: A Twenty-Year Perspective. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 219–245. [Google Scholar] [CrossRef] [Green Version]
- Todd, P.K.; Malter, J.S. Fragile X mental retardation protein in plasticity and disease. J. Neurosci. Res. 2002, 70, 623–630. [Google Scholar] [CrossRef]
- Zhang, A.; Shen, C.-H.; Ma, S.Y.; Ke, Y.; El Idrissi, A. Altered expression of Autism-associated genes in the brain of Fragile X mouse model. Biochem. Biophys. Res. Commun. 2009, 379, 920–923. [Google Scholar] [CrossRef]
- Ehyai, S.; Miyake, T.; Williams, D.; Vinayak, J.; A Bayfield, M.; McDermott, J.C. FMRP recruitment of β-catenin to the translation pre-initiation complex represses translation. Embo. Rep. 2018, 19, e45536. [Google Scholar] [CrossRef]
- Nishino, J.; Kim, S.; Zhu, Y.; Zhu, H.; Morrison, S.J. A network of heterochronic genes including Imp1 regulates temporal changes in stem cell properties. Elife 2013, 2, e00924. [Google Scholar] [CrossRef]
- Tsui, D.; Vessey, J.P.; Tomita, H.; Kaplan, D.R.; Miller, F.D. FoxP2 Regulates Neurogenesis during Embryonic Cortical Development. J. Neurosci. 2013, 33, 244–258. [Google Scholar] [CrossRef]
- Hornbeck, P.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- DeBoer, E.M.; Azevedo, R.; Vega, T.A.; Brodkin, J.; Akamatsu, W.; Okano, H.; Wagner, G.C.; Rasin, M.-R. Prenatal Deletion of the RNA-Binding Protein HuD Disrupts Postnatal Cortical Circuit Maturation and Behavior. J. Neurosci. 2014, 34, 3674–3686. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-R.; Parikh, H.; Park, Y. Stress resistance and lifespan enhanced by downregulation of antimicrobial peptide genes in the Imd pathway. Aging 2018, 10, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, D.J.; Phesse, T.J.; Barker, N.; Schwab, R.H.; Amin, N.; Malaterre, J.; Stange, D.E.; Nowell, C.J.; Currie, S.A.; Saw, J.T.; et al. Frizzled7 Functions as a Wnt Receptor in Intestinal Epithelial Lgr5+ Stem Cells. Stem Cell Rep. 2015, 4, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Lee, H.C.; Tsedensodnom, O.; Hartley, R.; Lim, Y.-S.; Yu, E.; Merle, P.; Wands, J.R. Functional interaction between Wnt3 and Frizzled-7 leads to activation of the Wnt/β-catenin signaling pathway in hepatocellular carcinoma cells. J. Hepatol. 2008, 48, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rašin, M.-R.; Gazula, V.-R.; Breunig, J.J.; Kwan, K.Y.; Johnson, M.B.; Liu-Chen, S.; Li, H.-S.; Jan, L.Y.; Jan, Y.-N.; Rakic, P.; et al. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat. Neurosci. 2007, 10, 819–827. [Google Scholar] [CrossRef]
- Mazumder, B.; Seshadri, V.; Fox, P.L. Translational control by the 3′-UTR: The ends specify the means. Trends Biochem. Sci. 2003, 28, 91–98. [Google Scholar] [CrossRef]
- Xue, S.; Tian, S.; Fujii, K.; Kladwang, W.; Das, R.; Barna, M. RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nat. Cell Biol. 2015, 517, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.T.; Wallingford, J.B. Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signalling in neuronal differentiation and development. Cell Tissue Res. 2015, 359, 215–223. [Google Scholar] [CrossRef]
- Pearson, C.A.; Moore, D.M.; Tucker, H.O.; Dekker, J.D.; Hu, H.; Miquelajáuregui, A.; Novitch, B.G. Foxp1 Regulates Neural Stem Cell Self-Renewal and Bias Toward Deep Layer Cortical Fates. Cell Rep. 2020, 30, 1964–1981. [Google Scholar] [CrossRef] [Green Version]
- Medvedeva, V.P.; A Rieger, M.; Vieth, B.; Mombereau, C.; Ziegenhain, C.; Ghosh, T.; Cressant, A.; Enard, W.; Granon, S.; Dougherty, J.D.; et al. Altered social behavior in mice carrying a cortical Foxp2 deletion. Hum. Mol. Genet. 2018, 28, 701–717. [Google Scholar] [CrossRef]
- Bacon, C.; Rappold, G.A. The distinct and overlapping phenotypic spectra of FOXP1 and FOXP2 in cognitive disorders. Qual. Life Res. 2012, 131, 1687–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Co, M.; Anderson, A.G.; Konopka, G. FOXP transcription factors in vertebrate brain development, function, and disorders. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e375. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.S.L.; Fisher, S.E.; Hurst, J.A.; Vargha-Khadem, F.; Monaco, A.P. A forkhead-domain gene is mutated in a severe speech and language disorder. Nat. Cell Biol. 2001, 413, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Meerschaut, I.; Rochefort, D.; Revençu, N.; Pètre, J.; Corsello, C.; A Rouleau, G.; Hamdan, F.F.; Michaud, J.L.; Morton, J.; Radley, J.; et al. FOXP1-related intellectual disability syndrome: A recognisable entity. J. Med. Genet. 2017, 54, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siper, P.M.; De Rubeis, S.; Trelles, M.D.P.; Durkin, A.; Di Marino, D.; Muratet, F.; Frank, Y.; Lozano, R.; Eichler, E.E.; Kelly, M.; et al. Prospective investigation of FOXP1 syndrome. Mol. Autism. 2017, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, M.E.; Bernis, M.E.; McLeod, F.; Podpolny, M.; Coullery, R.P.; Casadei, I.M.; Salinas, P.C.; Rosso, S.B. Wnt7b signalling through Frizzled-7 receptor promotes dendrite development by coactivating CaMKII and JNK. J. Cell Sci. 2018, 131, jcs216101. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
mRNA translations regulated by WNTs signaling. (1) Wnt3, secreted from thalamocortical axons, increases Rpl7 ribosomal protein within polysomes and promotes Foxp2 mRNA translation in the neocortex [48]. (2) Wnt3a stimulation induces the translational activation of mRNAs by sequestering β-catenin away (red arrow) from the β-catenin and FMRP complex that sustains the translational repression state (blue arrow) [97]. (3) Wnt1 signaling enhances RBP Imp1 expression during cortical development, which both translationally silences proneurogenic mRNAs and increases expression of pro-self-renewal mRNAs, to maintain NSCs in an undifferentiated state [98].
Figure 1.
mRNA translations regulated by WNTs signaling. (1) Wnt3, secreted from thalamocortical axons, increases Rpl7 ribosomal protein within polysomes and promotes Foxp2 mRNA translation in the neocortex [48]. (2) Wnt3a stimulation induces the translational activation of mRNAs by sequestering β-catenin away (red arrow) from the β-catenin and FMRP complex that sustains the translational repression state (blue arrow) [97]. (3) Wnt1 signaling enhances RBP Imp1 expression during cortical development, which both translationally silences proneurogenic mRNAs and increases expression of pro-self-renewal mRNAs, to maintain NSCs in an undifferentiated state [98].

Figure 2.
Colocalization of Wnt3 and Fzd7 with Foxp1 and Foxp2 expression at E16 and P0. (A,B). Immunohistochemistry (IHC) for Wnt3 (green), Fzd7 (red), Foxp1 (green), and Foxp2 (green) in WT E16 (A) and P0 (B) neocortices (n = 5 animals per age). Arrow denotes colocalized area between Wnt3 and Fzd7 in confocal microscope image using 20× objective lens, which was confirmed with 60× resolution (insert: higher magnification image of colocalization). DAPI shown in blue; CP: cortical plate; IZ: intermediate zone; VZ: ventricular zone; UL: upper layers; LL: lower layers; scale bar: 100 μm.
Figure 2.
Colocalization of Wnt3 and Fzd7 with Foxp1 and Foxp2 expression at E16 and P0. (A,B). Immunohistochemistry (IHC) for Wnt3 (green), Fzd7 (red), Foxp1 (green), and Foxp2 (green) in WT E16 (A) and P0 (B) neocortices (n = 5 animals per age). Arrow denotes colocalized area between Wnt3 and Fzd7 in confocal microscope image using 20× objective lens, which was confirmed with 60× resolution (insert: higher magnification image of colocalization). DAPI shown in blue; CP: cortical plate; IZ: intermediate zone; VZ: ventricular zone; UL: upper layers; LL: lower layers; scale bar: 100 μm.

Figure 3.
Interaction between Wnt3 and Fzd7 in neuroblastoma N2a cells. (A). IHC for GFP (green), Fzd7 (red), and Wnt3 (blue) in N2a cells after 3 days of each shRNA transfection containing GFP marker (Ctrl or Fzd7 shRNA) and following 4 h of Wnt3 (100 ng/mL) treatment (n = 3 separate transfections). Arrow: shRNA-transfected GFP cells (insert: higher magnification image); arrowhead: non-transfected cells; scale bar: 100 μm. (B). Fzd7 mRNA expression levels in total N2a cells determined by qRT-PCR (n = 10 repeats) when IHC was performed in A. Data of Fzd7 shRNA were normalized to Gapdh and then to Ctrl shRNA (100%). (C–E). Quantification of Fzd7 (C), Wnt3 (D), Gapdh (E)-positive cells among total GFP cells (251–356 counted), normalized to the Ctrl shRNA (n = 6–12 repeats). Data represent the mean and SEM. Statistics: Student’s t-test; *: p < 0.05; ***: p < 0.005; n.s.: not significant.
Figure 3.
Interaction between Wnt3 and Fzd7 in neuroblastoma N2a cells. (A). IHC for GFP (green), Fzd7 (red), and Wnt3 (blue) in N2a cells after 3 days of each shRNA transfection containing GFP marker (Ctrl or Fzd7 shRNA) and following 4 h of Wnt3 (100 ng/mL) treatment (n = 3 separate transfections). Arrow: shRNA-transfected GFP cells (insert: higher magnification image); arrowhead: non-transfected cells; scale bar: 100 μm. (B). Fzd7 mRNA expression levels in total N2a cells determined by qRT-PCR (n = 10 repeats) when IHC was performed in A. Data of Fzd7 shRNA were normalized to Gapdh and then to Ctrl shRNA (100%). (C–E). Quantification of Fzd7 (C), Wnt3 (D), Gapdh (E)-positive cells among total GFP cells (251–356 counted), normalized to the Ctrl shRNA (n = 6–12 repeats). Data represent the mean and SEM. Statistics: Student’s t-test; *: p < 0.05; ***: p < 0.005; n.s.: not significant.

Figure 4.
Foxp2 downregulation and Foxp1 upregulation with Fzd7 shRNA in primary neuronal cells. (A). IHC for GFP (Ctrl shRNA cells: green) and RFP (Fzd7 shRNA cells: red) with Foxp2 or Foxp1 immunostaining (blue) after four days in vitro (DIV) culture of primary neuronal cells in utero electroporated (IUE) at E13 (n = 3 animals of each GFP and RFP). GFP (arrow) and RFP (arrowhead) cells showing strong (++) intensity of Foxp2 (lower panel: Foxp1) protein are respectively represented. Insert: higher magnification images of same area; scale bar: 100 μm. (B,C). Quantification of Foxp2 (B) or Foxp1 (C) signal with −, +, and ++ intensities which were compared/normalized to the intensity (+) observed in non-transfected cell as an intrinsic control of immunostaining quality (n = 28–48 pictures counted with total 403–700 cells). Data (%) represent the mean and SEM of each intensity (total sum = 100%). Statistics: Student’s t-test; *: p < 0.05; ***: p < 0.005.
Figure 4.
Foxp2 downregulation and Foxp1 upregulation with Fzd7 shRNA in primary neuronal cells. (A). IHC for GFP (Ctrl shRNA cells: green) and RFP (Fzd7 shRNA cells: red) with Foxp2 or Foxp1 immunostaining (blue) after four days in vitro (DIV) culture of primary neuronal cells in utero electroporated (IUE) at E13 (n = 3 animals of each GFP and RFP). GFP (arrow) and RFP (arrowhead) cells showing strong (++) intensity of Foxp2 (lower panel: Foxp1) protein are respectively represented. Insert: higher magnification images of same area; scale bar: 100 μm. (B,C). Quantification of Foxp2 (B) or Foxp1 (C) signal with −, +, and ++ intensities which were compared/normalized to the intensity (+) observed in non-transfected cell as an intrinsic control of immunostaining quality (n = 28–48 pictures counted with total 403–700 cells). Data (%) represent the mean and SEM of each intensity (total sum = 100%). Statistics: Student’s t-test; *: p < 0.05; ***: p < 0.005.

Figure 5.
Foxp2 downregulation in UL and Foxp1 upregulation in LL with Fzd7 shRNA at P0 (E13–P0). (A). IHC for GFP cells (green) in P0 neocortices electroporated in utero with Ctrl or Fzd7 shRNA at E13 (n = 3 animals). Bin 1–5: upper layers (UL); Bin 6–10: lower layers (LL); scale bar: 100 μm. (B). Percentage (%) of GFP+ cells in each bin from total 10 bins (n = 13–15 pictures counted with total 1965–2500 GFP cells). GFP+ cells in the upper layers (Bin 1–5) of Fzd7 shRNA neocortices increased up to 26.9% compared to Ctrl shRNA, while decrease of 23.8% was shown in the lower layers (Bin 6–10). *: p < 0.05. (C). IHC for GFP (green), Foxp1 (blue), and Foxp2 (red) in P0 neocortices. Arrow and arrowhead: the areas showing bigger change in Foxp2 and Foxp1 expression, respectively, which are shown with higher magnification images in the lower panel. (D,E). Quantification of Foxp2 (D) and Foxp1 (E)-positive cells (n = 8–14 pictures counted with total 1752–2358 GFP cells), normalized to the Ctrl shRNA (100%). Data represent the mean and SEM. FL; full layers (Bin 1–10); statistics: Student’s t-test; *: p < 0.05; **: p < 0.01.
Figure 5.
Foxp2 downregulation in UL and Foxp1 upregulation in LL with Fzd7 shRNA at P0 (E13–P0). (A). IHC for GFP cells (green) in P0 neocortices electroporated in utero with Ctrl or Fzd7 shRNA at E13 (n = 3 animals). Bin 1–5: upper layers (UL); Bin 6–10: lower layers (LL); scale bar: 100 μm. (B). Percentage (%) of GFP+ cells in each bin from total 10 bins (n = 13–15 pictures counted with total 1965–2500 GFP cells). GFP+ cells in the upper layers (Bin 1–5) of Fzd7 shRNA neocortices increased up to 26.9% compared to Ctrl shRNA, while decrease of 23.8% was shown in the lower layers (Bin 6–10). *: p < 0.05. (C). IHC for GFP (green), Foxp1 (blue), and Foxp2 (red) in P0 neocortices. Arrow and arrowhead: the areas showing bigger change in Foxp2 and Foxp1 expression, respectively, which are shown with higher magnification images in the lower panel. (D,E). Quantification of Foxp2 (D) and Foxp1 (E)-positive cells (n = 8–14 pictures counted with total 1752–2358 GFP cells), normalized to the Ctrl shRNA (100%). Data represent the mean and SEM. FL; full layers (Bin 1–10); statistics: Student’s t-test; *: p < 0.05; **: p < 0.01.

Figure 6.
Foxp2 upregulation in UL and Foxp1 downregulation in LL with Fzd7 OE at P0 (E16–P0). (A,C). IHC for GFP cells (green) with Foxp2 (red in A) or Foxp1 (blue in C) immunostaining in P0 neocortices electroporated in utero with Ctrl or Fzd7 OE plasmid at E16 (n = 3 animals). Arrow in A: Foxp2 protein expressed in upper layer bins 1–3 with the Fzd7 overexpression; arrowhead area in C: Foxp1 expression reduced in lower layer bins 6–7 with the Fzd7 overexpression, which are shown with higher magnification images in the insert. DAPI shown in blue (A); Bin 1–5: upper layers (UL); Bin 6–10: lower layers (LL); scale bar: 100 μm. (B,D). Quantification of Foxp2- (B) or Foxp1- (D) positive cells (n = 10–20 pictures counted with total 1711–3358 GFP cells), normalized to the Ctrl shRNA (100%). Data represent the mean and SEM. FL; full layers (Bin 1–10); statistics: Student’s t-test; *: p < 0.05; n.s.: not significant.
Figure 6.
Foxp2 upregulation in UL and Foxp1 downregulation in LL with Fzd7 OE at P0 (E16–P0). (A,C). IHC for GFP cells (green) with Foxp2 (red in A) or Foxp1 (blue in C) immunostaining in P0 neocortices electroporated in utero with Ctrl or Fzd7 OE plasmid at E16 (n = 3 animals). Arrow in A: Foxp2 protein expressed in upper layer bins 1–3 with the Fzd7 overexpression; arrowhead area in C: Foxp1 expression reduced in lower layer bins 6–7 with the Fzd7 overexpression, which are shown with higher magnification images in the insert. DAPI shown in blue (A); Bin 1–5: upper layers (UL); Bin 6–10: lower layers (LL); scale bar: 100 μm. (B,D). Quantification of Foxp2- (B) or Foxp1- (D) positive cells (n = 10–20 pictures counted with total 1711–3358 GFP cells), normalized to the Ctrl shRNA (100%). Data represent the mean and SEM. FL; full layers (Bin 1–10); statistics: Student’s t-test; *: p < 0.05; n.s.: not significant.

Figure 7.
Translational reduction through Foxp2–3′UTR with Fzd7 shRNA. Quantification of translational effects (protein/mRNA ratio) was carried out using Luciferase-Foxp2_3′UTR construct in neuronal N2a cells, transfected with the Ctrl or Fzd7 shRNA plasmid and treated with mock (W–S–), Wnt3 (100 ng/mL) (W+S–), or Wnt3 + SFRP1 (100 ng/mL) (W+S+) for 48 h (n = 4 repeats). Translation was measured with Relative Luciferase light units (RLUs), and was then normalized to the Luciferase mRNA level (qRT-PCR). Data represent the mean and SEM. Statistics: Student’s t-test; **: p < 0.01; ***: p < 0.005; n.s.: not significant. Two-way ANOVA analysis with replication represents that the Fzd7 shRNA and Wnt3 treatments are significant and that the effects of them are dependent on each factor.
Figure 7.
Translational reduction through Foxp2–3′UTR with Fzd7 shRNA. Quantification of translational effects (protein/mRNA ratio) was carried out using Luciferase-Foxp2_3′UTR construct in neuronal N2a cells, transfected with the Ctrl or Fzd7 shRNA plasmid and treated with mock (W–S–), Wnt3 (100 ng/mL) (W+S–), or Wnt3 + SFRP1 (100 ng/mL) (W+S+) for 48 h (n = 4 repeats). Translation was measured with Relative Luciferase light units (RLUs), and was then normalized to the Luciferase mRNA level (qRT-PCR). Data represent the mean and SEM. Statistics: Student’s t-test; **: p < 0.01; ***: p < 0.005; n.s.: not significant. Two-way ANOVA analysis with replication represents that the Fzd7 shRNA and Wnt3 treatments are significant and that the effects of them are dependent on each factor.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Park, Y.; Lofton, M.; Li, D.; Rasin, M.-R. Extrinsic Regulators of mRNA Translation in Developing Brain: Story of WNTs. Cells 2021, 10, 253. https://doi.org/10.3390/cells10020253
AMA Style
Park Y, Lofton M, Li D, Rasin M-R. Extrinsic Regulators of mRNA Translation in Developing Brain: Story of WNTs. Cells. 2021; 10(2):253. https://doi.org/10.3390/cells10020253
Chicago/Turabian StylePark, Yongkyu, Midori Lofton, Diana Li, and Mladen-Roko Rasin. 2021. "Extrinsic Regulators of mRNA Translation in Developing Brain: Story of WNTs" Cells 10, no. 2: 253. https://doi.org/10.3390/cells10020253
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.