Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression
by
 ,
,
Jesse D. Armitage
1, 
Hannah V. Newnes
1,
Alison McDonnell
1,2,
Anthony Bosco
1,* and
Jason Waithman
1,* 1
Telethon Kids Institute, The University of Western Australia, Nedlands, WA 6009, Australia
2
National Centre for Asbestos Related Diseases, QEII Medical Centre, The University of Western Australia, Nedlands, WA 6009, Australia
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(1), 56; https://doi.org/10.3390/cells10010056
Submission received: 8 December 2020
/
Revised: 28 December 2020
/
Accepted: 30 December 2020
/
Published: 1 January 2021
(This article belongs to the Special Issue Tumor-Induced Immunosuppression)
Abstract
:Immunotherapy has revolutionised the treatment of cancers by harnessing the power of the immune system to eradicate malignant tissue. However, it is well recognised that some cancers are highly resistant to these therapies, which is in part attributed to the immunosuppressive landscape of the tumour microenvironment (TME). The contexture of the TME is highly heterogeneous and contains a complex architecture of immune, stromal, vascular and tumour cells in addition to acellular components such as the extracellular matrix. While understanding the dynamics of the TME has been instrumental in predicting durable responses to immunotherapy and developing new treatment strategies, recent evidence challenges the fundamental paradigms of how tumours can effectively subvert immunosurveillance. Here, we discuss the various immunosuppressive features of the TME and how fine-tuning these mechanisms, rather than ablating them completely, may result in a more comprehensive and balanced anti-tumour response.
Keywords:
tumour; immunosuppression; tumour microenvironment; mechanisms; immunotherapy; fine-tuning1. Introduction
The tumour microenvironment (TME) is a dynamic ecosystem that is manipulated by tumour cells to support its growth and subvert immune surveillance. Tumours exhibit varying degrees of inflammation that can be broadly categorised into immunologically ‘cold’ and ‘hot’ tumours. ‘Cold’ tumours are characterised by increased numbers of immunosuppressive cell types such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), M2-switched tumour-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs), as well as greater extracellular matrix (ECM) density, and poor oxygen and nutrient availability [1]. The ‘cold’ phenotype can be subcategorised into ‘excluded’ tumours, which display limited cytotoxic T lymphocyte (CTL) migration to the tumour periphery but not the tumour core, and ‘ignored’ tumours that are completely devoid of CTLs [2]. Conversely, ‘hot’ tumours present with improved infiltration of CTLs, natural killer (NK) cells, and M1 macrophages as well as increased pro-inflammatory and type-I interferon (IFN-I) signalling [1]. A major focus in the field is to develop new strategies that reprogram the TME toward the ‘hot’ phenotype, which is generally more responsive to immunotherapies including immune checkpoint blockade (ICB) and adoptive cell therapy (ACT). However, treatment-refractory patients commonly exhibit similar or even higher levels of tumour inflammation compared to those that do respond to therapy [3,4], suggesting that there are still significant deficiencies in our understanding of the molecular mechanisms underpinning tumour escape versus control. The TME is intrinsically tuned to resist natural and therapy-induced anti-tumour immune responses. Notably, many of these immunosuppressive mechanisms may also exert anti-tumour effects that are critically overlooked during drug development. This review will specifically highlight the common molecular drivers of the immunosuppressive TME, and the current strategies being investigated to target these pathways. We also discuss the role of these pathways in regulating the balance of inflammatory responses in the TME, highlighting their potential importance in promoting enhanced tumour control.
2. Cytokines
2.1. Interleukin-10
IL-10 is a homodimeric cytokine that is produced by various cell types including T cells, B cells, macrophages, mast cells, dendritic cells (DCs), granulocytes, and tumour cells [5,6]. IL-10 binds to IL-10 receptor-alpha, activating the Janus kinase (JAK)-Signal transducer and activator of transcription (STAT) and protein kinase B (Akt) signalling cascades, thereby phosphorylating the transcription factor STAT3 which then dimerises and translocates to the nucleus. Additionally, IL-10 signalling mediates the activation of MAFB, a transcription factor that regulates the polarisation of anti-inflammatory macrophages [7]. While recent work has shed light on its immune-stimulating activity [5], IL-10 was initially described as a potent inhibitor of pro-inflammatory cytokine production by activated macrophages and type-I T helper (Th1) cells [8,9]. Effective immune surveillance is governed by the presentation of tumour antigens by DCs to prime CTLs. However, IL-10 is known to interfere with this process by reducing the expression of co-stimulatory molecules or directly dampening T cell receptor (TCR) signalling strength through the re-arrangement of surface N-glycans [10,11,12]. Similar phenomena have also been observed in other antigen-presenting cells (APCs) such as monocytes and macrophages, where IL-10 downregulates the surface expression of CD86 and major histocompatibility complex (MHC)-II proteins. IL-10-treated DCs have been shown to promote CTL anergy toward melanoma-specific antigens, resulting in a loss of cytolytic activity in vitro [11]. Moreover, IL-10-producing monocytes/MDSCs subvert anti-tumour immunity by inhibiting macrophage-derived IL-12 and T cell proliferation in carcinoma models [13,14]. A meta-analysis of over 1700 patients from 21 published studies revealed that elevated serum IL-10 levels were associated with poor prognosis across most solid and haematological cancers [15]. Zhang et al. recently showed that increased IL-10+ TAMs in biopsies from gastric cancer patients were associated with poor clinical outcomes and response to chemotherapy [16]. In patients with lung cancer, IL-10 was not only shown to positively correlate with tumour diameter, but it was also demonstrated that IL-10 may counteract intra-tumoural programmed cell death protein 1 (PD-1)/programmed death-ligand 1 (PD-L1) signalling, highlighting the potential role of IL-10 in mediating resistance to immune checkpoint blockade [17]. Despite strong evidence suggesting that IL-10 plays an important role in promoting immune tolerance, numerous studies have also highlighted the anti-tumour potential of IL-10 [18]. This was exemplified by pre-clinical studies showing that IL-10 improves immune surveillance by augmenting the effector functions of intra-tumoural CTLs [19]. Moreover, a recent phase I clinical trial demonstrated that pegylated IL-10 in combination with anti-(α)PD-1 therapy elicited robust anti-tumour immune responses and an improvement in clinical outcomes in patients with renal and non-small-cell carcinoma [20]. Taken together, IL-10 plays a dual role in eliciting immune responses with pro- and anti-tumour properties that need to be precisely regulated.
2.2. Transforming Growth Factor-β
The TGF-β cytokine family is composed of three variants (TGF-β1, -β2 and -β3) each of which are initially produced as a latent form before being enzymatically converted to active TGF-β. Each variant interacts with type-I and type-II serine/threonine kinase receptors (TGF-βRI and TGF-βRII), which in turn activates the SMAD pathway. The phosphorylation of SMAD2/3 then facilitates binding to SMAD4 to form the transcriptional complex that accumulates in the nucleus and controls gene expression [21]. TGF-β plays a pivotal role in tumour immunosuppression by impairing the activation and production of cytolytic molecules by NK and CTLs [22,23,24]. Furthermore, TGF-β has been shown to suppress chemokine receptor expression on CTLs, rendering them incapable of trafficking to tumours [25]. TGF-β is also responsible for triggering immunosuppressive cascades by activating forkhead box P3 (Foxp3)+ Tregs, MDSCs and CAFs which also have distinct anti-inflammatory properties [26,27,28,29]. Indeed, elevated levels of TGF-β subtypes are associated with poor outcomes across numerous malignancies including cutaneous melanoma, lung, ovarian and triple-negative breast cancer [30,31,32,33]. Due to the major role that TGF-β plays in promoting tumourigenesis, there has been growing interest in therapeutically targeting TGF-β signalling to improve outcomes in cancer patients. Dodagatta-Marri et al. recently showed that resistance to αPD-1 therapy in mice with squamous cell carcinoma was mediated by the induction of immunosuppressive Tregs. However, combinatorial treatment using αPD-1/αTGF-β ablated this effect and was superior in promoting tumour rejection [34]. Similar findings were observed when using dual αPD-L1/LY364947 (a TGF-βRI kinase inhibitor) therapy, although the efficacy of this regimen was found to be more effective against the more immunogenic MC38 colon adenocarcinoma cell line, highlighting a further limitation of the strategy [35]. This synergistic effect with ICB has also led to the development of bifunctional agents that simultaneously block PD-L1 and TGF-βRII which has shown promising results in pre-clinical models and phase I clinical trials [36]. Although TGF-β blockade has shown promising results in combination with other immunotherapeutic strategies and in pre-clinical models, these findings have yet to be fully recapitulated across numerous clinical trials [21]. One possible explanation for this is the direct effect that TGF-β blockade has on cancer cells, given that the loss of TGF-β signalling promotes mesenchymal–epithelial transition—an important step during metastasis [37]. Secondly, TGF-β can be shuttled via extracellular vesicles, rendering them inaccessible to antibody-based therapies [38]. Lastly, TGF-β plays a crucial role in the formation and maintenance of CD8+ tissue resident memory T cells (TRM) that are a critical component of the anti-tumour response [39]. While TGF-β has been classically defined as an anti-inflammatory agent, contrary findings have shed light on its role in tempering tumour outgrowth and promoting immune homeostasis within the TME.
2.3. Type-II Inflammatory Cytokines
Type-II inflammation plays a critical role in modulating the anti-tumour immune response. An imbalance toward type-II inflammation has been associated with the ‘cold’ tumour phenotype compared to ‘hot’ tumours that are enriched with the more cytotoxic type-I inflammation across multiple cancers [40]. Alarmins such as thymic stromal lymphopoietin (TSLP) and IL-33 are essential upstream regulators of the type-II inflammatory response that are produced by stromal and endothelial cells in response to stress and cellular damage. TSLP signals through TSLP-receptor (TSLPR) that is found on group 2 innate lymphoid cells (ILC2s), T cells and APCs [41]. Similarly, IL-33 is a member of the IL-1 family and binds to the ST2 receptor found on ILC2s and T cells [42]. Tumour cell-derived TSLP reprograms myeloid DCs with Th2-polarising activity, and treatment of these mice with αTSLP antibodies not only diminished this effect but also significantly prolonged survival in humanised mice [43]. TSLP receptor (TSLPR)o/o mice engrafted with 4T1 breast tumour cells restored type-I immune responses resulting in slower 4T1 tumour growth and reduced lung metastasis [44]. Takahasi et al. also demonstrated that fibroblast-derived periostin could stimulate TSLP by keratinocytes and promote a Th2-dominant TME in a model for cutaneous T cell lymphoma [45]. Production of TSLP by CAFs within the TME and IL-4 by basophils in draining lymph nodes (dLNs) drives the polarisation of Th2 CD4+ T cells and M2 macrophages [46,47]. The IL-33/ST2 axis on Tregs is a key tumourigenic driver in both skin and colon cancer [48,49], while ST2 deletion enhanced type-I inflammatory responses that promote clearance of murine mammary carcinoma [50]. This pathway is also responsible for the polarisation of matrix metallopeptidase 9-secreting M2 macrophages which accelerate adenocarcinoma metastasis [51]. While the expression of alarmins in the TME have been classically associated with increased tumour growth and metastasis, contrary evidence has also positioned them as important regulators of anti-tumour immunity. TSLP elicits protection against skin carcinogenesis by enhancing dermal T memory cell immunity [52], which may not be surprising considering TSLPR naturally complexes with the IL7R to promote CTL memory formation and homeostasis [53,54]. A similar skin cancer model also demonstrates that TSLP enables Th2 formation which exerts an anti-tumour effect and improves tumour surveillance [55]. In an elegant model published by Demehri et al., transgenic K14 mice overexpressing dermal TSLP could successfully arrest breast and pancreatic tumour development, which was associated with an influx of GATA3+ Th2 cells to the primary tumour site [56]. Similarly, tumour growth was markedly reduced following IL-33 treatment, which was attributed to an increase in the migration and viability of cytotoxic eosinophils in a model for colorectal cancer [57]. In addition, IL-33 has been shown to drive anti-tumour CTL and NK cell activity that reduces melanoma tumour growth and metastasis [58]. Xia et al. further demonstrated that exogenous IL-33 recapitulates CTL effector functions in IL-33-deficient mice in a colon carcinoma model [59]. Further reports indicate that ST2 expression on CTLs is promptly upregulated following polarisation by type-I inflammation, while IL-33 and IL-12 (a canonical type-I inflammatory cytokine) act synergistically to augment the effector functions of CTLs [60]. This critical finding suggests that a balance of type-I and type-II inflammation is essential for optimal immune function. Canonical type-II inflammatory cytokines, including IL-4, IL-5, IL-13 are primarily produced by Th2 cells and ILC2s. Type-II inflammatory cytokines, including alarmins in the TME is tightly linked with the induction of M2 macrophages and Tregs [61,62]. Additionally, ILC2 numbers have been shown to correlate with infiltrating MDSCs in recurrent bladder cancer, where ILC2-derived IL-13 upregulated the expression of T cell-suppressing genes including ARG1 (encoding arginase-I; ARG1) and NOS2 (encoding inducible nitric oxide synthase; iNOS) [63]. Durable responses to ICB correlate with improved Th1/Th17 activity and a concomitant suppression of Th2 immunity [64]. One novel approach to circumvent the type-II-skewed TME is to utilise Inverted Cytokine Receptor (ICR)-modified CAR T cells that arm, rather than attenuate the cancer-killing mechanisms of T cells in the presence of Th2 cytokines. Using this strategy, IL-4/21 ICR (where the ectodomain of the IL-4R is fused to the endodomain of the IL-21R) CAR T cells were significantly more effective at eradicating IL-4+ tumours in vivo [65]. It is noteworthy that type-II inflammatory cytokines may be beneficial in eliciting anti-tumour immune responses. For instance, recent work has demonstrated that targeted knockdown of TGF-βRII expression on CD4+ T cells augments anti-tumour responses and vascular remodelling in an IL-4-dependent manner [66,67]. A recent study by Moral et al. supports this further by showing the ILC2s in pancreatic ductal adenocarcinoma (PDAC) cells are highly receptive to PD-1 blockade and augment tissue-specific tumour immunity [68]. Remarkably, adoptive transfer of tumour-reactive Th2 cells were successful in promoting tumour rejection [69]. This is corroborated by other work demonstrating that memory Th2 cells potently activate cytotoxic NK cells to slow tumour growth [70], suggesting that a complete ablation of type-II inflammation within the TME may be detrimental to immune surveillance.
3. Metabolites
3.1. Tryptophan and Kynurenine
Catabolism of the essential amino acid tryptophan (Trp) by indoleamine 2,3-dioxygenase-1 (IDO1) is the first and rate-limiting step in the synthesis of nicotinamide adenine dinucleotide (NAD) which is a critical co-factor involved in glycolysis and oxidative phosphorylation [71]. Immune suppression is triggered by a two-fold effect of IDO activity by first depleting Trp, and the accumulation of the immunosuppressive metabolite kynurenine (Kyn). IDO1 is an inducible enzyme produced by a broad range of myeloid cells (including DCs and macrophages), endothelial cells, mesenchymal stromal cells and fibroblasts [72]. Increased IDO1 has been shown to stifle the activation of T cells, inhibit NK cell function, stimulate Tregs, induce tolerogenic DCs, expand MDSCs and neovascularisation [73]. The Trp starvation theory proposes that depletion of Trp facilitates accumulation of uncharged tRNA and activation of the general control nonderepressible 2 (GCN2) pathway and subsequent T cell dysfunction [74]. Trp depletion has been shown to inhibit mammalian target of rapamycin (mTOR) and protein kinase C in cancer cells and enhance autophagy and Treg development [75]. Furthermore, a loss of mTOR signalling may shift the balance of the CTL compartment toward short-lived and suboptimal cytolytic responses [76]. Accumulation of Kyn is important in maintaining peripheral homeostasis and tempering inflammatory lymphocyte activity. The Aryl hydrocarbon receptor (AhR) pathway is activated by Kyn which is toxic to lymphocytes and induces CD4+ T cell differentiation into Tregs [72,77]. AhR dimerises with the AhR nuclear translocator (ARNT) protein to bind multiple transcriptional co-factors to drive transcription of IL-10 in DCs and NK cells and IL-6 in macrophages and cancer cells [78,79]. As previously described, IL-10 has a dichotomous role in the TME by eliciting both pro- and anti-tumour responses. Interestingly, tumour cells expressing high levels of IDO were observed to have a slower growth rate compared to low IDO expressing cells [80]. Furthermore, higher intra-tumoural expression of IDO has been correlated with longer survival in multiple cancer types [81,82,83] but also worse prognosis in several other cancers [84,85,86,87,88]. The effect of IDO as an immunosuppressive, pro-tumour factor has been queried due to a lack of efficacy reported in a recent phase III clinical trial combining the potent IDO inhibitor epacadostat with αPD-1 therapy in patients with non-resectable and metastatic melanoma [89].
3.2. Adenosine
Purinergic signalling is tightly regulated by the activity of surface ectonucleotidases, CD39 and CD73. The pair of enzymes are responsible for the conversion of extracellular adenosine triphosphate (eATP) to adenosine. The release of eATP is a common consequence of cellular stress including inflammation, hypoxia or ischemia which triggers adenosine accumulation. Adenosine elicits immunosuppression predominantly through the type-I purinergic receptors, A2A and A2B as part of a regulatory negative feedback loop [90]. Indeed, the accumulation of adenosine and the expression of CD39 and CD73 are well described features of the TME [91]. A broad range of immune cells express adenosine receptors including T cells, NK cells, natural killer T cells (NKTs), macrophages, DCs, neutrophils, mast cells and B cells [92]. The A2A receptor has been shown to promote proliferation and immunosuppressive function of Tregs [93,94], inhibit T cell proliferation, cytotoxicity and inflammatory cytokine production [95]. The CD39/CD73 axis fine-tunes macrophage differentiation and activity by promoting M2 polarisation [96], while A2B agonists promoted infiltration of MDSC in melanoma-bearing mice which was diminished following A2B blockade [97]. Furthermore, CD73-deficient mice are resistant to carcinogenesis, while metastasis of CD73+ tumours is significantly impaired following A2A blockade [98,99]. However, in contrast to the established role of adenosine in generating an immunosuppressive TME, other work has highlighted its importance in T cell differentiation. Deletion of A2A receptors increased tumour growth and impaired CTL and differentiation in a B16F10 tumour model [100]. This is supported in other studies which demonstrate that adenosine signalling favours the generation of long-lived memory T cell precursors that are protected against ATP-induced apoptosis [101,102]. Increased expression of CD73 has been observed in a wide range of cancers and correlated with a worse prognosis [103]. However, contradictory to such evidence, Ineoue et al. [104] found that tumour CD73 and A2A protein expression in lung adenocarcinoma associated with a better prognosis. Further confusing the prognostic value of CD73, studies have shown expression correlated with better prognosis in ovarian and breast cancer [105,106]. Nevertheless, the dual-blockade of CD39 and CD73, in combination with αPD-1/αCytotoxic T lymphocyte-associated protein-4 (CTLA-4) was successful in promoting robust anti-tumour T cell immunity in known therapy-resistant cancer models [107]. The authors also noted that the accumulation of eATP following CD39/CD73 blockade promoted the activation and maturation of DCs and M1 macrophages [107]. This has also led to the development of clinical trials targeting the A2A receptor in patients with refractory renal cell cancer [108]. In summary, blocking adenosine signalling is a feasible strategy that favourably re-shapes the immune landscape of the TME, although contradictory findings do suggest that the preservation of this pathway might be important in regulating T cell homeostasis and the maintenance of long-lived memory T cell precursors.
3.3. Nitric Oxide
Inducible nitric oxide synthase (iNOS) is a key enzyme involved in the production of nitric oxide (NO) and is expressed by an array of cells including macrophages, MDSCs, DCs, NK cells, tumour cells and endothelial cells [109]. NO is critical in many physiological functions but also has been shown to drive a dual role in tumour development. iNOS is promptly upregulated upon exposure to external stimuli (such as lipopolysaccharide; LPS), hypoxia and proinflammatory cytokines (IL-1, IFN-gamma; IFNγ), tumour necrosis factor-alpha; TNFα) resulting in the production of large quantities of NO [110]. Over-expression of iNOS has been associated with poor prognosis in a series of human cancers [111,112,113]. Furthermore, elevated iNOS gene expression in patients has been demonstrated in numerous cancer types, which was contrasted by a lower expression in surrounding healthy tissue [114,115]. However, other attempts to correlate iNOS expression with patient outcomes has led to contradictory results, and additional studies have questioned its value as a prognostic marker [116,117]. Work by various groups have clearly demonstrated the suppressive effects of NO on T cell function via different mechanisms including the inhibition of the critical JAK3/STAT5 signalling pathway, inhibition of MHC class II expression and induction of T cell apoptosis [118,119,120]. Moreover, NO crucially recruits MDSCs, Tregs, M2 macrophages and Th2 cells to the TME to propagate the ‘cold’ tumour niche [121]. Increased expression of NO mediates the upregulation of vascular endothelial growth factor (VEGF) signalling in the TME which promotes tumour growth and invasiveness [110,122]. Xiong et al. demonstrated that NO also inhibited the production of IL-12 in DCs and M1 macrophages [123]. Multiple groups have demonstrated increased type-I immunity and IL-12 production in iNOS knockout mouse models after bacterial infection. Intriguing work from Marigo et al. demonstrated the local iNOS-expressing DCs cooperate with adoptively-transferred CTLs to orchestrate tumour killing. This study also demonstrated that the expression of the iNOS-encoding gene, NOS2 correlated with improved T cell density in tumours and disease-free survival in patients with colorectal cancer [124]. Klug et al. indicated the ability of low dose irradiation in vivo to polarise iNOS+ M1 macrophages, which promoted type-I immunity and improved CTL infiltration via NO-dependent vascular remodelling [125]. Notably, these findings challenge other lines of evidence showing that the presence of reactive nitrogen species in tumours, such as NO can impair CTL infiltration via the nitration of T cell-attracting chemokines [126]. Expression of iNOS in CD4+ T cells has also been reported to suppress Treg accumulation in pre-clinical cancer models and disrupt tumour tolerance by inhibiting production of TGF-β1 [127]. NO has been classically recognised as a myeloid-derived immunosuppressive molecule that inhibits T cell survival, function, and migration. However, conflicting reports also show that NO is indispensable to anti-tumour immunity which is likely determined by its precise concentration and spatiotemporal abundance in the TME.
3.4. L-Arginine
Arginase-1/2 (ARG1/2) promotes the catabolism of the amino acid arginine (L-Arg) into urea and ornithine, which is subsequently broken down into proline and polyamines to drive collagen synthesis and cell proliferation, respectively [128]. Regulatory myeloid cells such as M2 macrophages and MDSCs are recognised as the primary regulators of L-Arg metabolism through the expression of ARG1 during infection and inflammation [129]. Expression of ARG1 is promptly upregulated in these cells in response to Th2 and anti-inflammatory cytokines (including IL-4, IL-13, IL-10 and TGFβ) to assist in resolution of inflammation and promote tissue repair [130]. Many studies have in fact correlated overexpression of ARG1/2 and poor prognosis in a variety of cancers types [131,132,133,134,135,136]. Notably, the deprivation of L-Arg has been shown to have a direct detrimental effect on tumour growth by promoting autophagy, apoptosis and cell cycle arrest [137]. However, it is also well described that the loss of L-Arg metabolism has a profound effect on anti-tumour immunity. Deprivation of L-Arg from the microenvironment by regulatory myeloid cells has a profound effect on the local immune landscape. ARG1 expression by MDSCs favours the generation of IDO-expressing, tolerogenic DCs [138], while L-Arg deficiency compromises CD3 zeta chain (CD3ζ) expression on T cells, subsequently impairing TCR signalling, proliferation and IFNγ production [139]. Furthermore, L-Arg availability shifts the metabolic programs of T cells toward oxidative phosphorylation to promote the generation of a central memory phenotype that are endowed with improved survival and anti-tumour activity in a B16 melanoma model [140]. Indeed, the inhibition of ARG1/2 activity has yielded positive results across numerous cancer models by reducing myeloid-driven immune suppression [134,141]. However, adoptive transfer of Arg2−/− CTLs was also more efficient at clearing tumours and synergised with PD-1 blockade, suggesting that CTL-intrinsic ARG2 activity contributes to the suppression of their anti-tumour activity [142]. To date, phase I clinical trials testing arginase inhibitors have shown some promise in boosting anti-tumour immunity. However, its progress to the clinic has been limited partly due to the essential role of ARG in metabolising ammonia which is highly toxic [143,144].
3.5. Prostaglandin-E2
Prostaglandin-E2 (PGE2) is a bioactive lipid generated by cycloxygenase-2 (COX-2) following the enzymatic conversion from arachidonic acid. In the TME, PGE2 is predominantly synthesised by myeloid, stromal and cancer cells, and signals through the G protein-coupled receptor group, EP1-EP4 [145]. PGE2 is recognised as a pan-immunosuppressive mediator as it inhibits CTLs, NK cells and type-I inflammation, while promoting Treg, MDSC expansion and type-II inflammation [146]. Indeed, pilot data from a phase I clinical trial has shown that small-molecule inhibitors of EP4 are well tolerated and slowed disease progression in a proportion of patients with advanced cancers [147], while pre-clinical studies have demonstrated that COX-2 inhibition synergises with ICB to improve tumour eradication, highlighting its potential as a therapeutic adjuvant [148]. Despite strong evidence for its role in dampening anti-tumour inflammation, PGE2 has been implicated in regulating the memory T cell compartment which is an important axis of tumour control. For instance, PGE2 treatment of umbilical cord blood (UCB) cells lead to the expansion of TCF7- and eomesodermin (EOMES)-expressing CTLs that display a stem-like phenotype, thereby improving the immune reconstitution efficiency of UCB transplantation [149]. In contrast to what has been previously reported, PGE2 selectively inhibits the expansion of certain Treg subtypes, revealing a pro-inflammatory role for PGE2 [150,151]. Furthermore, PGE2 has been shown to restore CCR7-dependent migration of DCs to dLNs, which subsequently improves CTL-DC crosstalk in a prostate cancer model [152]. This is complicated further by more recent data demonstrating that TLR agonists in combination with PGE2 promotes mature, cytokine producing DCs with impaired antigen cross-presentation activity [153]. These divergent findings may suggest that nominal PGE2 signalling, possibly through certain EP receptors may be beneficial in certain aspects of anti-tumour immunity. Collectively, PGE2, in combination with other soluble factors play a fundamental role in creating an immunosuppressive TME (Figure 1).
4. Impairment of IFN-I Signalling
Type-I interferons (IFN-Is) are a functionally diverse family of cytokines that play a crucial role in generating potent innate and adaptive immune responses against cancer [154]. IFN-Is are indispensable to anti-tumour immunity by enhancing intra-tumoural CTL-DC crosstalk [155], as well as the augmentation of NK and M1 macrophage activity in the TME [156,157]. ‘Cold’ tumours are generally characterised by poor immune cell infiltration and an accumulation of immunosuppressive factors within the TME. Consistent with this theme, ‘cold’ tumours also restrict endogenous IFN-I activity. In biopsies from patients with triple-negative breast cancer, tumour-associated plasmacytoid DCs (pDCs) produced significantly lower amounts of IFNα, which correlated with the accumulation of intra-tumoural Tregs [158]. IFN-I production by pDCs has also been shown to be compromised in the presence of TGF-β and TNFα within breast tumours [159]. Katlinski et al. demonstrated that the hypoxia-induced downregulation of IFN alpha receptor-1 (IFNAR1) in the TME was a central mechanism that impedes the viability of CTLs, generating ‘cold’ tumour niches [160]. Higher expression of interferon regulatory factor-7 (IRF7) gene signatures in primary tumours has also been linked to prolonged bone metastasis-free survival in breast cancer [161]. Indeed, the efficacy of current cancer therapies such as radiotherapy, chemotherapy and immunotherapy rely on intact IFN-I signalling within tumours [162,163,164]. Accordingly, agents that induce IFN-I responses (such as poly-I:C and stimulator of IFN genes; STING agonists) are used widely as adjuvants for current therapies with moderate success [165,166]. Despite this, there has been mounting evidence that IFN-I signalling also exerts a detrimental effect on anti-tumour immunity. Persistent tumour IFN signalling has been shown to drive adaptive resistance to ICB, which was diminished when mice were pre-treated with JAK inhibitors to block downstream IFN signalling [167]. Furthermore, inflammatory breast cancer (which is a rare and aggressive form of breast cancer) is frequently associated with hyper-activation of IFN-I pathways [168]. More recently, Effern et al. noted that recurring tumours with significant loss of antigen expression were associated with more intense IFN signalling [169], suggesting that tumour dedifferentiation as a mechanism of immune evasion may be driven by overexuberant anti-tumour responses within the TME. This may also be confounded by the functional heterogeneity of IFN-Is, where specific IFNα subtypes are clearly more potent primers of the anti-tumour immune response, although this has only been explored in murine models [154].
5. Hypoxia
The establishment of a hypoxic TME is a hallmark of solid cancer progression and dramatically re-shapes the immune contexture of the TME. The cellular response to hypoxia is driven largely by hypoxia-inducible factors (HIF-1α, -2α, -3α) which are oxygen-sensitive transcription factors that are stabilised in the presence of low oxygen concentrations [170]. The relationship between hypoxia and immune suppression in the TME is well established and is strongly linked to numerous mechanisms described above, including the impairment of IFN-I signalling, upregulation of immune checkpoint molecules and the upregulation of extracellular adenosine [170]. Elevated hypoxic gene signatures are a major prognostic indicator in cancer patients and are frequently associated with immune-privileged (‘cold’) tumour niches [171,172]. Hypoxia and the induction of lactate metabolism has been reported to promote M2 polarisation of TAMs via the activation of HIF-1, Hedgehog and mTOR pathways [173,174]. In addition, tumour cell-derived exosomes under hypoxic conditions were also able to reprogram macrophages toward an M2 phenotype [175]. While it has been demonstrated that elevated HIF-1α signalling in DCs results in Th2-biased activation, decreased antigen uptake, and decreased CTL expansion [176,177,178], other work has shown that hypoxic stress improved dLN trafficking of DCs via CCR7 and promoted a highly pro-inflammatory gene expression profile [179,180]. These divergent findings may be at least partially explained by the activation of HIF-1α-independent pathways and the prevalence of other metabolic confounders in the TME that subvert cellular function (i.e., the depletion of glucose, low pH, etc.). Of note, HIF-1α also acts a negative regulator for pDC development, limiting IFNα production [181]. Hypoxic zones within the TME are significantly more resistant to CTL penetrance, which represents a major challenge in T cell-based immunotherapies [182]. The exclusion of CTLs from these regions rely heavily on the accumulation of immunosuppressive myeloid cells and Tregs that are dependent on various cytokines such as CCL28, CCL5 and TGF-β [183,184,185]. Knockdown of hypoxia-induced triggering receptors expressed on myeloid cells (TREM)+PD-L1+ TAMs in advanced hepatocellular carcinoma (HCC) could successfully rescue CTLs from an exhaustive state and restore their cytolytic activity, which was largely attributed to a reduced recruitment of Tregs to the TME [186]. This is supported by other studies showing that the use of oxygen supplementation or hypoxia-disrupting drugs in combination with ICB can also successfully mitigate this effect by reducing MDSC density [182,187]. Hypoxic stress, mediated by HIF signalling has also been shown to directly regulate T cell differentiation by favouring glycolytic metabolism and effector transition that eventually leads to exhaustion [188]. CART cells, under hypoxic conditions in vitro displayed significantly less proliferative and cytokine-producing activity compared to those under normoxia [189]. Moreover, hypoxia has been shown to induce defects in mitochondrial function that lead to CTL exhaustion [190]. Conversely, Palazon et al. demonstrated that intrinsic HIF-1α signalling was essential for CTL infiltration and effector function, highlighting the importance of HIF-1α in the adaptation of CTLs to the hypoxic TME [191]. While this seemingly contradicts the detrimental role of hypoxia and HIF signalling in CTLs, it is important to consider that tumour cells can out-manoeuvre this metabolic adaptation by also depleting glucose from the TME, thereby diminishing the activity of CTLs that rely heavily on glycolysis for energy production [192]. Interestingly, CTLs reprogrammed to utilise fatty acid catabolism under oxygen and glucose deprivation could recapitulate their cytolytic activity, rendering them more responsive to ICB [193]. These metabolic changes in the TME also have a direct effect on tumour cells and their ability to evade immune responses, as combined oxygen and glucose starvation decreases their presentation of tumour antigens on MHC class I to avoid recognition by CTLs [194]. Hypoxia and various mechanisms of immune suppression interface heavily and mutually support each other to promote tumour growth, highlighting the enormous challenge presented when designing new therapies that successfully address both issues.
6. Extracellular Vesicles
Extracellular vesicles (EVs) are lipid bilayered nanoparticles that shuttle various bioactive components, such as RNAs, proteins and lipids between cells. Tumour-derived EVs (T-EVs) bearing Fas ligand (FasL) and TNF-related apoptosis-inducing ligand promote CTL apoptosis [195,196,197]. Similarly, T-EVs expressing membrane-associated TGF-β1 enhanced Treg function while impairing NK and CTL activation [198]. Other lines of evidence demonstrate that T-EVs promote M2 macrophage polarisation which accelerates cancer growth and metastasis [199,200]. Moreover, exosomes produced by these macrophages have been shown to contain micro-RNAs (miRNAs) that dysregulate the balance of Treg:Th17 cells in ovarian cancer [201]. EVs produced by other anti-inflammatory cells such as Tregs and MDSCs also dampen CTL and type-I inflammatory responses by delivering various miRNAs or immunomodulatory proteins [202,203,204], suggesting that EVs produced by both tumour and immune cells in the TME help propagate the ‘cold’ tumour niche to suppress anti-tumour immunity. EVs within the TME are also major mediators of therapy resistance in cancer patients. Richards et al. showed that CAFs-derived EVs could elicit chemotherapy resistance in PDAC cells via the upregulation of the Snail pathway [205], while similar resistance to chemotherapy has been demonstrated via the delivery of macrophage-derived EVs containing miR-21 and miR-365 [206,207]. In a melanoma model, tumour cells shedding EVs mediated chemotherapy resistance by promoting M2 macrophage polarisation and upregulating IL10 and ARG1 expression in stromal cells [208]. Additionally, PD-L1+ EVs produced by cancer cells are a prominent mechanism of immunotherapy resistance by acting as off-target decoys for αPD-1 monoclonal antibodies that are used to reinvigorate anti-tumour T cell immunity [209]. Although targeting EVs within the TME may be a plausible approach to ameliorating tumour immunosuppression, a critical study by Wolfers et al. showed that T-EVs deliver tumour antigens to DCs to enable CTL cross-priming [210]. Other work has highlighted the role of DC-derived EVs in delivering peptide-loaded MHC and co-stimulatory molecules to cancer cells, improving their immunogenicity [211].
7. Exclusion of T Cells from the Tumour Bed and Disruption of T Cell Homeostasis
T cells, in particular CTLs are considered one of the key effectors in mediating anti-tumour immunity. Indeed, a defining characteristic of ‘cold’ tumours is the exclusion of CTLs from the tumour bed. Several factors contribute to the impairment of CTL infiltration into tumours, including mechanisms described above such as hypoxia and the accumulation of anti-inflammatory cells. These conditions that are hostile to CTLs also disrupt the chemokine signalling pathways that are essential to CTL trafficking. TME-residing MDSCs promote the nitration of CCL2 via the production of reactive nitrogen species, impairing the trafficking of CTLs to the tumour site and trapping them in the surrounding stroma [126]. Additionally, increased concentrations of CCL27, CCL5 and CXCL10 in tumours have been associated with better mobilisation of CTLs to the TME [212,213,214]. Tumour cell-derived galectins have been shown to impair the activities of IFNγ-induced chemokines, CXCL9/10/11 by decorating ECM glycans and subsequently trapping intra-tumoural IFNγ [215]. Conversely, CAFs can directly impede CTL trafficking by secreting CXCL12 which, at high concentrations deters CTL migration [216]. The tumour vasculature also undergoes significant remodelling to stifle the migration of CTLs to the tumour bed. Upregulation of VEGF, IL-10 and PGE2 at the tumour site cooperatively promotes Fas ligand expression on tumour endothelial to elicit apoptosis of CTLs, but not Tregs [217]. Furthermore, VEGF signalling and local NO production induces defects in the structural arrangement of adhesion molecules on tumour endothelial cells to impair CTL extravasation [218]. Lastly, the ECM architecture laid out by CAFs physically constrains CTLs to areas of lower collagen and fibronectin density, which was reversed following collagenase treatment [219]. Collectively, tumour cells can manipulate its local milieu to suppress multiple mechanisms of CTL migration to the TME (Figure 2).
CTLs that do successfully migrate into the TME are required to integrate an array of pro- and anti-inflammatory signals to appropriately endow them with cancer-killing activity. Infiltrating CTLs that maintain memory and stem-like properties are superior in mediating long-term anti-tumour immunity, while durable response to immunotherapy relies on the expansion of these subsets [220,221]. While it is indisputable that ‘hot’ tumours are skewed toward a Th1, pro-inflammatory phenotype that supports the effector functions of CTLs, there is emerging evidence that many “pro-tumour” factors play a critical role in regulating this protective T cell niche in the TME. For instance, IL-10 and TGF-β have been implicated in the maintenance of TRM populations in tumours [222,223,224], while the induction of TCF7 gene expression (a key transcription factor that regulates T cell stemness and longevity) has also been shown to be controlled by a core transcription factor group that includes the master Th2 regulator, GATA3 [225]. Moreover, type-II inflammatory cytokines such as TSLP regulate the balance of antigen-specific memory CTLs by cooperating with IL-7 signalling [54]. Conversely, cytokines such as IL-12 and IFN-I can counteract the generation of long-lived memory T cells to favour terminal differentiation and eventual exhaustion when produced in excess [226,227,228], suggesting strong pro-inflammatory cues within the TME may drive the formation of short-lived effector CTL responses that are unfavourable in establishing anti-tumour immunity (Figure 3).
8. Inhibitory Receptors
8.1. T Cell Immunoglobulin and Mucin Domain-Containing 3 (TIM-3)
T cell immunoglobulin and mucin domain-containing 3 (TIM-3) is a type-I transmembrane protein and member of the immunoglobulin (Ig) superfamily that is upregulated on activated T cells and associated with a terminally differentiated effector state. TIM-3 has been shown to interact with several ligands including galectin-9 (Gal-9), phosphatidylserine (PtdSer), high mobility group box protein B1 (HMGB1) and carcinoembryonic antigen related cell adhesion molecule 1 (CEACAM-1) [229]. Interaction of TIM-3 with its ligand’s triggers phosphorylation of two tyrosine residues, releasing human leukocyte antigen B (HLA-B)-associated transcript 3 (BAT3) and allowing TIM-3 to exert its inhibitory function [230]. TIM-3 expression is dependent on the type-I master regulator, T-box transcription factor 21 (T-BET) [231]. Under inflammatory conditions, IL-12 and IL-27 induces TIM-3 expression and T cell dysfunction via T-BET and nuclear factor, interleukin-3 regulated (NFIL3), respectively [232,233]. In CTLs, signalling via the nuclear factor of activated T cells (NFAT) has been shown to play a role in also regulating TIM-3 expression and subsequent exhaustion [234]. It has recently been demonstrated that the interaction of TIM-3 with CEACAM1 is required for the T cell inhibitory function of TIM-3 in cancer [235]. In addition, TIM-3 is also expressed at high levels on tumour-infiltrating Tregs [236,237,238] and their presence is associated with advanced disease and the nodal metastasis in patients with NSCLC [236]. High levels of TIM-3 expression in the TME correlate with suppression of T cell responses and T cell dysfunction in cancer [239,240]. In line with this, a recent meta-analysis demonstrates that TIM-3 expression is significantly associated with worse overall survival in patients with solid cancer [241]. Consistent with a role in the negative regulation of anti-tumour immunity, there is extensive pre-clinical data demonstrating the therapeutic benefit of blocking TIM-3 signalling, mostly in conjunction with PD-1 blockade [242]. These encouraging pre-clinical results have led to development of TIM-3 blocking antibodies for clinical use, with several studies reporting early clinical findings and strong safety profiles when used in combination with αPD-1/PD-L1 or decitabine [243,244,245].
8.2. Lymphocyte Activation Gene-3 (LAG-3)
Lymphocyte activation gene-3 (LAG-3; CD223) is member of the Ig superfamily of receptors with structural similarities to CD4. LAG-3 conventionally binds to MHC II. however, there is also evidence of its interaction with galectin-3 (Gal-3), LSECtin, and fibrinogen like protein 1 (FGL1) [246]. LAG-3 acts as a TCR co-receptor that is upregulated following antigen exposure [247]. In line with this, IL-12 a potent inducer of IFNγ, is known to upregulate LAG-3 expression on activated human T cells along with IL-2 and IL-7 [248]. In the TME, chronic antigen stimulation and inflammation maintains LAG-3 expression on T cells and where it is commonly co-expressed with other immune checkpoints such as PD-1 and Tim-3 [249,250]. In addition, LAG-3 is constitutively expressed on suppressive Tregs [251] where it interacts with MHC II on DCs inhibiting proliferation and maturation [252]. LAG-3+ Tregs in the TME secrete high levels of immunosuppressive cytokines, IL10 and TGFβ, which act to dampen the anti-tumour immune response and magnify Treg activity [252]. In murine studies, blockade of LAG-3 alone or in combination with anti-PD-1 has been shown to enhance the anti-tumour CTL response and inhibit tumour growth [253,254,255]. In humans, a meta-analysis revealed that elevated LAG-3 expression is consistently associated with poor prognosis across multiple cancers [256]. Due to the fundamental role of LAG-3 plays in T cell dysfunction, early in-human clinical trials have demonstrated promising results of LAG-3 blockade in combination with αPD-1 in patients with advanced malignancies [257,258,259]. In addition to its membrane-bound form, LAG-3 can be shed from the surface by proteases, yielding soluble LAG-3 (sLAG-3). Notably, sLAG-3 has been demonstrated to elicit an immunostimulatory effect on APCs which promotes robust type-I and tumour-reactive CTL responses [260,261,262]. Indeed, high levels of sLAG-3 is associated with improved prognosis in patients with gastric cancer and correlates with serum levels of IL-12 and IFNγ [263]. Harnessing this feature of sLAG-3, clinical trials utilising recombinant sLAG-3 together with ICB have also shown encouraging results in early clinical trials [264,265], highlighting its potential role as a therapeutic adjuvant.
8.3. T cell Immunoreceptor with Ig and ITIM Domain (TIGIT)
T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif (ITIM) domains (TIGIT) belongs to the family of poliovirus receptor (PVR)-like proteins and is expressed on activated T cells, Tregs and NK cells [266]. TIGIT, which normally binds CD155 and CD112 on DCs and tumour cells, exerts its immunosuppressive function through numerous mechanisms, such as its direct binding to tumour expressing CD155 to trigger T/NK cell inhibition, outcompeting its co-stimulatory counterpart, CD226 on the T/NK cell surface and by indirect means including the activation of immunosuppressive DCs and Tregs following CD155-CD226 recognition [267]. Indeed, overexpression of TIGIT has been associated with poor prognosis in numerous cancers including bladder, gastric, lung adenocarcinoma and HCC [268,269,270,271]. Like other inhibitory receptors, TIGIT expression is a hallmark of T cell exhaustion [272]. Kurtulus et al. highlighted that elevated TIGIT demarcates a highly dysfunctional CTL subset in tumours, while demonstrating that TIGIT signalling orchestrates the expansion of Tregs that dampen local anti-tumour immunity [273]. Notably, tumour CD155 (the binding partner for TIGIT) expression has been linked with resistance to αPD-1 therapy in patients with metastatic melanoma [274], while expression of CD155 on cancer cells ablates CTL activation via CD226 degradation [275]. Overcoming this, dual blockade of PD-1/TIGIT could successfully recapitulate anti-tumour CTL and NK cell responses in pre-clinical mouse models [276,277,278]. Collectively, encouraging pre-clinical data has prompted the commencement of numerous clinical trials utilising αTIM-3, αLAG-3 and αTIGIT as a new class of checkpoint inhibitors for patients with treatment-refractory cancers (Table 1).
8.4. Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA-4)
CTLA-4 (CD152) is a member of the Ig superfamily and is highly expressed on activated T cells and constitutively expressed on Tregs [279]. The primary function of CTLA-4 is to outcompete and block CD28 co-stimulation by binding the B7 molecules on APCs (CD80/CD86) to elicit downstream T cell inhibition [280]. Cell-extrinsic mechanisms also come into play by stripping B7 molecules from APCs following CTLA-4 engagement via trans-endocytosis [281], and reverse CTLA-4-B7 signalling that activates immunosuppressive IDO activity in DCs [282]. Furthermore, CTLA-4 plays a pivotal role in maintenance of Tregs and their immunosuppressive activity [283]. In humans, the correlation between high levels of CTLA-4 expression and poor outcome is well established in several types of cancer [284]. In pre-clinical studies, blockade of CTLA-4 led to rejection of lymphoma, colorectal, renal and fibrosarcoma cancer cell lines in mice [285] and activation of human tumour-specific CTLs [286]. These studies led to the development of ipilimumab, a fully human αCTLA-4 IgG1 monoclonal antibody (mAb), that demonstrated improved survival outcomes in a key phase 3 study of patients with previously treated metastatic melanoma [287]. This pivotal trial led to Ipilimumab being the first immune checkpoint blockade therapy approved by the Food and Drug Administration (FDA) in 2011 for patients with advanced melanoma, a disease stage for which there was no previous standard of care therapy that prolonged survival. Since then, αCTLA-4 blockade has been examined extensively in pre-clinical studies and clinical trials as a single agent and in combination with chemotherapy [288,289,290], radiotherapy [291,292,293], cancer vaccination [294], and other immunotherapies [295,296,297]. However, it is the combination of αPD-1/αCTLA-4 blockade that has demonstrated the greatest therapeutic efficacy in the clinic, with ipilimumab (αCTLA-4) and nivolumab (αPD-1) approved for treatment of patients with advanced solid tumours (Table 2).
8.5. Programmed Cell Death Protein 1 (PD-1)
PD-1 (CD279) is another membrane-bound co-inhibitory receptor with Ig-like domains. However, unlike CTLA-4, PD-1 expression is more broadly expressed across hematopoietic and non-hematopoietic cells. PD-1 binds its cognate ligands, PD-L1 and PD-L2 which are membrane proteins also found on numerous cell types including APCs, endothelial cells, cancer cells, mast cells and lymphocytes [298]. In T cells, PD-1 engagement by PD-L1/PD-L2 triggers several immunosuppressive mechanisms by interfering with both downstream TCR and CD28 signalling, while directly inducing the expression of regulatory transcription factors such as basic leucine zipper transcriptional factor ATF-like (BATF) [299]. In the TME, PD-1 is highly expressed on tumour-infiltrating lymphocytes, where it is commonly associated with a population of dysfunctional or “exhausted” T cells that co-express multiple immune checkpoint molecules and display a unique epigenetic landscape compared with effector and memory T cells [300]. The expression of PD-L1 on tumour cells has been studied extensively as a mechanism of subverting T cell immunity, which has been reported in a variety of solid cancers [301] and often associated with poor overall survival [302]. The success of targeting the PD-1/PD-L1 signalling axis in pre-clinical studies lead to the prompt development mAbs against this pathway in the clinic. Pembrolizumab (Keytruda; Merck) is a humanized IgG4 αPD-1 monoclonal antibody that was the first PD-1 targeting immunotherapy to receive FDA approval in 2014 for treatment of patient with advanced melanoma [303,304]. Since then, PD-1/PD-L1 inhibitors, commonly paired with CTLA-4 blockade (ipilimumab) have been used extensively across many advanced solid cancers with modest clinical efficacy (Table 2). Despite its success, resistance to ICB is routinely observed among cancer patients, where lower levels of mutation burden, and reduced intra-tumoural PD-1/PD-L1 and MHC-I expression are common drivers of ICB resistance [305]. In addition, the use of αPD-1/PD-L1 upregulates alternative checkpoint molecules such as TIM-3 which may also be targetable in combination therapies [306]. Interestingly, chronic IFN signalling in the TME leads to the upregulation of tumour-derived inhibitory molecules such as PD-L1 which act to resist ICB and promote CTL dysfunction [307]. While IFNs have proven to be a critical axis in eliciting anti-tumour immunity, it is conceivable that sustained IFN signalling confers several adaptive resistance programs to ICB due its ability to exert strong selective pressures.
9. Conclusions
Solid tumours display remarkable heterogeneity both within and across various cancer types, which reflects the diversity of response rates to immunotherapy between patients [308]. Current advances in precision medicine have enabled the stratification of those that are more likely to benefit from immunotherapy. For instance, the quantitative measurement of CD3+ T cells within the tumour core and invasive margin has been useful in predicting clinical responses to treatment in patients with colorectal cancer [309], while other work has shown that an absence of intra-tumoural PD-L1 expression is more likely to blunt the therapeutic effects of αPD-1 ICB [310]. Another well-recognised predictor of treatment response also includes tumour mutational burden (TMB), where cancer types that exhibit higher rates of TMB, such as melanoma and cutaneous squamous cell tend to benefit the most from ICB [308]. These metrics do have their limitations and still cannot account for the vast majority of individuals that fail to respond to therapy. However, with the advent of next generation sequencing (NGS), new insights can now be garnered by unveiling the complex intercellular networks at exquisite single-cell resolution in the TME. Although it is indisputable that some molecular pathways clearly subvert tumour immunosurveillance (i.e., hypoxia and inhibitory receptor expression); many have both pro- and anti-tumour activities. It is therefore intuitive that a ‘hot’ and ‘cold’ TME is an overly simplistic binary representation of the local immune contexture and that additional tumour subtypes across this spectrum need to be investigated further (Figure 4). Our review highlights the necessity of balancing pro- and anti-inflammation in the TME to mobilise the host immune system against cancer and establish long-term anti-tumour immunity. With the use of modern molecular profiling techniques, the identification of patients with varying degrees of tumour inflammation, including those that are potentially ‘overheated’ will lead to the improvement of more personalised therapeutics that maximise clinical responses for patients with advanced solid tumours.
Funding
The preparation of this review article received no external funding. This work was supported by Australian Government Research Training Program Scholarship at The University of Western Australia (scholarship to H.V.N.), the Cancer Council Western Australia (fellowships to A.M. and J.W.), Telethon Kids Supporting Research Leaders Scheme (grant funding to A.B.) and Brady Cancer Support Foundation Inc (grant funding to J.W.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the preparation or writing of the manuscript or in the decision to publish this review.
References
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, L.L.; Gorris, M.A.J.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Damotte, D.; Warren, S.; Arrondeau, J.; Boudou-Rouquette, P.; Mansuet-Lupo, A.; Biton, J.; Ouakrim, H.; Alifano, M.; Gervais, C.; Bellesoeur, A.; et al. The tumor inflammation signature (TIS) is associated with anti-PD-1 treatment benefit in the CERTIM pan-cancer cohort. J. Transl. Med. 2019, 17, 357. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Huang, R.-R.; Wen, D.-R.; Paul, E.; Wünsch, P.H.; Cochran, A.J. IL-10 expression by primary tumor cells correlates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011, 24, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Balakrishnan, L.; Sharma, K.; Khan, A.A.; Advani, J.; Gowda, H.; Tripathy, S.P.; Suar, M.; Pandey, A.; Gandotra, S.; et al. A network map of Interleukin-10 signaling pathway. J. Cell Commun. Signal. 2016, 10, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, D.F.; Zlotnik, A.; Vieira, P.; Mosmann, T.R.; Howard, M.; Moore, K.W.; Garra, A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 1991, 146, 3444. [Google Scholar]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815. [Google Scholar]
- Kim, B.-G.; Joo, H.-G.; Chung, I.-S.; Chung, H.Y.; Woo, H.-J.; Yun, Y.-S. Inhibition of interleukin-10 (IL-10) production from MOPC 315 tumor cells by IL-10 antisense oligodeoxynucleotides enhances cell-mediated immune responses. Cancer Immunol. Immunother. 2000, 49, 433–440. [Google Scholar] [CrossRef]
- Steinbrink, K.; Jonuleit, H.; Müller, G.; Schuler, G.; Knop, J.R.; Enk, A.H. Interleukin-10–Treated Human Dendritic Cells Induce a Melanoma-Antigen–Specific Anergy in CD8+ T Cells Resulting in a Failure to Lyse Tumor Cells. Blood 1999, 93, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 Directly Inhibits CD8(+) T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 2018, 48, 299–312.e295. [Google Scholar] [CrossRef] [PubMed]
- Loercher, A.E.; Nash, M.A.; Kavanagh, J.J.; Platsoucas, C.D.; Freedman, R.S. Identification of an IL-10-Producing HLA-DR-Negative Monocyte Subset in the Malignant Ascites of Patients with Ovarian Carcinoma That Inhibits Cytokine Protein Expression and Proliferation of Autologous T Cells. J. Immunol. 1999, 163, 6251. [Google Scholar] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, D.; Wu, P.; Wang, Z.; Huang, J. Serum IL-10 Predicts Worse Outcome in Cancer Patients: A Meta-Analysis. PLoS ONE 2015, 10, e0139598. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, R.; Cao, Y.; Gu, Y.; Lin, C.; Liu, X.; Lv, K.; He, X.; Fang, H.; Jin, K.; et al. Poor Clinical Outcomes and Immunoevasive Contexture in Intratumoral IL-10-Producing Macrophages Enriched Gastric Cancer Patients. Ann. Surg. 2020. [Google Scholar] [CrossRef]
- Vahl, J.M.; Friedrich, J.; Mittler, S.; Trump, S.; Heim, L.; Kachler, K.; Balabko, L.; Fuhrich, N.; Geppert, C.-I.; Trufa, D.I.; et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br. J. Cancer 2017, 117, 1644–1655. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Zhang, L.; Yang, X.; Li, H.; Ma, B.; Walton, S.; Wu, X.; Yuan, J.; Wang, T.; Liu, X. Targeting interleukin-10 signalling for cancer immunotherapy, a promising and complicated task. Hum. Vaccines Immunother. 2020, 16, 2328–2332. [Google Scholar] [CrossRef]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 Elicits IFNg-Dependent Tumor Immune Surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef] [Green Version]
- Naing, A.; Wong, D.J.; Infante, J.R.; Korn, W.M.; Aljumaily, R.; Papadopoulos, K.P.; Autio, K.A.; Pant, S.; Bauer, T.M.; Drakaki, A.; et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): A multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019, 20, 1544–1555. [Google Scholar] [CrossRef]
- Teixeira, A.F.; ten Dijke, P.; Zhu, H.-J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-b directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Fox, N.; Phillips, M.; Alice, A.; Blair, T.; Whiteford, M.; O’Brien, D.; Ahmad, R.; et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020, 11, 1749. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 Is Essential for TGF-β to Convert Naive CD4+CD25- Cells to CD25+Foxp3+ Regulatory T Cells and for Expansion of These Cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.R.; Lee, W.; Cho, S.K.; Park, S.G. Characterization of Multiple Cytokine Combinations and TGF-β on Differentiation and Functions of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2018, 19, 869. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-R.; Kwak, Y.; Yang, T.; Han, J.H.; Park, S.-H.; Ye, M.B.; Lee, W.; Sim, K.-Y.; Kang, J.-A.; Kim, Y.-C.; et al. Myeloid-Derived Suppressor Cells Are Controlled by Regulatory T Cells via TGF-b during Murine Colitis. Cell Rep. 2016, 17, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.R.; Wang, Y.X.; Guo, S.; Han, S.Y.; Li, H.H.; Jin, S.F. Prognostic significance of in situ and plasma levels of transforming growth factor β1, -2 and -3 in cutaneous melanoma. Mol. Med. Rep. 2015, 11, 4508–4512. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shen, C.; Wang, X.; Lai, Y.; Zhou, K.; Li, P.; Liu, L.; Che, G. Prognostic value of TGF-β in lung cancer: Systematic review and meta-analysis. BMC Cancer 2019, 19, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Jiang, W.; Huang, W.; Ye, M.; Zhu, X. Prognostic Values of Transforming Growth Factor-Beta Subtypes in Ovarian Cancer. BioMed Res. Int. 2020, 2020, 2170606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, J.; Mao, K.; Deng, H.; Yang, Y.; Zhou, E.; Liu, J. Role of transforming growth factor-β1 in triple negative breast cancer patients. Int. J. Surg. 2017, 45, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef]
- Sow, H.S.; Ren, J.; Camps, M.; Ossendorp, F.; ten Dijke, P. Combined Inhibition of TGF-β Signaling and the PD-L1 Immune Checkpoint Is Differentially Effective in Tumor Models. Cells 2019, 8, 320. [Google Scholar] [CrossRef] [Green Version]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.M.; Palena, C.; Schlom, J. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Biswas, T.; Gu, X.; Yang, J.; Ellies, L.G.; Sun, L.-Z. Attenuation of TGF-β signaling supports tumor progression of a mesenchymal-like mammary tumor cell line in a syngeneic murine model. Cancer Lett. 2014, 346, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. OncoImmunology 2017, 6, e1362530. [Google Scholar] [CrossRef]
- Corgnac, S.; Boutet, M.; Kfoury, M.; Naltet, C.; Mami-Chouaib, F. The Emerging Role of CD8+ Tissue Resident Memory T (TRM) Cells in Antitumor Immunity: A Unique Functional Contribution of the CD103 Integrin. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef] [Green Version]
- Kabata, H.; Flamar, A.-L.; Mahlakõiv, T.; Moriyama, S.; Rodewald, H.-R.; Ziegler, S.F.; Artis, D. Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol. 2020, 13, 626–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.-S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza-Gonzalez, A.; Xu, K.; Wu, T.-C.; Aspord, C.; Tindle, S.; Marches, F.; Gallegos, M.; Burton, E.C.; Savino, D.; Hori, T.; et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J. Exp. Med. 2011, 208, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, R.B.; Gartner, J.G.; Leonard, W.J.; Ellison, C.A. Lack of Functional TSLP Receptors Mitigates Th2 Polarization and the Establishment and Growth of 4T1 Primary Breast Tumours but has Different Effects on Tumour Quantities in the Lung and Brain. Scand. J. Immunol. 2013, 78, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Sugaya, M.; Suga, H.; Oka, T.; Kawaguchi, M.; Miyagaki, T.; Fujita, H.; Sato, S. Thymic Stromal Chemokine TSLP Acts through Th2 Cytokine Production to Induce Cutaneous T-cell Lymphoma. Cancer Res. 2016, 76, 6241–6252. [Google Scholar] [CrossRef] [Green Version]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- De Monte, L.; Wörmann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M.; et al. Basophil Recruitment into Tumor-Draining Lymph Nodes Correlates with Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [Green Version]
- Ameri, A.H.; Moradi Tuchayi, S.; Zaalberg, A.; Park, J.H.; Ngo, K.H.; Li, T.; Lopez, E.; Colonna, M.; Lee, R.T.; Mino-Kenudson, M.; et al. IL-33/regulatory T cell axis triggers the development of a tumor-promoting immune environment in chronic inflammation. Proc. Natl. Acad. Sci. USA 2019, 116, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Pastille, E.; Wasmer, M.-H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, I.; Radosavljevic, G.; Mitrovic, M.; Lisnic Juranic, V.; McKenzie, A.N.J.; Arsenijevic, N.; Jonjic, S.; Lukic, M.L. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur. J. Immunol. 2011, 41, 1902–1912. [Google Scholar] [CrossRef] [Green Version]
- Andersson, P.; Yang, Y.; Hosaka, K.; Zhang, Y.; Fischer, C.; Braun, H.; Liu, S.; Yu, G.; Liu, S.; Beyaert, R.; et al. Molecular mechanisms of IL-33–mediated stromal interactions in cancer metastasis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Piazza, M.; Nowell, C.S.; Koch, U.; Durham, A.-D.; Radtke, F. Loss of Cutaneous TSLP-Dependent Immune Responses Skews the Balance of Inflammation from Tumor Protective to Tumor Promoting. Cancer Cell 2012, 22, 479–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochman, Y.; Leonard, W.J. The Role of Thymic Stromal Lymphopoietin in CD8+ T Cell Homeostasis. J. Immunol. 2008, 181, 7699–7705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shane, H.L.; Klonowski, K.D. A Direct and Nonredundant Role for Thymic Stromal Lymphopoietin on Antiviral CD8 T Cell Responses in the Respiratory Mucosa. J. Immunol. 2014, 192, 2261–2270. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Turkoz, A.; Manivasagam, S.; Yockey, L.J.; Turkoz, M.; Kopan, R. Elevated Epidermal Thymic Stromal Lymphopoietin Levels Establish an Antitumor Environment in the Skin. Cancer Cell 2012, 22, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Cunningham, T.J.; Manivasagam, S.; Ngo, K.H.; Moradi Tuchayi, S.; Reddy, R.; Meyers, M.A.; DeNardo, D.G.; Yokoyama, W.M. Thymic stromal lymphopoietin blocks early stages of breast carcinogenesis. J. Clin. Investig. 2016, 126, 1458–1470. [Google Scholar] [CrossRef] [Green Version]
- Kienzl, M.; Hasenoehrl, C.; Valadez-Cosmes, P.; Maitz, K.; Sarsembayeva, A.; Sturm, E.; Heinemann, A.; Kargl, J.; Schicho, R. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. OncoImmunology 2020, 9, 1776059. [Google Scholar] [CrossRef]
- Gao, X.; Wang, X.; Yang, Q.; Zhao, X.; Wen, W.; Li, G.; Lu, J.; Qin, W.; Qi, Y.; Xie, F.; et al. Tumoral Expression of IL-33 Inhibits Tumor Growth and Modifies the Tumor Microenvironment through CD8+ T and NK Cells. J. Immunol. 2015, 194, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Ohno, T.; Nishii, N.; Bhingare, A.; Tachinami, H.; Kashima, Y.; Nagai, S.; Saito, H.; Nakae, S.; Azuma, M. Endogenous IL-33 exerts CD8+ T cell antitumor responses overcoming pro-tumor effects by regulatory T cells in a colon carcinoma model. Biochem. Biophys. Res. Commun. 2019, 518, 331–336. [Google Scholar] [CrossRef]
- Yang, Q.; Li, G.; Zhu, Y.; Liu, L.; Chen, E.; Turnquist, H.; Zhang, X.; Finn, O.J.; Chen, X.; Lu, B. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur. J. Immunol. 2011, 41, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-C.; Hwang, Y.-S.; Chen, Y.-Y.; Liu, C.-L.; Shen, C.-N.; Hong, W.-H.; Lo, S.-M.; Shen, C.-R. Interleukin-4 Supports the Suppressive Immune Responses Elicited by Regulatory T Cells. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaynagetdinov, R.; Sherrill, T.P.; Gleaves, L.A.; McLoed, A.G.; Saxon, J.A.; Habermann, A.C.; Connelly, L.; Dulek, D.; Peebles, R.S.; Fingleton, B.; et al. Interleukin-5 Facilitates Lung Metastasis by Modulating the Immune Microenvironment. Cancer Res. 2015, 75, 1624–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salomé, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.-S.; et al. ILC2-modulated T cell–to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.; Mulders, P.; Punt, C.J.; et al. PD-1 blockade augments Th1 and Th17 and suppresses Th2 responses in peripheral blood from patients with prostate and advanced melanoma cancer. J. Immunother. 2012, 35, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, M.; Do, M.H.; Chou, C.; Stamatiades, E.G.; Nixon, B.G.; Shi, W.; Zhang, X.; Li, P.; Gao, S.; et al. Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature 2020, 587, 121–125. [Google Scholar] [CrossRef]
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-β suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120. [Google Scholar] [CrossRef]
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135. [Google Scholar] [CrossRef]
- Lorvik, K.B.; Hammarström, C.; Fauskanger, M.; Haabeth, O.A.W.; Zangani, M.; Haraldsen, G.; Bogen, B.; Corthay, A. Adoptive Transfer of Tumor-Specific Th2 Cells Eradicates Tumors by Triggering an In Situ Inflammatory Immune Response. Cancer Res. 2016, 76, 6864–6876. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, M.; Ito, T.; Tumes, D.J.; Endo, Y.; Onodera, A.; Hashimoto, K.; Motohashi, S.; Yamashita, M.; Nishimura, T.; Ziegler, S.F.; et al. Memory type 2 helper T cells induce long-lasting antitumor immunity by activating natural killer cells. Cancer Res. 2011, 71, 4790–4798. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [Green Version]
- Wagage, S.; John, B.; Krock, B.L.; Hall, A.O.H.; Randall, L.M.; Karp, C.L.; Simon, M.C.; Hunter, C.A. The Aryl Hydrocarbon Receptor Promotes IL-10 Production by NK Cells. J. Immunol. 2014, 192, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ye, Z.; Kijlstra, A.; Zhou, Y.; Yang, P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2014, 177, 521–530. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Riesenberg, R.; Weiler, C.; Spring, O.; Eder, M.; Buchner, A.; Popp, T.; Castro, M.; Kammerer, R.; Takikawa, O.; Hatz, R.A.; et al. Expression of Indoleamine 2,3-Dioxygenase in Tumor Endothelial Cells Correlates with Long-term Survival of Patients with Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 6993–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishio, T.; Goto, S.; Tahara, K.; Tone, S.; Kawano, K.; Kitano, S. Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2004, 19, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martine, E.D.C.; Arjan, A.v.d.L.; Corine, J.H.; Jeroen, J.W.M.J.; Adri, Z.; Ruud, D.; Peter, J.M.V.; Bob, L.; Gert, J.O. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. [Google Scholar] [CrossRef]
- Astigiano, S.; Morandi, B.; Costa, R.; Mastracci, L.; D’Agostino, A.; Ratto, G.B.; Melioli, G.; Frumento, G. Eosinophil Granulocytes Account for Indoleamine 2,3-Dioxygenase-Mediated Immune Escape in Human Non Small Cell Lung Cancer. Neoplasia 2005, 7, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Suda, T.; Furuhashi, K.; Suzuki, M.; Fujie, M.; Hahimoto, D.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer 2010, 67, 361–365. [Google Scholar] [CrossRef]
- Feder-Mengus, C.; Wyler, S.; Hudolin, T.; Ruszat, R.; Bubendorf, L.; Chiarugi, A.; Pittelli, M.; Weber, W.P.; Bachmann, A.; Gasser, T.C.; et al. High expression of indoleamine 2,3-dioxygenase gene in prostate cancer. Eur. J. Cancer 2008, 44, 2266–2275. [Google Scholar] [CrossRef] [Green Version]
- Ino, K.; Yoshida, N.; Kajiyama, H.; Shibata, K.; Yamamoto, E.; Kidokoro, K.; Takahashi, N.; Terauchi, M.; Nawa, A.; Nomura, S.; et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br. J. Cancer 2006, 95, 1555–1561. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandapathil, M.; Hilldorfer, B.; Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Ren, J.; Lang, S.; Jackson, E.K.; Gorelik, E.; Whiteside, T.L. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J. Biol. Chem. 2010, 285, 7176–7186. [Google Scholar] [CrossRef] [Green Version]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Ryzhov, S.; Novitskiy, S.V.; Goldstein, A.E.; Biktasova, A.; Blackburn, M.R.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Adenosinergic Regulation of the Expansion and Immunosuppressive Activity of CD11b+Gr1+ Cells. J. Immunol. 2011, 187, 6120–6129. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.P.; Möller, A.; Darcy, P.K.; Smyth, M.J. CD73-Deficient Mice Are Resistant to Carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Linden, J. Adenosine A2A Receptors Intrinsically Regulate CD8+ T Cells in the Tumor Microenvironment. Cancer Res. 2014, 74, 7239–7249. [Google Scholar] [CrossRef] [Green Version]
- Flores-Santibáñez, F.; Fernández, D.; Meza, D.; Tejón, G.; Vargas, L.; Varela-Nallar, L.; Arredondo, S.; Guixé, V.; Rosemblatt, M.; Bono, M.R.; et al. CD73-mediated adenosine production promotes stem cell-like properties in mouse Tc17 cells. Immunology 2015, 146, 582–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Yan, J.; Putheti, P.; Wu, Y.; Sun, X.; Toxavidis, V.; Tigges, J.; Kassam, N.; Enjyoji, K.; Robson, S.C.; et al. Isolated CD39 Expression on CD4+ T Cells Denotes both Regulatory and Memory Populations. Am. J. Transplant. 2009, 9, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.; Lin, X.; Gao, Y.; Zhu, Y. Prognositic value of CD73-adenosinergic pathway in solid tumor: A meta-analysis and systematic review. Oncotarget 2017, 8, 57327–57336. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Yoshimura, K.; Kurabe, N.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; Funai, K.; Shinmura, K.; et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget 2017, 8, 8738. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.K.; Sin, J.I.; Choi, J.; Park, S.H.; Lee, T.S.; Choi, Y.S. Overexpression of CD73 in epithelial ovarian carcinoma is associated with better prognosis, lower stage, better differentiation and lower regulatory T cell infiltration. J. Gynecol. Oncol. 2012, 23, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Supernat, A.; Markiewicz, A.; Welnicka-Jaskiewicz, M.; Seroczynska, B.; Skokowski, J.; Sejda, A.; Szade, J.; Czapiewski, P.; Biernat, W.; Zaczek, A. CD73 expression as a potential marker of good prognosis in breast carcinoma. Appl. Immunohistochem. Mol. Morphol. 2012, 20, 103–107. [Google Scholar] [CrossRef]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e2419. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Ekmekcioglu, S.; Grimm, E.A.; Roszik, J. Targeting iNOS to increase efficacy of immunotherapies. Hum. Vaccines Immunother. 2017, 13, 1105–1108. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: The two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef]
- Garrido, P.; Shalaby, A.; Walsh, E.M.; Keane, N.; Webber, M.; Keane, M.M.; Sullivan, F.J.; Kerin, M.J.; Callagy, G.; Ryan, A.E.; et al. Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 2017, 8, 80568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-N.; Hsieh, F.-J.; Cheng, Y.-M.; Chang, K.-J.; Lee, P.-H. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in angiogenesis and clinical outcome of human gastric cancer. J. Surg. Oncol. 2006, 94, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Amunni, G.; Villanucci, A.; Boddi, V.; Baroni, G.; Taddei, A.; Taddei, G.L. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in ovarian cancer: Correlation with clinical outcome. Gynecol. Oncol. 2004, 92, 806–812. [Google Scholar] [CrossRef]
- Ambs, S.; Merriam, W.G.; Bennett, W.P.; Felley-Bosco, E.; Ogunfusika, M.O.; Oser, S.M.; Klein, S.; Shields, P.G.; Billiar, T.R.; Harris, C.C. Frequent nitric oxide synthase-2 expression in human colon adenomas: Implication for tumor angiogenesis and colon cancer progression. Cancer Res. 1998, 58, 334–341. [Google Scholar] [PubMed]
- Chhatwal, V.J.S.; Ngoi, S.S.; Chan, S.T.F.; Chia, Y.W.; Moochhala, S.M. Aberrant expression of nitric oxide synthase in human polyps, neoplastic colonic mucosa and surrounding peritumoral normal mucosa. Carcinogenesis 1994, 15, 2081–2085. [Google Scholar] [CrossRef] [PubMed]
- Anttila, M.A.; Voutilainen, K.; Merivalo, S.; Saarikoski, S.; Kosma, V.-M. Prognostic significance of iNOS in epithelial ovarian cancer. Gynecol. Oncol. 2007, 105, 97–103. [Google Scholar] [CrossRef]
- Ropponen, K.M.; Kellokoski, J.K.; Lipponen, P.K.; Eskelinen, M.J.; Alanne, L.; Alhava, E.M.; Kosma, V.M. Expression of Inducible Nitric Oxide Synthase in Colorectal Cancer and Its Association with Prognosis. Scand. J. Gastroenterol. 2000, 35, 1204–1211. [Google Scholar] [CrossRef]
- Bingisser, R.M.; Tilbrook, P.A.; Holt, P.G.; Kees, U.R. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J. Immunol. 1998, 160, 5729–5734. [Google Scholar]
- Olivier, H.; James, K.L. Inhibition of MHC II Gene Transcription by Nitric Oxide and Antioxidants. Curr. Pharm. Des. 2004, 10, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Rivoltini, L.; Carrabba, M.; Huber, V.; Castelli, C.; Novellino, L.; Dalerba, P.; Mortarini, R.; Arancia, G.; Anichini, A.; Fais, S.; et al. Immunity to cancer: Attack and escape in T lymphocyte–tumor cell interaction. Immunol. Rev. 2002, 188, 97–113. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Ambs, S.; Merriam, W.G.; Ogunfusika, M.O.; Bennett, W.P.; Ishibe, N.; Hussain, S.P.; Tzeng, E.E.; Geller, D.A.; Billiar, T.R.; Harris, C.C. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat. Med. 1998, 4, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Zhu, C.; Li, F.; Hegazi, R.; He, K.; Babyatsky, M.; Bauer, A.J.; Plevy, S.E. Inhibition of interleukin-12 p40 transcription and NF-kappaB activation by nitric oxide in murine macrophages and dendritic cells. J. Biol. Chem. 2004, 279, 10776–10783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, I.; Zilio, S.; Desantis, G.; Mlecnik, B.; Agnellini, A.H.; Ugel, S.; Sasso, M.S.; Qualls, J.E.; Kratochvill, F.; Zanovello, P.; et al. T Cell Cancer Therapy Requires CD40-CD40L Activation of Tumor Necrosis Factor and Inducible Nitric-Oxide-Synthase-Producing Dendritic Cells. Cancer Cell 2016, 30, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an iNOS+/M1 Phenotype that Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, P.; Alfarano, M.G.; Svider, P.F.; Parikh, F.; Lu, G.; Kidwai, S.; Xiong, H.; Sikora, A.G. iNOS expression in CD4+ T cells limits Treg induction by repressing TGFβ1: Combined iNOS inhibition and Treg depletion unmask endogenous antitumor immunity. Clin. Cancer Res. 2014, 20, 6439–6451. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Munder, M.; Eichmann, K.; Morán, J.M.; Centeno, F.; Soler, G.; Modolell, M. Th1/Th2-Regulated Expression of Arginase Isoforms in Murine Macrophages and Dendritic Cells. J. Immunol. 1999, 163, 3771–3777. [Google Scholar]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bron, L.; Jandus, C.; Andrejevic-Blant, S.; Speiser, D.E.; Monnier, P.; Romero, P.; Rivals, J.-P. Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int. J. Cancer 2013, 132, E85–E93. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Oguro, S.; Shimada, K.; Kosuge, T.; Zavada, J.; Kanai, Y.; Hiraoka, N. Arginase II Expressed in Cancer-Associated Fibroblasts Indicates Tissue Hypoxia and Predicts Poor Outcome in Patients with Pancreatic Cancer. PLoS ONE 2013, 8, e55146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef]
- Ma, Z.; Lian, J.; Yang, M.; Wuyang, J.; Zhao, C.; Chen, W.; Liu, C.; Zhao, Q.; Lou, C.; Han, J.; et al. Overexpression of Arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathol. Res. Pract. 2019, 215, 152383. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chen, W.; Chen, J.; Zheng, Q.; Dong, J.; Zhu, Y. The Oncogenic Role of ARG1 in Progression and Metastasis of Hepatocellular Carcinoma. BioMed Res. Int. 2018, 2018, 2109865. [Google Scholar] [CrossRef]
- Patil, M.D.; Bhaumik, J.; Babykutty, S.; Banerjee, U.C.; Fukumura, D. Arginine dependence of tumor cells: Targeting a chink in cancer’s armor. Oncogene 2016, 35, 4957–4972. [Google Scholar] [CrossRef]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef]
- Zea, A.H.; Rodriguez, P.C.; Culotta, K.S.; Hernandez, C.P.; DeSalvo, J.; Ochoa, J.B.; Park, H.-J.; Zabaleta, J.; Ochoa, A.C. l-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes. Cell. Immunol. 2004, 232, 21–31. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813. [Google Scholar] [CrossRef] [Green Version]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Martí i Líndez, A.-A.; Dunand-Sauthier, I.; Conti, M.; Gobet, F.; Núñez, N.; Hannich, J.T.; Riezman, H.; Geiger, R.; Piersigilli, A.; Hahn, K.; et al. Mitochondrial arginase-2 is a cell-autonomous regulator of CD8+ T cell function and antitumor efficacy. JCI Insight 2020, 4. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Tsai, F.Y.-C.; Bauer, T.M.; Muigai, L.; Liang, Y.; Bennett, M.K.; Orford, K.W.; Fu, S. CX-1158-101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. J. Clin. Oncol. 2017, 35, 3005. [Google Scholar] [CrossRef]
- Yau, T.; Cheng, P.N.; Chan, P.; Chan, W.; Chen, L.; Yuen, J.; Pang, R.; Fan, S.T.; Poon, R.T. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Investig. New Drugs 2013, 31, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Markovič, T.; Jakopin, Ž.; Dolenc, M.S.; Mlinarič-Raščan, I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today 2017, 22, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Kalinski, P. Regulation of Immune Responses by Prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Parikh, A.; Shapiro, G.I.; Varga, A.; Naing, A.; Meric-Bernstam, F.; Ataman, Ö.; Reyderman, L.; Binder, T.A.; Ren, M.; et al. First-in-human phase I study of immunomodulatory E7046, an antagonist of PGE2-receptor E-type 4 (EP4), in patients with advanced cancers. J. Immunother. Cancer 2020, 8, e000222. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Kim, H.T.; Nellore, A.; Patsoukis, N.; Petkova, V.; McDonough, S.; Politikos, I.; Nikiforow, S.; Soiffer, R.; Antin, J.H.; et al. Prostaglandin E2 promotes survival of naive UCB T cells via the Wnt/β-catenin pathway and alters immune reconstitution after UCBT. Blood Cancer J. 2014, 4, e178. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Guan, K.; Zhou, Y.; Wu, J.; Wang, Y.; Wang, W. Prostaglandin E2 signal inhibits T regulatory cell differentiation during allergic rhinitis inflammation through EP4 receptor. World Allergy Organ. J. 2019, 12, 100090. [Google Scholar] [CrossRef] [Green Version]
- Hooper, K.M.; Kong, W.; Ganea, D. Prostaglandin E2 inhibits Tr1 cell differentiation through suppression of c-Maf. PLoS ONE 2017, 12, e0179184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youlin, K.; Weiyang, H.; Simin, L.; Xin, G. Prostaglandin E(2) Inhibits Prostate Cancer Progression by Countervailing Tumor Microenvironment-Induced Impairment of Dendritic Cell Migration through LXRα/CCR7 Pathway. J. Immunol. Res. 2018, 2018, 5808962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierlich, P.; Lex, V.; Technau, A.; Keupp, A.; Morper, L.; Glunz, A.; Sennholz, H.; Rachor, J.; Sauer, S.; Marcu, A.; et al. Prostaglandin E2 in a TLR3- and 7/8-agonist-based DC maturation cocktail generates mature, cytokine-producing, migratory DCs but impairs antigen cross-presentation to CD8+ T cells. Cancer Immunol. Immunother. 2020, 69, 1029–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzai, A.C.; Wagner, T.; Audsley, K.M.; Newnes, H.V.; Barrett, L.W.; Barnes, S.; Wylie, B.C.; Stone, S.; McDonnell, A.; Fear, V.S.; et al. Diverse Anti-Tumor Immune Potential Driven by Individual IFNα Subtypes. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Hayakawa, Y.; Zerafa, N.; Sheehan, K.C.F.; Scott, B.; Schreiber, R.D.; Hertzog, P.; Smyth, M.J. Type I IFN Contributes to NK Cell Homeostasis, Activation, and Antitumor Function. J. Immunol. 2007, 178, 7540–7549. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-α Production by Plasmacytoid Dendritic Cells Favors Regulatory T-cell Expansion That May Contribute to Breast Cancer Progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [Green Version]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast cancer-derived transforming growth factor-β and tumor necrosis factor-α compromise interferon-α production by tumor-associated plasmacytoid dendritic cells. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The Efficacy of Radiotherapy Relies upon Induction of Type I Interferon–Dependent Innate and Adaptive Immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [Green Version]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, S.; Zhou, M.; Pan, Z.; Sun, R.; Chen, M.; Jiang, H.; Shi, B.; Luo, H.; Li, Z. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Su, T.; Zhang, Y.; Valerie, K.; Wang, X.Y.; Lin, S.; Zhu, G. STING activation in cancer immunotherapy. Theranostics 2019, 9, 7759–7771. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e1512. [Google Scholar] [CrossRef] [Green Version]
- Provance, O.K.; Lewis-Wambi, J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res. 2019, 21, 59. [Google Scholar] [CrossRef] [Green Version]
- Effern, M.; Glodde, N.; Braun, M.; Liebing, J.; Boll, H.N.; Yong, M.; Bawden, E.; Hinze, D.; van den Boorn-Konijnenberg, D.; Daoud, M.; et al. Adoptive T Cell Therapy Targeting Different Gene Products Reveals Diverse and Context-Dependent Immune Evasion in Melanoma. Immunity 2020, 53, 564–580.e569. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Brooks, J.M.; Menezes, A.N.; Ibrahim, M.; Archer, L.; Lal, N.; Bagnall, C.J.; von Zeidler, S.V.; Valentine, H.R.; Spruce, R.J.; Batis, N.; et al. Development and Validation of a Combined Hypoxia and Immune Prognostic Classifier for Head and Neck Cancer. Clin. Cancer Res. 2019, 25, 5315–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Wu, S.; Chen, X.; Ye, Y.; Weng, Y.; Pan, Y.; Chen, Z.; Chen, L.; Qiu, X.; Qiu, S. Characterization of Hypoxia Signature to Evaluate the Tumor Immune Microenvironment and Predict Prognosis in Glioma Groups. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, B.; Wang, X.; Guan, G.; Xin, Y.; Sun, Y.D.; Wang, J.H.; Guo, Y.; Zang, Y.J. Macrophage transcriptome modification induced by hypoxia and lactate. Exp. Ther. Med. 2019, 18, 4811–4819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Hammami, A.; Charpentier, T.; Smans, M.; Stäger, S. IRF-5-Mediated Inflammation Limits CD8+ T Cell Expansion by Inducing HIF-1α and Impairing Dendritic Cell Functions during Leishmania Infection. PLOS Pathog. 2015, 11, e1004938. [Google Scholar] [CrossRef]
- Yang, M.; Ma, C.; Liu, S.; Sun, J.; Shao, Q.; Gao, W.; Zhang, Y.; Li, Z.; Xie, Q.; Dong, Z.; et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology 2009, 128, e237–e249. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Chen, K.; Cheng, Y.; Liu, S.; Xia, M.; Chen, Y.; Zhu, H.; Li, Z.; Cao, X. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1α-Mediated Glycolysis. Immunity 2019, 50, 600–615.e615. [Google Scholar] [CrossRef] [Green Version]
- Blengio, F.; Raggi, F.; Pierobon, D.; Cappello, P.; Eva, A.; Giovarelli, M.; Varesio, L.; Bosco, M.C. The hypoxic environment reprograms the cytokine/chemokine expression profile of human mature dendritic cells. Immunobiology 2013, 218, 76–89. [Google Scholar] [CrossRef]
- Weigert, A.; Weichand, B.; Sekar, D.; Sha, W.; Hahn, C.; Mora, J.; Ley, S.; Essler, S.; Dehne, N.; Brüne, B. HIF-1α is a negative regulator of plasmacytoid DC development in vitro and in vivo. Blood 2012, 120, 3001–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Yu, Y.; Wang, L.; Zhu, Z.; Lu, R.; Yao, Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 2016, 7, 75763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhong, M.; Wang, C.; Xu, Y.; Gao, W.-Q.; Zhang, Y. CCL5-deficiency enhances intratumoral infiltration of CD8+ T cells in colorectal cancer. Cell Death Dis. 2018, 9, 766. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Vuillefroy de Silly, R.; Dietrich, P.-Y.; Walker, P.R. Hypoxia and antitumor CD8+ T cells: An incompatible alliance? OncoImmunology 2016, 5, e1232236. [Google Scholar] [CrossRef] [Green Version]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells In Vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-N.; Yang, J.-F.; Huang, D.-J.; Ni, H.-H.; Zhang, C.-X.; Zhang, L.; He, J.; Gu, J.-M.; Chen, H.-X.; Mai, H.-Q.; et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8+ T cells. Sci. Immunol. 2019, 4, eaap9520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγ receptor signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [Green Version]
- Peng, P.; Yan, Y.; Keng, S. Exosomes in the ascites of ovarian cancer patients: Origin and effects on anti-tumor immunity. Oncol. Rep. 2011, 25, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, S.; Zheng, X.; Zheng, P.; Fu, Y.; Wu, C.; Lu, B.; Ju, J.; Jiang, J. Immune suppressed tumor microenvironment by exosomes derived from gastric cancer cells via modulating immune functions. Sci. Rep. 2020, 10, 14749. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human Tumor-Derived Exosomes Selectively Impair Lymphocyte Responses to Interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Jian, S.-F.; Lin, Y.-S.; Pan, Y.-C.; Wu, C.-Y.; Kuo, P.-L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-Containing T-Regulatory-Cell-Derived Exosomes Suppress Pathogenic T Helper 1 Cells. Immunity 2014, 41, 89–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.M.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 6065. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Borin, T.F.; Ara, R.; Piranlioglu, R.; Achyut, B.R.; Korkaya, H.; Liu, Y.; Arbab, A.S. The critical immunosuppressive effect of MDSC-derived exosomes in the tumor microenvironment. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.-C.; Guo, S.-C. Role of extracellular vesicles in tumour microenvironment. Cell Commun. Signal. 2020, 18, 163. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- De Sousa Andrade, L.N.; Otake, A.H.; Cardim, S.G.B.; da Silva, F.I.; Ikoma Sakamoto, M.M.; Furuya, T.K.; Uno, M.; Pasini, F.S.; Chammas, R. Extracellular Vesicles Shedding Promotes Melanoma Growth in Response to Chemotherapy. Sci. Rep. 2019, 9, 14482. [Google Scholar] [CrossRef]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Romagnoli, G.G.; Zelante, B.B.; Toniolo, P.A.; Migliori, I.K.; Barbuto, J.A.M. Dendritic Cell-Derived Exosomes may be a Tool for Cancer Immunotherapy by Converting Tumor Cells into Immunogenic Targets. Front. Immunol. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pivarcsi, A.; Müller, A.; Hippe, A.; Rieker, J.; van Lierop, A.; Steinhoff, M.; Seeliger, S.; Kubitza, R.; Pippirs, U.; Meller, S.; et al. Tumor immune escape by the loss of homeostatic chemokine expression. Proc. Natl. Acad. Sci. USA 2007, 104, 19055–19060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, J.M.; Gomez, A.C.; Aguilar, A.; Salgado, R.; Balko, J.M.; Bravo, L.; Doimi, F.; Bretel, D.; Morante, Z.; Flores, C.; et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci. Rep. 2018, 8, 4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, F.; Ping, Y.; Wang, L.; Chen, X.; Wang, D.; Cao, L.; Zhao, S.; Li, B.; Kalinski, P.; et al. Local production of the chemokines CCL5 and CXCL10 attracts CD8 + T lymphocytes into esophageal squamous cell carcinoma. Oncotarget 2015, 6, 24978. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Alonso, M.; Hirsch, T.; Wildmann, C.; van der Bruggen, P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat. Commun. 2017, 8, 793. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Froemming, A.; Vater, A. Increasing tumor-infiltrating T cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Bouzin, C.; Brouet, A.; De Vriese, J.; DeWever, J.; Feron, O. Effects of Vascular Endothelial Growth Factor on the Lymphocyte-Endothelium Interactions: Identification of Caveolin-1 and Nitric Oxide as Control Points of Endothelial Cell Anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.A.; Darrah, P.A.; Foulds, K.E.; Hoffer, E.; Caffrey-Carr, A.; Norenstedt, S.; Perbeck, L.; Seder, R.A.; Kedl, R.M.; Loré, K. Monocytes Acquire the Ability to Prime Tissue-Resident T Cells via IL-10-Mediated TGF-b Release. Cell Rep. 2019, 28, 1127–1135.e1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Bevan, M.J. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, A.P.; Braun, A.; Ritchie, S.C.; Carbone, F.R.; Mackay, L.K.; Gebhardt, T.; Inouye, M. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 2019, 14, e0210495. [Google Scholar] [CrossRef]
- Harly, C.; Kenney, D.; Wang, Y.; Ding, Y.; Zhao, Y.; Awasthi, P.; Bhandoola, A. A Shared Regulatory Element Controls the Initiation of Tcf7 Expression During Early T Cell and Innate Lymphoid Cell Developments. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016, 1, eaai8593. [Google Scholar] [CrossRef] [Green Version]
- Danilo, M.; Chennupati, V.; Silva, J.G.; Siegert, S.; Held, W. Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 2018, 22, 2107–2117. [Google Scholar] [CrossRef] [Green Version]
- Wiesel, M.; Crouse, J.; Bedenikovic, G.; Sutherland, A.; Joller, N.; Oxenius, A. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur. J. Immunol. 2012, 42, 320–329. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; Lord, G.M.; Dardalhon, V.; Lee, D.H.; Sabatos-Peyton, C.A.; Glimcher, L.H.; Kuchroo, V.K. T-bet, a Th1 transcription factor regulates the expression of Tim-3. Eur. J. Immunol. 2010, 40, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sakuishi, K.; Xiao, S.; Sun, Z.; Zaghouani, S.; Gu, G.; Wang, C.; Tan, D.J.; Wu, C.; Rangachari, M.; et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 2015, 6, 6072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The transcription factor NFAT promotes exhaustion of activated CD8⁺ T cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 Expression Characterizes Regulatory T Cells in Tumor Tissues and Is Associated with Lung Cancer Progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Ngiow, S.F.; Sullivan, J.M.; Teng, M.W.L.; Kuchroo, V.K.; Smyth, M.J.; Anderson, A.C. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. OncoImmunology 2013, 2, e23849. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, Y.; Zhang, J.-P.; Liang, J.; Li, L.; Zheng, L. Tim-3 Expression Defines Regulatory T Cells in Human Tumors. PLoS ONE 2013, 8, e58006. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Qin, S.; Dong, B.; Yi, M.; Chu, Q.; Wu, K. Prognostic Values of TIM-3 Expression in Patients With Solid Tumors: A Meta-Analysis and Database Evaluation. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Patnaik, A.; Moreno, V.; Stein, M.; Jankowska, A.M.; Mendizabal, N.V.d.; Liu, Z.T.; Koneru, M.; Calvo, E. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J. Clin. Oncol. 2019, 37, 12. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, W.M.D.; Forde, P.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, S.; et al. Abstract CT183: Phase (Ph) I/II study of MBG453± spartalizumab (PDR001) in patients (pts) with advanced malignancies. Cancer Res. 2019, 79, CT183. [Google Scholar] [CrossRef]
- Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2019, 134, 570. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.-m.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, e001014. [Google Scholar] [CrossRef] [PubMed]
- Maçon-Lemaître, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005, 115, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Bruniquel, D.; Borie, N.; Hannier, S.; Triebel, F. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 1998, 48, 116–124. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [Green Version]
- Zelba, H.; Bedke, J.; Hennenlotter, J.; Mostböck, S.; Zettl, M.; Zichner, T.; Chandran, A.; Stenzl, A.; Rammensee, H.-G.; Gouttefangeas, C. PD-1 and LAG-3 Dominate Checkpoint Receptor–Mediated T-cell Inhibition in Renal Cell Carcinoma. Cancer Immunol. Res. 2019, 7, 1891–1899. [Google Scholar] [CrossRef]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.B.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 Expression Defines a Subset of CD4+CD25highFoxp3+ Regulatory T Cells That Are Expanded at Tumor Sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.-W.; Mao, L.; Yu, G.-T.; Bu, L.-L.; Ma, S.-R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.-F.; Sun, Z.-J. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. OncoImmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.R.; Peinado, P.; Fuentes-Antrás, J.; Pérez-Segura, P.; Pandiella, A.; Amir, E.; Ocaña, A. Prognostic Value of Lymphocyte-Activation Gene 3 (LAG3) in Cancer: A Meta-Analysis. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.; Ulahannan, S.V.; Chu, Q.; Patel, M.; George, B.; Auguste, A.; Leo-Kress, T.; Stadermann, K.B.; Kraemer, N.; Elgadi, M.; et al. Abstract 779: A phase I study of BI 754111, an anti-LAG-3 monoclonal antibody (mAb), in combination with BI 754091, an anti-PD-1 mAb: Biomarker analyses from the microsatellite stable metastatic colorectal cancer (MSS mCRC) cohort. Cancer Res. 2020, 80, 779. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Lakhani, N.J.; Johnson, M.L.; Park, H.; Wang, D.; Yap, T.A.; Dowlati, A.; Maki, R.G.; Lynce, F.; Ulahannan, S.V.; et al. First-in-human study of REGN3767 (R3767), a human LAG-3 monoclonal antibody (mAb), ± cemiplimab in patients (pts) with advanced malignancies. J. Clin. Oncol. 2019, 37, 2508. [Google Scholar] [CrossRef]
- Casati, C.; Camisaschi, C.; Rini, F.; Arienti, F.; Rivoltini, L.; Triebel, F.; Parmiani, G.; Castelli, C. Soluble Human LAG-3 Molecule Amplifies the In vitro Generation of Type 1 Tumor-Specific Immunity. Cancer Res. 2006, 66, 4450–4460. [Google Scholar] [CrossRef] [Green Version]
- Prigent, P.; El mir, S.; Dréano, M.; Triebel, F. Lymphocyte activation gene-3 induces tumor regression and antitumor immune responses. Eur. J. Immunol. 1999, 29, 3867–3876. [Google Scholar] [CrossRef]
- El mir, S.; Triebel, F. A Soluble Lymphocyte Activation Gene-3 Molecule Used as a Vaccine Adjuvant Elicits Greater Humoral and Cellular Immune Responses to Both Particulate and Soluble Antigens. J. Immunol. 2000, 164, 5583–5589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Jilisihan, B.; Wang, W.; Tang, Y.; Keyoumu, S. Soluble LAG3 acts as a potential prognostic marker of gastric cancer and its positive correlation with CD8+T cell frequency and secretion of IL-12 and INF-γ in peripheral blood. Cancer Biomark. 2018, 23, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, V.; Khattak, A.; Haydon, A.; Eastgate, M.; Roy, A.; Prithviraj, P.; Mueller, C.; Brignone, C.; Triebel, F. Eftilagimod alpha, a soluble lymphocyte activation gene-3 (LAG-3) protein plus pembrolizumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001681. [Google Scholar] [CrossRef] [PubMed]
- Legat, A.; Maby-El Hajjami, H.; Baumgaertner, P.; Cagnon, L.; Abed Maillard, S.; Geldhof, C.; Iancu, E.M.; Lebon, L.; Guillaume, P.; Dojcinovic, D.; et al. Vaccination with LAG-3Ig (IMP321) and Peptides Induces Specific CD4 and CD8 T-Cell Responses in Metastatic Melanoma Patients—Report of a Phase I/IIa Clinical Trial. Clin. Cancer Res. 2016, 22, 1330–1340. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Q.; Wang, Z.; Zhang, H.; Zeng, H.; Huang, Q.; Chen, Y.; Jiang, W.; Lin, Z.; Qu, Y.; et al. Intratumoral TIGIT+ CD8+ T-cell infiltration determines poor prognosis and immune evasion in patients with muscle-invasive bladder cancer. J. Immunother. Cancer 2020, 8, e000978. [Google Scholar] [CrossRef]
- Xu, D.; Zhao, E.; Zhu, C.; Zhao, W.; Wang, C.; Zhang, Z.; Zhao, G. TIGIT and PD-1 may serve as potential prognostic biomarkers for gastric cancer. Immunobiology 2020, 225, 151915. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, J.; Chen, Y.; Cui, J.; Lei, Y.; Cui, Y.; Jiang, N.; Jiang, W.; Chen, L.; Chen, Y.; et al. Combined evaluation of the expression status of CD155 and TIGIT plays an important role in the prognosis of LUAD (lung adenocarcinoma). Int. Immunopharmacol. 2020, 80, 106198. [Google Scholar] [CrossRef]
- Duan, X.; Liu, J.; Cui, J.; Ma, B.; Zhou, Q.; Yang, X.; Lu, Z.; Du, Y.; Su, C. Expression of TIGIT/CD155 and correlations with clinical pathological features in human hepatocellular carcinoma. Mol. Med. Rep. 2019, 20, 3773–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroumov, D.; Duong, S.; Wingerath, J.; Woller, N.; Manns, M.P.; Timrott, K.; Kleine, M.; Ramackers, W.; Roessler, S.; Nahnsen, S.; et al. Transcriptome profiling identifies TIGIT as a marker of T cell exhaustion in liver cancer. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus, S.; Sakuishi, K.; Ngiow, S.-F.; Joller, N.; Tan, D.J.; Teng, M.W.L.; Smyth, M.J.; Kuchroo, V.K.; Anderson, A.C. TIGIT predominantly regulates the immune response via regulatory T cells. J. Clin. Investig. 2015, 125, 4053–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepletier, A.; Madore, J.; O’Donnell, J.S.; Johnston, R.L.; Li, X.-Y.; McDonald, E.; Ahern, E.; Kuchel, A.; Eastgate, M.; Pearson, S.-A.; et al. Tumor CD155 expression is associated with resistance to anti-PD1 immunotherapy in metastatic melanoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Aguilera, A.R.; Sundarrajan, A.; Corvino, D.; Stannard, K.; Krumeich, S.; Das, I.; Lima, L.G.; Meza Guzman, L.G.; Li, K.; et al. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8(+) T Cells. Immunity 2020, 53, 805–823.e815. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-h.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Stamper, C.C.; Zhang, Y.; Tobin, J.F.; Erbe, D.V.; Ikemizu, S.; Davis, S.J.; Stahl, M.L.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 2001, 410, 608–611. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4–Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Liu, Q.; Deng, G.; Zhang, J.; Liang, N.; Xie, J.; Zhang, J. The prognostic value of cytotoxic T-lymphocyte antigen 4 in cancers: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 42913. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Salmons, J.; Nowak, A.K.; Rozali, E.N.; Khong, A.; Dick, I.M.; Harken, J.A.; Robinson, B.W.; Lake, R.A. Synergistic Effect of CTLA-4 Blockade and Cancer Chemotherapy in the Induction of Anti-Tumor Immunity. PLoS ONE 2013, 8, e61895. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kim, D.W.; Bassett, R.L.; Cain, S.; Washington, E.; Hwu, W.J.; Kim, K.B.; Papadopoulos, N.E.; Homsi, J.; Hwu, P.; et al. A phase II study of ipilimumab plus temozolomide in patients with metastatic melanoma. Cancer Immunol. Immunother. 2017, 66, 1359–1366. [Google Scholar] [CrossRef]
- Qian, J.M.; Martin, A.M.; Martin, K.; Hammoudeh, L.; Catalano, P.J.; Hodi, F.S.; Cagney, D.N.; Haas-Kogan, D.A.; Schoenfeld, J.D.; Aizer, A.A. Response rate and local recurrence after concurrent immune checkpoint therapy and radiotherapy for non-small cell lung cancer and melanoma brain metastases. Cancer 2020, 126, 5274–5282. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Chaput-Gras, N.; Lanoy, E.; Larive, A.; Mateus, C.; Routier, E.; Sun, R.; Tao, Y.G.; Massard, C.; Bahleda, R.; et al. Dose escalation phase 1 study of radiotherapy in combination with anti-cytotoxic-T-lymphocyte-associated antigen 4 monoclonal antibody ipilimumab in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, A.A.; Yu, T.F.-Y.; Leach, D.R.; Allison, J.P. CTLA-4 blockade synergizes with tumor-derived granulocyte– macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 10067–10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, M.A.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J.P. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kojima, Y.; Uno, T.; Hayakawa, Y.; Teng, M.W.L.; Yoshizawa, H.; Yagita, H.; Gejyo, F.; Okumura, K.; Smyth, M.J. Combination Therapy of Established Tumors by Antibodies Targeting Immune Activating and Suppressing Molecules. J. Immunol. 2010, 184, 5493–5501. [Google Scholar] [CrossRef] [Green Version]
- Redmond, W.L.; Linch, S.N.; Kasiewicz, M.J. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol. Res. 2014, 2, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Zak, K.M.; Grudnik, P.; Magiera, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: Meta-analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandini, S.; Massi, D.; Mandalà, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2016, 100, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M. Pembrolizumab: First Global Approval. Drugs 2014, 74, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Shergold, A.L.; Millar, R.; Nibbs, R.J.B. Understanding and overcoming the resistance of cancer to PD-1/PD-L1 blockade. Pharmacol. Res. 2019, 145, 104258. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- López-Soto, A.; Gonzalez, S.; Folgueras, A.R. IFN Signaling and ICB Resistance: Time is on Tumor’s Side. Trends Cancer 2017, 3, 161–163. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2019, 26, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The immune circuitry within ‘cold’ tumour niches is governed by soluble factors such as cytokines, growth factors and enzyme-regulated metabolites. Immune and stromal cell types send and receive signals within the tumour milieu which culminates in the generation of an immune privileged TME that impairs anti-tumour immunity. Abbreviations: T2-C: type-II inflammatory cytokines, IDO: indoleamine 2,3-dioxygenase, TGF-B: transforming growth factor β, ADO: adenosine, ARG1: arginase-1, iNOS: inducible nitric oxide synthase, and IL-10: interleukin-10.
Figure 1.
The immune circuitry within ‘cold’ tumour niches is governed by soluble factors such as cytokines, growth factors and enzyme-regulated metabolites. Immune and stromal cell types send and receive signals within the tumour milieu which culminates in the generation of an immune privileged TME that impairs anti-tumour immunity. Abbreviations: T2-C: type-II inflammatory cytokines, IDO: indoleamine 2,3-dioxygenase, TGF-B: transforming growth factor β, ADO: adenosine, ARG1: arginase-1, iNOS: inducible nitric oxide synthase, and IL-10: interleukin-10.

Figure 2.
The TME perturbs multiple mechanisms of T cell migration to avoid immune surveillance. Cancer cells can impede with CTL trafficking to the tumour bed at multiple levels including the loss of extravasation capacity, disrupted chemokine gradients and physical constraints including increased ECM deposition and poor oxygen availability.
Figure 2.
The TME perturbs multiple mechanisms of T cell migration to avoid immune surveillance. Cancer cells can impede with CTL trafficking to the tumour bed at multiple levels including the loss of extravasation capacity, disrupted chemokine gradients and physical constraints including increased ECM deposition and poor oxygen availability.

Figure 3.
Tumours with intense inflammation may perturb the homeostatic balance of memory-effector T cell populations in the TME. ‘Cold’ tumour niches contain an abundance of canonical immunosuppressive factors that create an immune-privileged TME. Conversely, ‘hot’ tumours that contain excessive amounts of pro-inflammatory factors may disrupt the balance of effector-memory CTL populations, resulting in short-lived effector responses. In contrast, a balance of pro- and anti-inflammatory signals in the TME may endow CTLs with improved cytolytic responses that are long lived.
Figure 3.
Tumours with intense inflammation may perturb the homeostatic balance of memory-effector T cell populations in the TME. ‘Cold’ tumour niches contain an abundance of canonical immunosuppressive factors that create an immune-privileged TME. Conversely, ‘hot’ tumours that contain excessive amounts of pro-inflammatory factors may disrupt the balance of effector-memory CTL populations, resulting in short-lived effector responses. In contrast, a balance of pro- and anti-inflammatory signals in the TME may endow CTLs with improved cytolytic responses that are long lived.

Figure 4.
The cellular composition of ‘hot’ and ‘cold’ tumours. Anti-tumour immunity in the TME depends on the presence of CTLs that are activated by endogenous IFN-I and other pro-inflammatory stimuli produced by neighbouring cells. However, these can be subverted by an imbalance of anti-inflammatory factors that also reduces their trafficking to the tumour site. Conversely, too much inflammation may impair the cytolytic activities of CTLs, triggering immune escape. This intense inflammation mediated by conventional anti-tumour factors such as IFN-Is also upregulate T cell inhibitory molecules (TCIMs) on tumour cells that drive the adaptive resistance to immunotherapy.
Figure 4.
The cellular composition of ‘hot’ and ‘cold’ tumours. Anti-tumour immunity in the TME depends on the presence of CTLs that are activated by endogenous IFN-I and other pro-inflammatory stimuli produced by neighbouring cells. However, these can be subverted by an imbalance of anti-inflammatory factors that also reduces their trafficking to the tumour site. Conversely, too much inflammation may impair the cytolytic activities of CTLs, triggering immune escape. This intense inflammation mediated by conventional anti-tumour factors such as IFN-Is also upregulate T cell inhibitory molecules (TCIMs) on tumour cells that drive the adaptive resistance to immunotherapy.


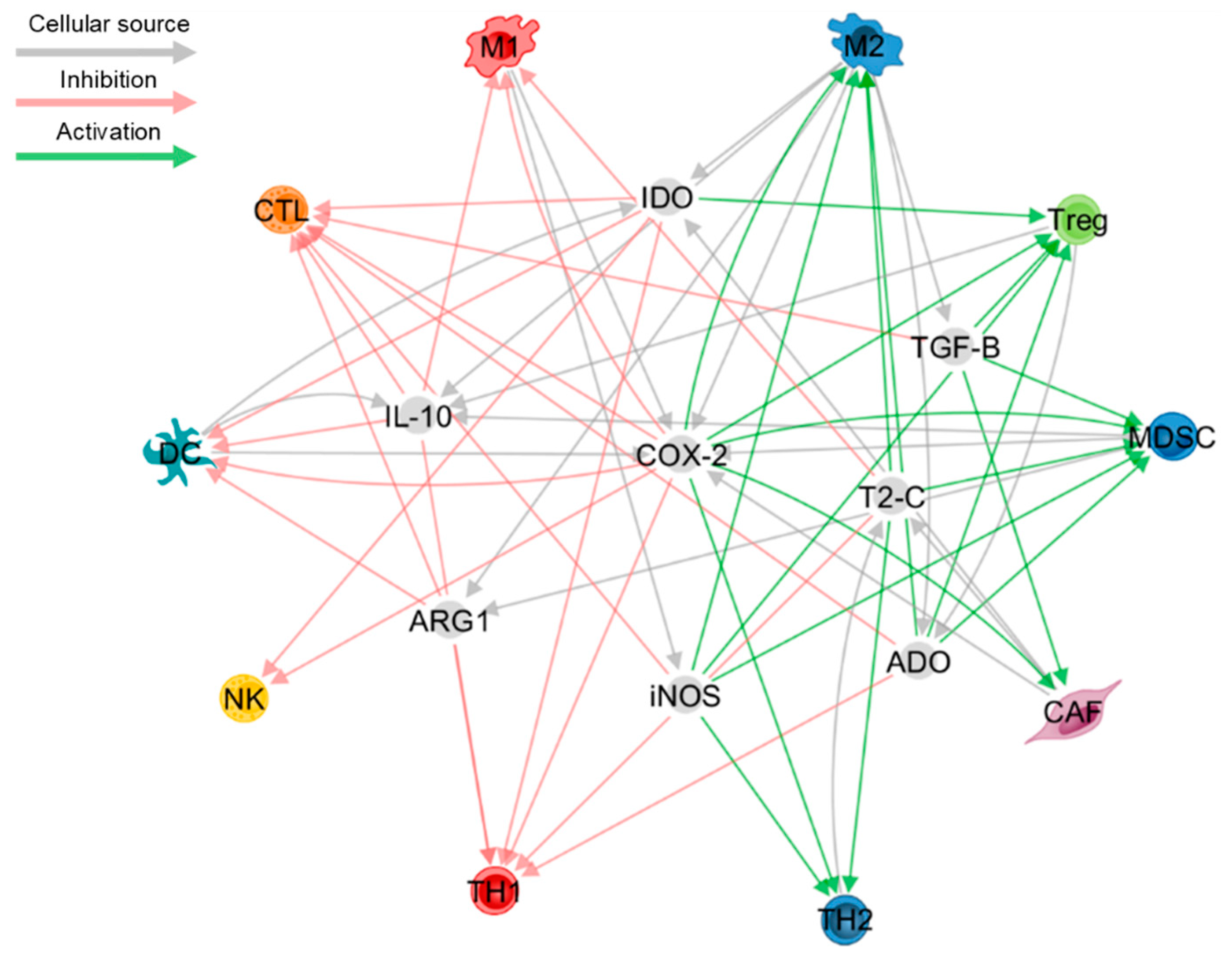{kind=link}
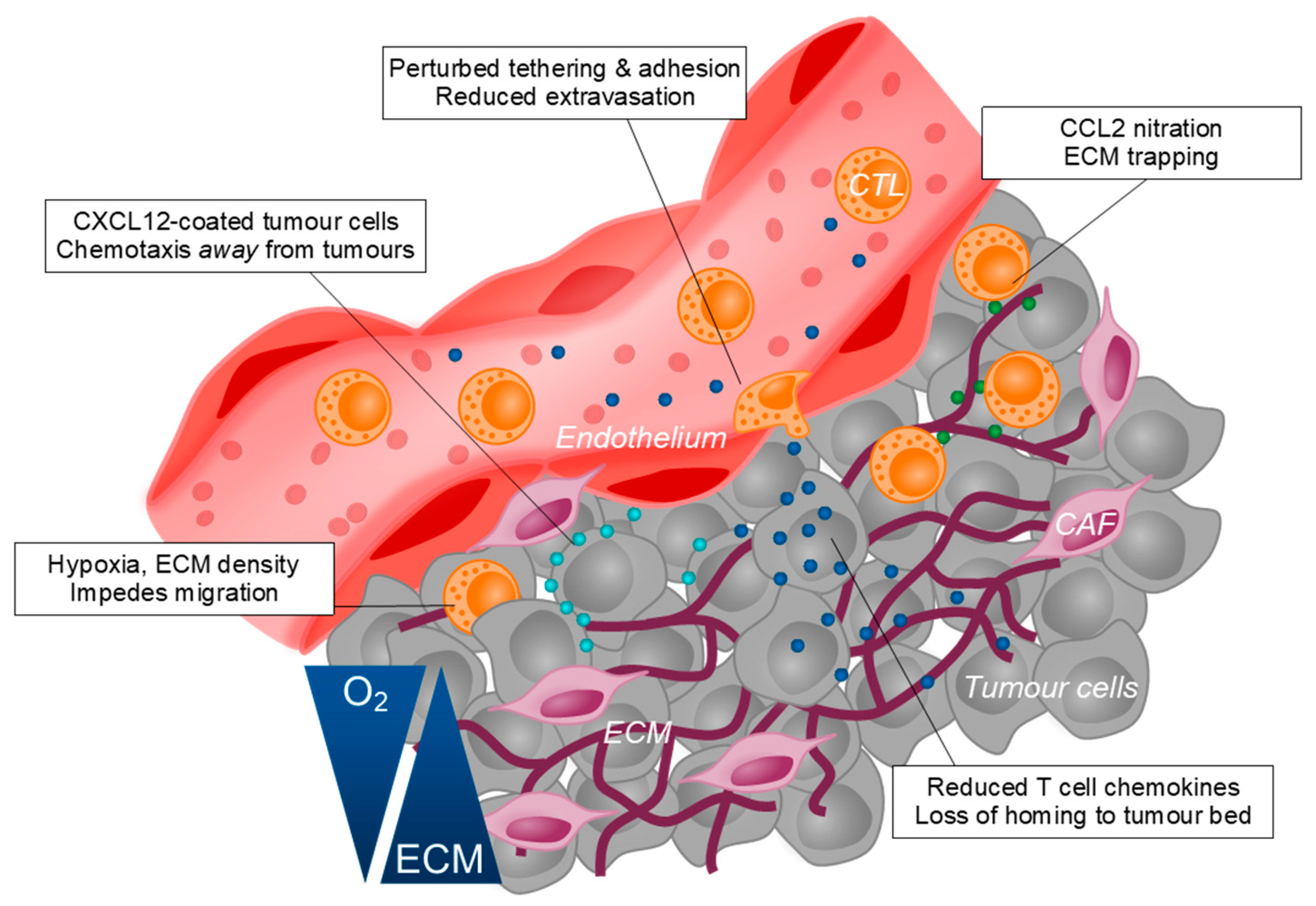{kind=link}
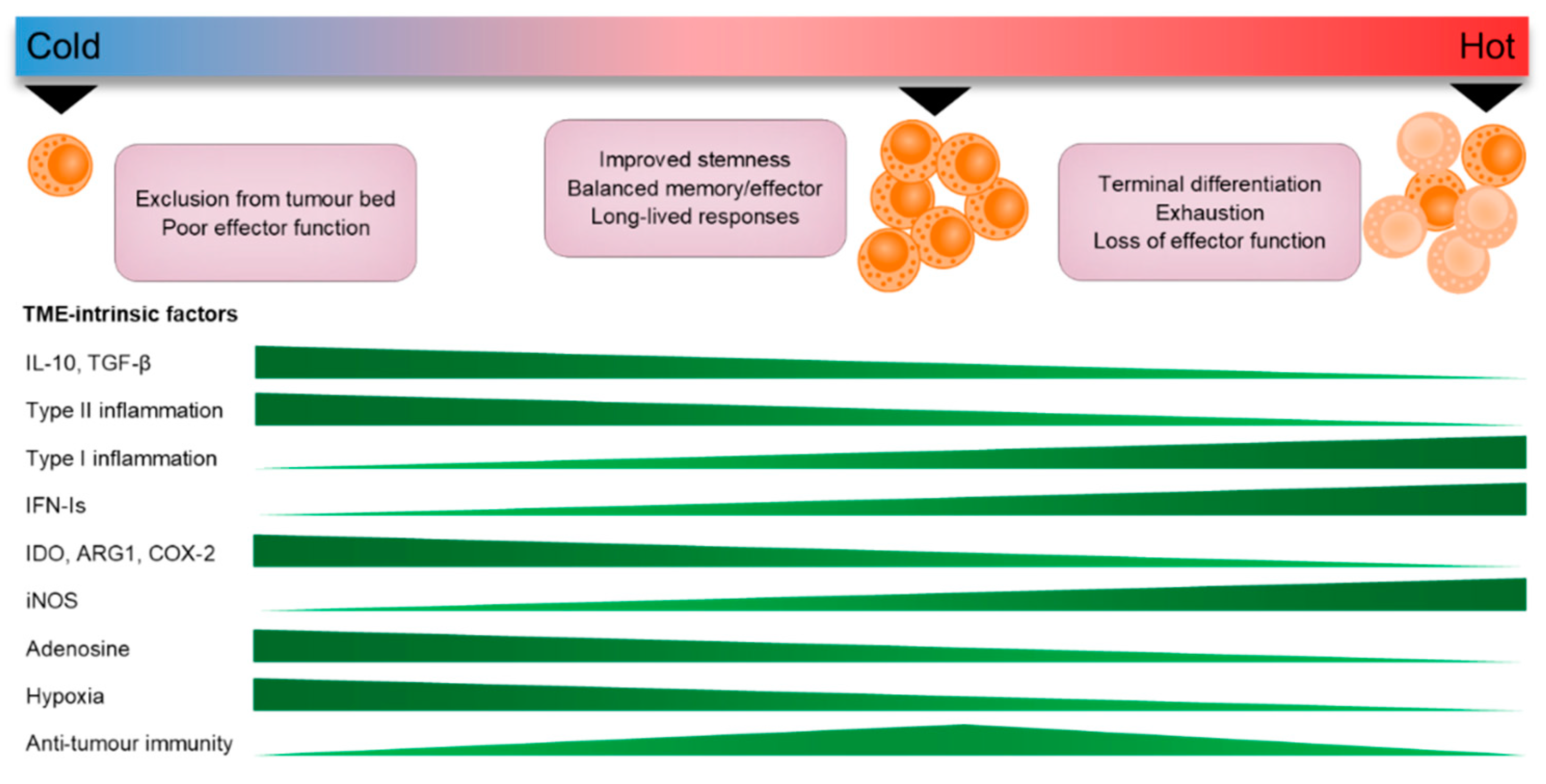{kind=link}
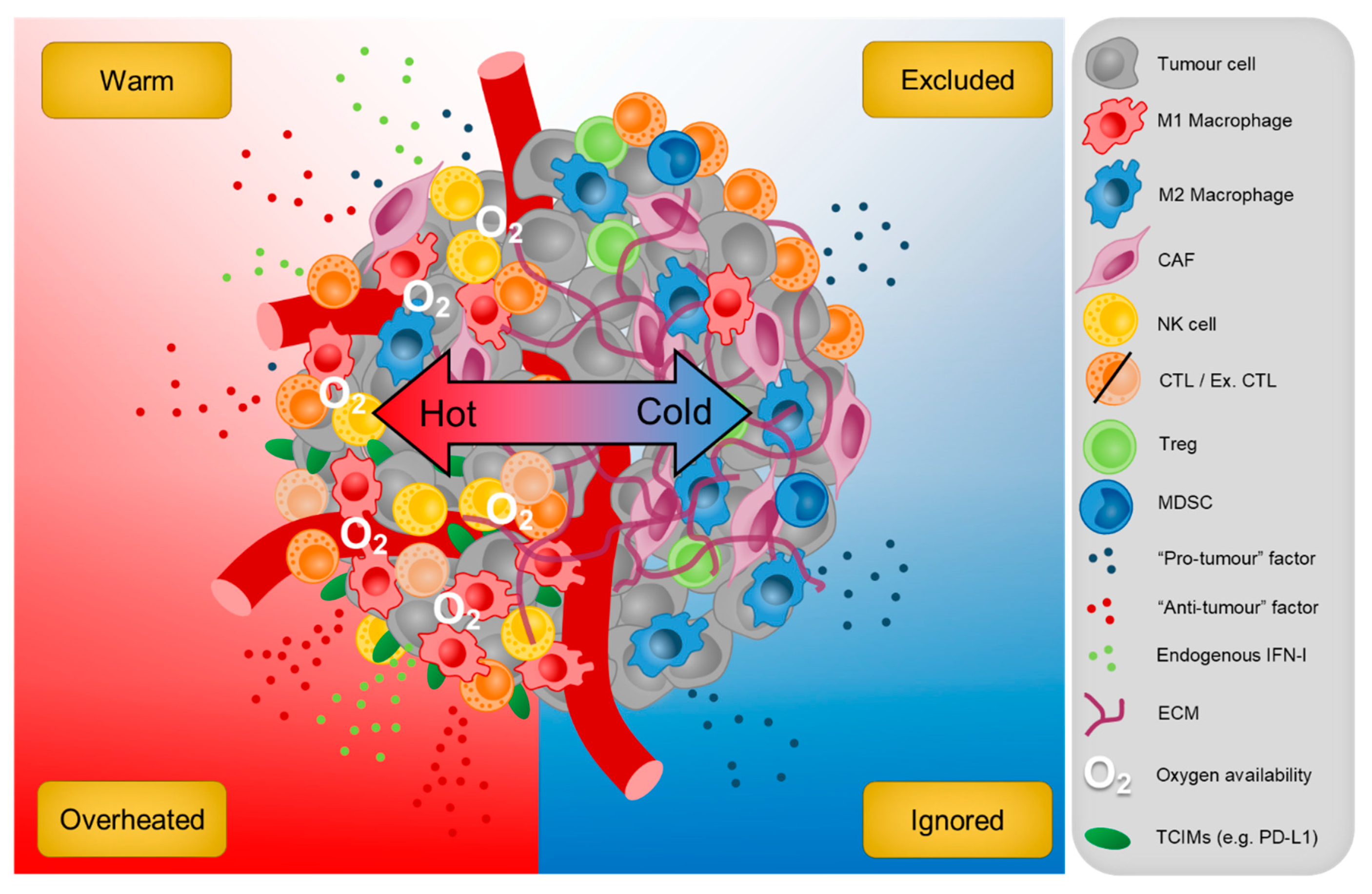{kind=link}
Table 1.
Clinical trials underway using therapeutics against TIM-3, LAG-3 and TIGIT (clinicaltrials.gov).
Table 1.
Clinical trials underway using therapeutics against TIM-3, LAG-3 and TIGIT (clinicaltrials.gov).
| Product | Description | Clinical Stage | Cancer(s) | Trial No. |
|---|---|---|---|---|
| A. TIM-3 | ||||
| Sym023 (Symphogen) | IgG1 mAb | 1 | Advanced solid cancer or lymphoma | NCT03489343 NCT03311412 |
| TSR-022 (Tesaro) | Humanised IgG4 mAb | 2 | HCC, melanoma, advanced solid cancers | NCT03680508 NCT02817633 NCT03307785 |
| LY3321367 (Eli Lilly and Co) | IgG1k, Fc silent | 1 | Melanoma, MSI-H and advanced solid cancer | NCT03099109 NCT02791334 |
| MBG453 (Novartis) | Humanised IgG4 mAb | 2 | AML, CMML-2, MDS, GBM | NCT04150029 NCT04266301 NCT03946670 NCT03066648 NCT03940352 NCT02608268 NCT03961971 |
| BGB-A425 (BeiGene) | IgG1, variant, Fc silent | 1/2 | Advanced/ Metastatic solid cancer | NCT03744468 |
| ICAGN02390 (Incyte) | IgG1k, N297A, Fc silent | 1 | Advanced solid cancer, melanoma | NCT03652077 NCT04370704 |
| BMS-986258 (Bristol-Myers Squibb) | IgG1, Fc silent | 1/2 | Advanced solid cancer | NCT03446040 |
| RO7121661 (Hoffman-La Roche) | PD-1/TIM-3 bispecific Ab | 1 | Melanoma, NSCLC, SCLC, ESCC, urothelial cancer | NCT03708328 NCT03869190 |
| B. LAG-3 | ||||
| IMP321 * (Immunopet) | LAG-3-Ig fusion protein | 1/2 | Breast cancer, advanced solid cancers, NSCLC, HNSCC | NCT02614833 NCT03252938 NCT03625323 |
| Relatlimab # (Bristol-Myers Squibb) | IgG4 mAb | 2 | Uveal melanoma, CRC, sarcoma, melanoma | NCT04552223 NCT03642067 NCT04095208 NCT03470922 |
| LAG525 (Novartis) | IgG4 mAb | 2 | Breast cancer, advanced solid cancer, melanoma | NCT03499899 NCT02460224 NCT03742349 NCT03484923 |
| MK-4280 (Merck) | Humanised IgG4 | 1/2 | HL, NHL, BCL, NSCLC, RCC, advanced solid cancer | NCT03598608 NCT02720068 NCT03516981 NCT04626479 NCT04626518 |
| REGN3767 (Regeneron) | IgG4 mAb | 1 | BC, advanced cancers | NCT03005782 NCT01042379 |
| TSR-033 (Tesaro) | Humanised IgG4 mAb | 1 | Advanced solid cancers | NCT03250832 NCT02817633 |
| Sym022 (Symphogen) | Fc inert mAb | 1 | Advanced solid cancer, lymphoma | NCT04641871 NCT03311412 |
| INCAGN02385 (InCyte) | Fc-engineered IgG1k | 1/2 | Melanoma | NCT04370704 |
| MGD013 (MacroGenics) | Humanised LAG3-PD-1 bispecific Ab | 1/2 | GC, HCC, advanced solid cancers | NCT04212221 NCT03219268 NCT04178460 |
| FS118 (F-star) | Humanised LAG3-PD-L1 bispecific Ab | 1 | Advanced solid cancer | NCT03440437 |
| C. TIGIT | ||||
| BMS-986207 (Bristol-Myers Squibb) | IgG1 mAb, FcγR null | 1/2 | EC, OC, Myeloma, advanced solid cancer, | NCT04570839 NCT02913313 NCT04150965 |
| IBI939 (Innovent) | IgG1 mAb | 1 | Advanced cancer | NCT04353830 |
| BGB-A1217 (BeiGene) | Humanized IgG1 mAb | 1 | Metastatic solid tumours | NCT04047862 |
| Tiragolumab † (Genentech) | IgG1 mAb | 1–3 | NSCLC, SCLC, ESCC, BC | NCT03563716 NCT04300647 NCT04543617 NCT04584112 |
| Etigilmab (Mereo BioPharma) | Humanised IgG1 mAb | 1 | Advanced solid cancer | NCT03119428 |
| Vibostolimab (Merck) | Humanised IgG1 mAb | 1/2 | GC, NSCLC, PC, melanoma, | NCT02964013 NCT02861573 NCT04305054 NCT04303169 NCT04165070 NCT04305041 |
| Domvanalimab (Arcus Biosciences) | Humanised IgG1 mAb | 1 | Advanced solid cancer, NSCLC | NCT03628677 NCT04262856 |
| ASP8374 (Potenza) | IgG4 mAb | 1 | Advanced solid cancer | NCT03260322 NCT03945253 |
# 28 studies recruiting/active, not recruiting, 4 not yet recruiting, 1 withdrawn; * 3 studies recruiting/active, not recruiting, 6 completed, 1 not yet recruiting; † 13 studies recruiting, 1 active, not recruiting, 1 not yet recruiting. Abbreviations: HCC: heptocellular carcinoma; MSI-H: microsatellite instability high; AML: acute myeloid leukemia; CMML: chronic myelomonocytic leukemia; MDS: myelodysplastic syndrome; GBM: glioblastoma multiforme; NSCLC: non-small-cell lung cancer; SCLC: small-cell lung cancer; ESCC: esophageal squamous cell carcinoma; HNSCC: head and neck squamous cell carcinoma; CRC: colorectal cancer; HL: Hodgkin’s lymphoma, NHL: non-Hodgkin’s lymphoma; BCL: B cell lymphoma; RCC: renal cell carcinoma; BC: breast cancer; GC: gastric cancer; EC: endometrial cancer; OC: ovarian cancer; PC: prostate cancer.
Table 2.
Clinical applications of αCTLA-4/αPD-1 drugs combinations and the seminal studies that lead to their FDA approval (clinicaltrials.gov and fda.gov).
Table 2.
Clinical applications of αCTLA-4/αPD-1 drugs combinations and the seminal studies that lead to their FDA approval (clinicaltrials.gov and fda.gov).
| Product | Combination(s) | Cancer Type(s) | Seminal Study |
|---|---|---|---|
| A. CTLA-4 | |||
| Ipilimumab | Nil | Melanoma | NCT00094653 |
| (Bristol-Myers Squibb) | Surgery | Melanoma | NCT00636168 |
| Nivolumab | Melanoma | NCT01844505 | |
| Nivolumab | RCC | NCT02231749 | |
| Nivolumab | MSI-H/dMMR CRC | NCT02060188 | |
| Nivolumab | HCC | NCT01658878 | |
| Nivolumab and limited Chemotherapy | NSCLC | NCT03215706 | |
| Nivolumab | Malignant pleural mesothelioma | NCT02899299 | |
| B. PD-1 | |||
| Cemiplimab (Regeneron) | Nil | Cutaneous SCC | NCT02760498 |
| Pembrolizumab | Nil | Melanoma | NCT01866319 |
| (Merck) | Surgery | Melanoma | NCT02362594 |
| Nil | NSCLC | NCT01295827 | |
| NCT02220894 | |||
| Doublet platinum-based Chemotherapy | NSCLC | NCT02039674 | |
| Carboplatin and paclitaxel | NSCLC | NCT02775435 | |
| Axitinib | RCC | NCT02853331 | |
| Nil | MSI-H/dMMR CRC | NCT02563002 | |
| Nil | SCLC | NCT02054806 | |
| NCT02628067 | |||
| Platinum and FU | HNSCC | NCT01848834 | |
| Nil | HNSCC | NCT02358031 | |
| Nil | Gastric cancer | NCT02335411 | |
| Nil | ESCC | NCT02564263 | |
| Nil | HCC | NCT02702414 | |
| Nil | MCC | NCT02267603 | |
| Lenvatinib | Endometrial cancer | NCT02501096 | |
| Nil | cSCC | NCT03284424 | |
| Paclitaxel or gemcitabine and carboplatin | TNBC | NCT02819518 | |
| Nil | Urothelial cancer | NCT02335424 | |
| NCT02625961 | |||
| Nil | PMBCL | NCT02576990 | |
| Nil | Classical HL | NCT02453594 | |
| NCT02684292 | |||
| Nivolumab | Nil | NSCLC | NCT01642004 |
| (Bristol-Myers Squibb) | Nil | RCC | NCT01668784 |
| Nil | Classical HL | NCT02181738 | |
| NCT01592370 | |||
| Nil | HNSCC | NCT02105636 | |
| Nil | Urothelial Carcinoma | NCT02387996 | |
| Nil | MSI-H/dMMR CRC | NCT02060188 | |
| Nil | HCC | NCT01658878 | |
| Surgery | Melanoma | NCT02388906 | |
| Ipilimumab | SCLC | NCT01928394 | |
| Nil | ESCC | NCT02569242 |
NSCLC: non-small-cell lung cancer; RCC: renal cell carcinoma; MSI-H: microsatellite instability high; dMMR: mismatch repair deficient; CRC: colorectal cancer; SCLC: small-cell lung cancer; HNSCC: head and neck squamous cell carcinoma; ESCC: esophageal squamous cell carcinoma; MCC: Merkel cell carcinoma; cSSC: cutaneous squamous cell carcinoma; TNBC: triple-negative breast cancer; PBMCL: Primary mediastinal large B-cell lymphoma; HL: Hodgkin’s lymphoma: TMB-H: tumour mutational burden high.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Armitage, J.D.; Newnes, H.V.; McDonnell, A.; Bosco, A.; Waithman, J. Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression. Cells 2021, 10, 56. https://doi.org/10.3390/cells10010056
AMA Style
Armitage JD, Newnes HV, McDonnell A, Bosco A, Waithman J. Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression. Cells. 2021; 10(1):56. https://doi.org/10.3390/cells10010056
Chicago/Turabian StyleArmitage, Jesse D., Hannah V. Newnes, Alison McDonnell, Anthony Bosco, and Jason Waithman. 2021. "Fine-Tuning the Tumour Microenvironment: Current Perspectives on the Mechanisms of Tumour Immunosuppression" Cells 10, no. 1: 56. https://doi.org/10.3390/cells10010056
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.