Gas-Phase Epoxidation of Propylene to Propylene Oxide on a Supported Catalyst Modified with Various Dopants


Abstract
:1. Introduction

2. Results and Discussion
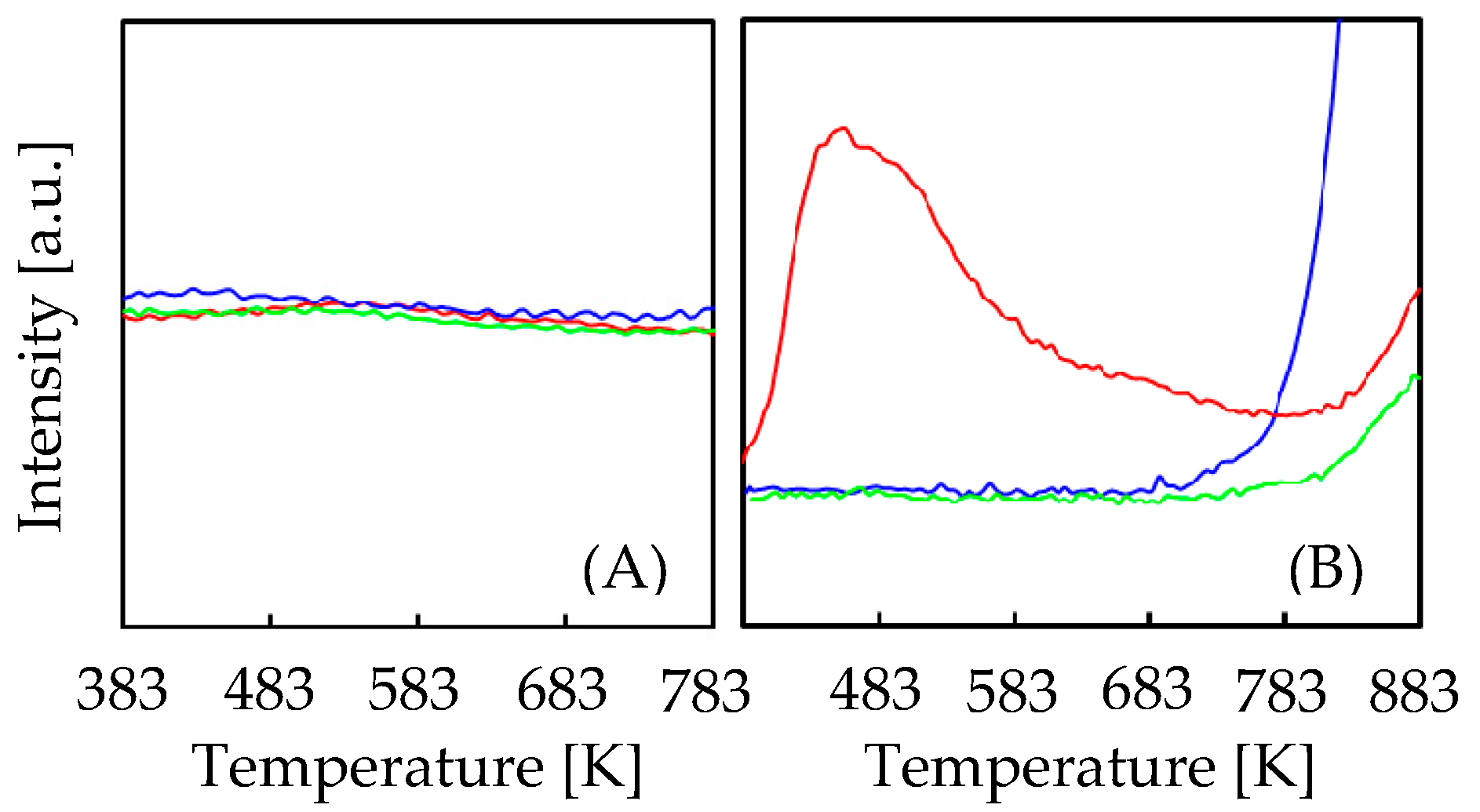
2.1. Catalytic Performances on MCM-41 Doped and Undoped with Al and/or Ti
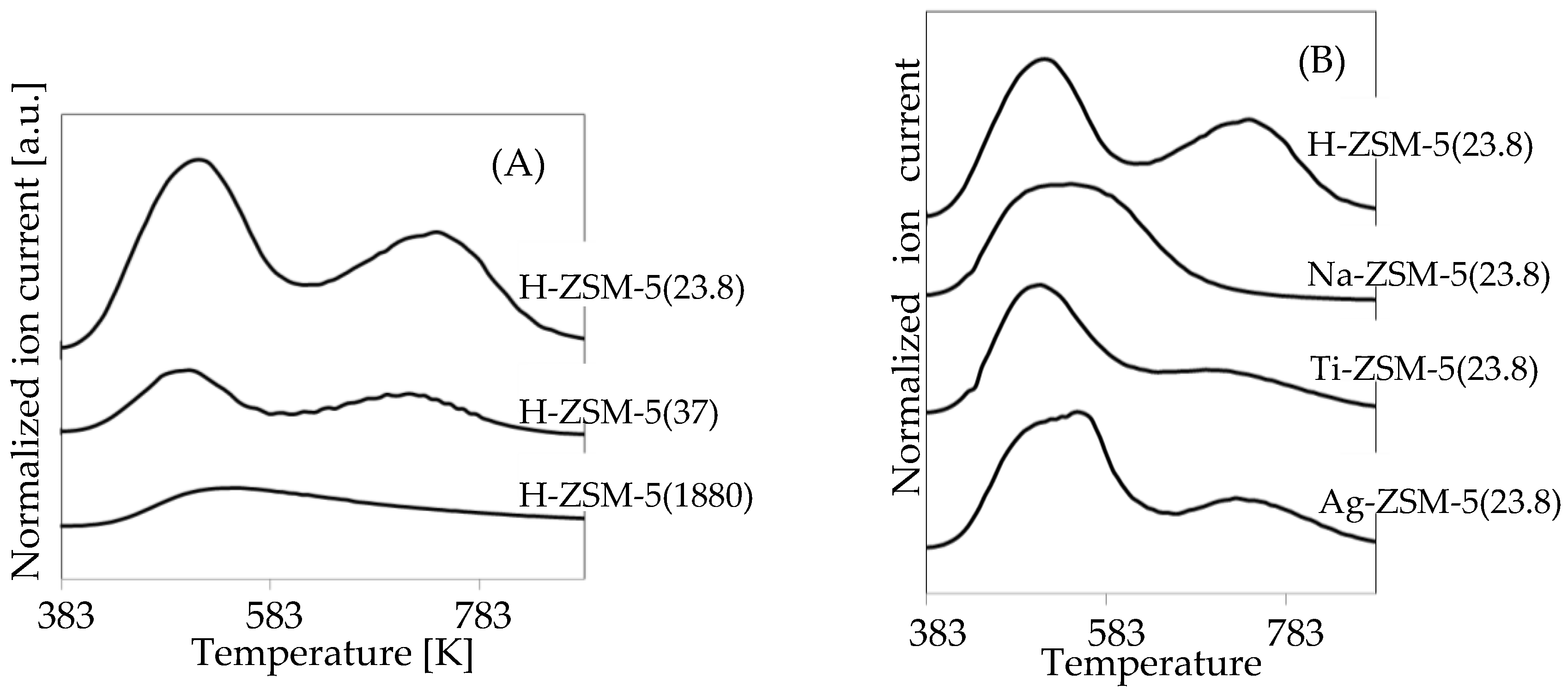
2.2. Catalytic Performances on H-ZSM-5 Doped and Undoped with Na, Ti, or Ag
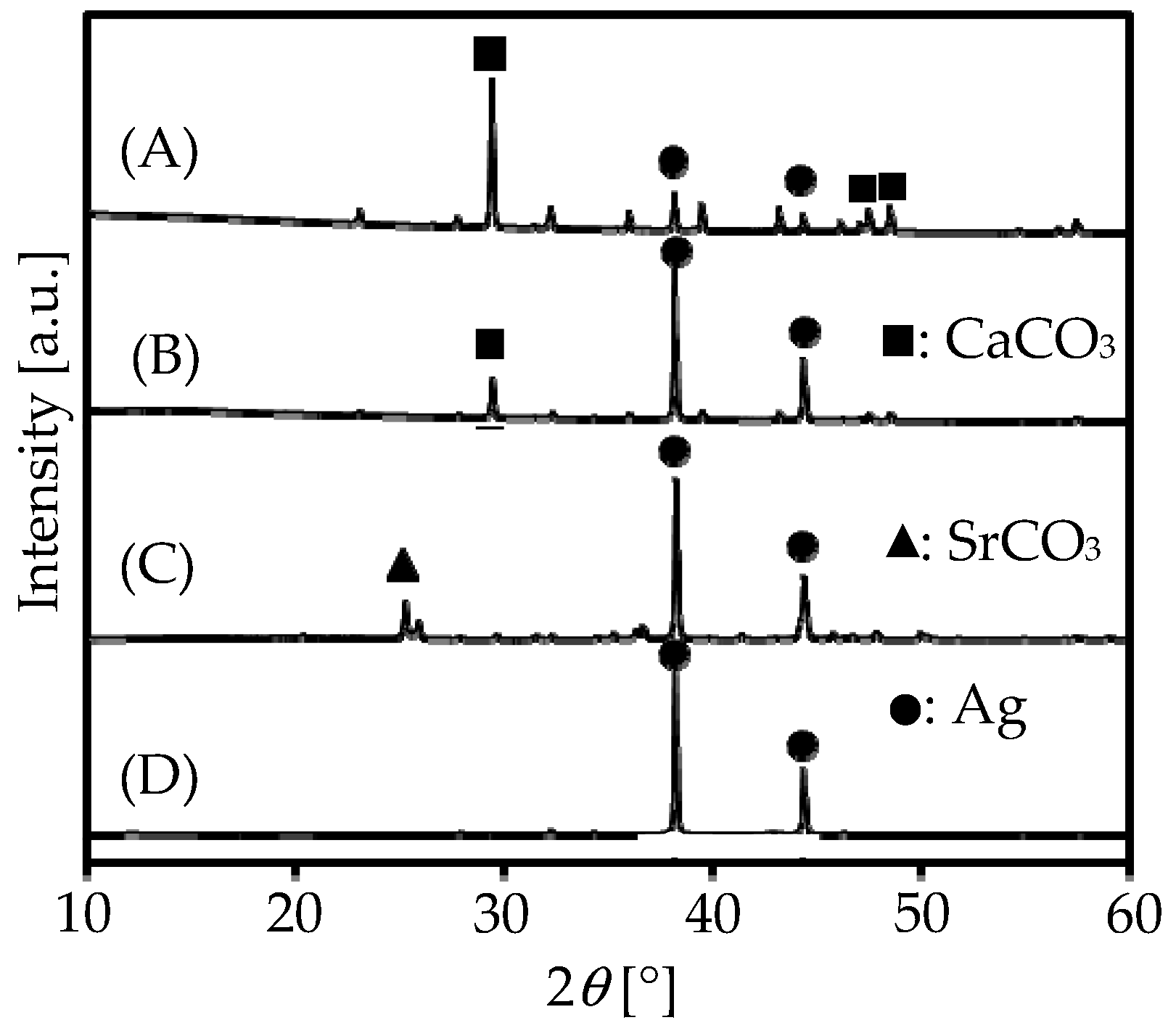
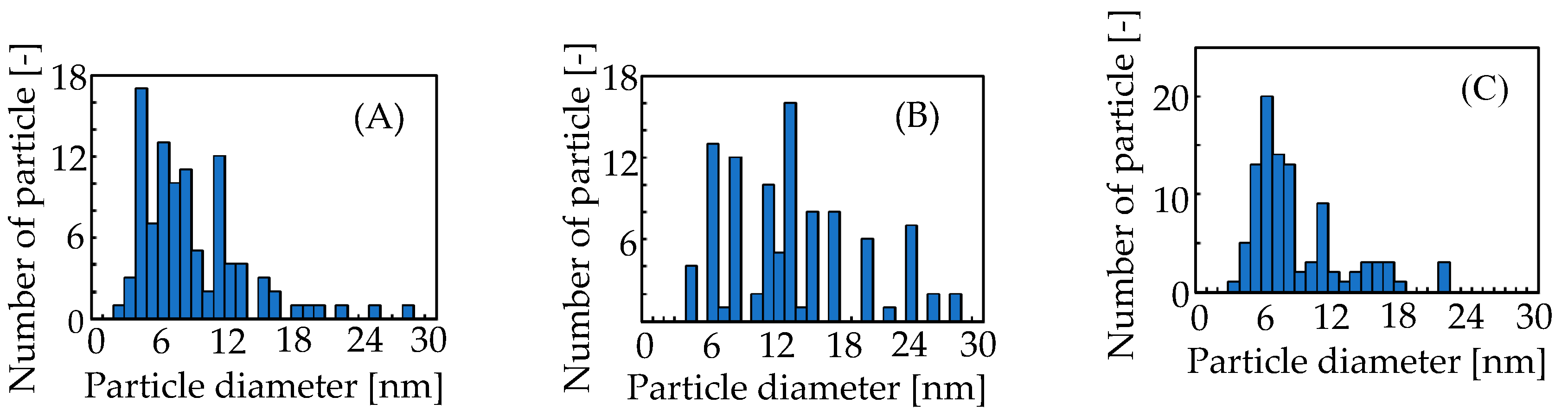
2.3. Catalytic Performances on Ag Metal Doped on Carbonates
3. Materials and Methods
3.1. Materials and Reagents
3.2. Preparation of Catalysts
3.3. Evaluation of Catalytic Performances
3.4. Characterization of Catalysts
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nijhuis, T.A.; Makkee, M.; Moulijin, J.A.; Weckhuysen, B.M. The production of propene oxide: Catalytic processes and recent development. Ind. Eng. Chem. Res. 2006, 45, 3447–3459. [Google Scholar] [CrossRef]
- Tsuji, J.; Yamamoto, J.; Ishino, M.; Oku, N. Development of new propylene oxide process. Sumitomo Kagaku 2006, 1, 4–10. [Google Scholar]
- Palermo, A.; Husain, A.; Tikhov, M.S.; Lambert, R.M. Ag-Catalyzed epoxidation of propene and ethene: An investigation using electrochemical promotion of the effects of alkali, NOx, and chlorine. J. Catal. 2002, 207, 331–340. [Google Scholar] [CrossRef]
- Harris, J.W.; Arvay, J.; Mitchell, G.; Delgass, W.N.; Ribeiro, F.H. Propylene oxide inhibits propylene epoxidation over Au/TS-1. J. Catal. 2018, 365, 105–114. [Google Scholar] [CrossRef]
- Ghosh, S.; Acharyya, S.S.; Tiwari, R.; Sarkar, B.; Singha, R.K.; Pendem, C.; Sasaki, T.; Bal, R. Selective oxidation of propylene to propylene oxide over silver-supported Tungsten oxide nanostructure with molecular oxygen. ACS Catal. 2014, 4, 2169–2174. [Google Scholar] [CrossRef]
- Lei, Y.; Chen, X.; Xu, C.; Dai, Z.; Wei, K. Enhanced catalytic performance in the gas-phase epoxidation of propylene over Ti-modified MoO3-Bi2SiO5/SiO2 catalysts. J. Catal. 2015, 321, 100–112. [Google Scholar] [CrossRef]
- Held, A.; Kowalska-Kuś, J.; Lapiński, A.; Nowińska, K. Vanadium species supported on inorganic oxides as catalysts for propene epoxidation in the presence of N2O as an oxidant. J. Catal. 2013, 306, 1–10. [Google Scholar] [CrossRef]
- Murata, K.; Kiyozumi, Y. Oxidation of propene by molecular oxygen over Ti-modified silicate catalysts. Chem. Commun. 2001, 15, 1356–1357. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Miura, N.; Inaba, M. Direct vapor phase oxidation of propylene by molecular oxygen over MCM-41 or MCM-22 based catalysts. Catal. Commun. 2003, 4, 385–391. [Google Scholar] [CrossRef]
- Murata, K.; Liu, Y.; Inaba, M.; Mimura, N. Effect of Ti-modified mesoporous materials on the direct epoxidation of propylene by molecular oxygen. Catal. Taday 2004, 91, 39–42. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Mimura, N. Syntheses of Ti- and Al-containing hexagonal mesoporous silicas for gas-phase epoxidation of propylene by molecular oxygen. Appl. Catal. A Gen. 2006, 309, 91–105. [Google Scholar] [CrossRef]
- Mimura, N.; Tsubota, S.; Murata, K.; Bando, K.; Bravo-Suárez, J.J.; Haruta, M.; Oyama, S.T. Gas-phase radical generation by Ti oxide clusters supported on silica: application to the direct epoxidation of propylene to propylene oxide using molecular oxygen as an oxidant. Catal. Lett. 2006, 110, 47–51. [Google Scholar] [CrossRef]
- Zemichael, F.W.; Palermo, A.; Tikhov, M.S.; Lambert, R.M. Propene epoxidation over K-promoted Ag/CaCO3 catalysts: the effect of metal particle size. Catal. Lett. 2002, 80, 93–98. [Google Scholar] [CrossRef]
- Ishizawa, N. On the thermal decomposition of calcite. Annu. Rep. Adv. Ceram. Res. Cent. Nagoya Inst. Technol. 2014, 2, 19–24. [Google Scholar]
- Sawada, Y.; Uematsu, K.; Mizutani, N.; Kato, M. Thermal decomposition of basic magnesium carbonates. J. Chem. Soc. Jpn. 1979, 1, 57–63. [Google Scholar]
- Kobayashi, Y.; Sumi, K.; Asai, T.; Kato, E. Fabrication of dense celsian ceramics by reaction of kaolin with SrCO3 and BaCO3. J. Ceram. Soc. Jpn. 1999, 107, 657–661. [Google Scholar] [CrossRef]
- Van Santen, R.A.; Kuipers, H.P.C.E. The mechanism of ethylene epoxidation. Adv. Catal. 1987, 35, 265–321. [Google Scholar]
- Kestenbaum, H.; Lange de Oliveira, A.L.; Schmidt, W.; Schüth, F.; Ehrfeld, W.; Gebauer, K.; Löwe, H.; Richter, T.; Lebiedz, D.; Untiedt, I.; et al. Silver-catalyzed oxidation of ethylene to ethylene oxide in a microreaction system. Ind. Eng. Chem. Res. 2002, 41, 710–719. [Google Scholar] [CrossRef]
- Lei, Y.; Mehmood, F.; Lee, S.; Greeley, J.; Lee, B.; Seifert, S.; Winans, R.E.; Elam, J.W.; Meyer, R.J.; Redfern, P.C.; et al. Increased Silver Activity for Direct Propylene Epoxidation via Subnanometer Size Effects. Science 2010, 328, 224–228. [Google Scholar] [CrossRef]
- Anderson, J.R.; Mole, T.; Christov, V. Mechanism of some conversions over ZSM-5 Catalyst. J. Catal. 1980, 61, 477–484. [Google Scholar] [CrossRef]
- Kitagawa, H.; Sendoda, Y.; Ono, Y. Transformation of propane into aromatic hydrocarbons over ZSM-5 zeolite. J. Catal. 1986, 101, 12–18. [Google Scholar] [CrossRef]
- Hoang, T.Q.; Zhu, X.; Sooknoi, T.; Resasco, D.E.; Mallinson, R.G. A comparison of the reactivities of propanal and propylene on HZSM-5. J. Catal. 2010, 271, 201–208. [Google Scholar] [CrossRef]
- Lu, J.; Bravo-Suárez, J.J.; Haruta, M.; Oyama, S.T. Direct propylene epoxidation over modified Ag/CaCO3 catalysts. Appl. Catal. A Gen. 2006, 302, 283–295. [Google Scholar] [CrossRef]
- Cooker, B.; Gaffney, A.M.; Jewson, J.D.; Kahn, A.P.; Pitchai, R. Epoxidation Process Using Supported Silver Catalysts Pretreated with Organic Chloride. U.S. Patent No. 5,770,746, 20 May 1998. [Google Scholar]
- Lu, J.; Bravo-Suárez, J.J.; Takahashi, A.; Haruta, M.; Oyama, S.T. In situ UV-vis studies of the effect of particle size on the epoxidation of ethylene and propylene on supported silver catalysts with molecular oxygen. J. Catal. 2005, 232, 85–95. [Google Scholar] [CrossRef]
- Chongterdtoonskul, A.; Schwank, J.W.; Chavadej, S. Effects of oxide supports on ethylene epoxidation activity over Ag-based catalysts. J. Mol. Catal. A Chem. 2012, 358, 58–66. [Google Scholar] [CrossRef]
- Iwamoto, M.; Abe, T.; Tachibana, Y. Control of bandgap of iron oxide through its encapsulation into SiO2-based mesoporous materials. J. Mol. Catal. A Chem. 2000, 155, 143–153. [Google Scholar] [CrossRef]
- Ehiro, T.; Itagaki, A.; Misu, H.; Nakagawa, K.; Katoh, M.; Katou, Y.; Ninomiya, W.; Sugiyama, S. Effects of acid treatment on the acidic properties and catalytic activity of MCM-41 for the oxidative dehydrogenation of isobutane. J. Chem. Eng. Jpn. 2016, 49, 152–160. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catal. 1 | Conv. [%] | Selectivity [%] | Yield [%] | |||||
|---|---|---|---|---|---|---|---|---|
| C3H6 | COx | PO | PA | PO | PA | |||
| A | 9.6 | 97.7 | 0 | 0 | 0 | 0 | ||
| B | 4.0 | 97.0 | 0 | 1.4 | 0 | 0.054 | ||
| C | 7.0 | 28.2 | 0.37 | 1.0 | 0.026 | 0.069 | ||
| D | 5.1 | 88.2 | 0.06 | 1.8 | 0.003 | 0.093 | ||
| E | 7.2 | 30.9 | 0.35 | 1.7 | 0.025 | 0.122 | ||
| Catal. 1 | 2θ from (100) 2 [°] | Surface Area 3 [m2/g] | Total Pore Volume 4 [cm3/g] | Acidity 5 [mmol/g] |
|---|---|---|---|---|
| A | 2.52 | 908 | 0.82 | 0.006 |
| B | - | 335 | 0.47 | 0.026 |
| C | - | 490 | 0.69 | 0.179 |
| D | - | 137 | 0.39 | 0.164 |
| E | - | 479 | 0.78 | 0.070 |
| Catalyst | Conv. [%] | Select. [%] | Yield [%] | Acid Amount | Temp. 3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| C3H6 | COx | PO | PO | [mmol/g] | [K] | |||||
| H-ZSM-5(23.8) 1 | 90.7 | - | 0.71 | 0.64 | 0.803 0.686 | 513 743 | ||||
| H-ZSM-5(23.8) 2 | 91.5 | 8.2 | 0.73 | 0.67 | ||||||
| H-ZSM-5(37) 1 | 11.4 | - | 1.12 | 0.13 | 0.428 0.431 | 505 713 | ||||
| H-ZSM-5(1880) 1 | 13.8 | - | 1.35 | 0.19 | 0.296 | 528 | ||||
| Na-ZSM-5(23.8) 2 | 6.5 | 36.9 | 1.05 | 0.07 | 0.857 | 555 | ||||
| Ti-ZSM-5(23.8) 2 | 68.3 | 3.2 | 0.84 | 0.57 | 0.395 0.082 | 510 694 | ||||
| Ag-ZSM-5(23.8) 2 | 5.1 | - | 1.31 | 0.07 | 0.341 0.094 | 550 726 | ||||
| Catalyst | Conversion [%] | Selectivity [%] | Yield [%] | |||
|---|---|---|---|---|---|---|
| C3H6 | COx | PO | PO | |||
| Ag(56)/CaCO3 | 33.1 | 99.0 | 0.1 | 0.03 | ||
| Ag(56)-Na(1)/CaCO3 | 10.6 | 87.7 | 12.2 | 1.29 | ||
| Ag(5.6)-Na(1)/CaCO3 | 3.5 | 88.0 | 11.7 | 0.41 | ||
| Ag(56)-Na(0.5)/CaCO3 | 13.2 | 92.9 | 6.6 | 0.87 | ||
| Ag(56)-Na(2)/CaCO3 | 11.2 | 91.4 | 8.2 | 0.92 | ||
| Ag(56)-Na(1)/SrCO3 | 17.4 | 86.9 | 12.5 | 2.17 | ||
| Ag(56)-Na(1)/MgCO3 | 22.0 | 98.0 | 0.7 | 0.15 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugiyama, S.; Sakuwa, Y.; Ogino, T.; Sakamoto, N.; Shimoda, N.; Katoh, M.; Kimura, N. Gas-Phase Epoxidation of Propylene to Propylene Oxide on a Supported Catalyst Modified with Various Dopants. Catalysts 2019, 9, 638. https://doi.org/10.3390/catal9080638
Sugiyama S, Sakuwa Y, Ogino T, Sakamoto N, Shimoda N, Katoh M, Kimura N. Gas-Phase Epoxidation of Propylene to Propylene Oxide on a Supported Catalyst Modified with Various Dopants. Catalysts. 2019; 9(8):638. https://doi.org/10.3390/catal9080638
Chicago/Turabian StyleSugiyama, Shigeru, Yasuhiro Sakuwa, Tomoyasu Ogino, Naotaka Sakamoto, Naohiro Shimoda, Masahiro Katoh, and Nobuhiro Kimura. 2019. "Gas-Phase Epoxidation of Propylene to Propylene Oxide on a Supported Catalyst Modified with Various Dopants" Catalysts 9, no. 8: 638. https://doi.org/10.3390/catal9080638