All manipulations were performed under normal conditions, and the solvents were dried when necessary, by refluxing over the appropriate drying reagents and distilling under nitrogen prior to use. Elemental analyses were determined with an Elementar Vario EL III Elemental Analyzer (Elementar AnalysensystemeGmbH, Germany). Infrared spectra (4000–400 cm
−1) were recorded on a Jasco FT/IR-430 instrument (Jasco, Tokyo, Japan) in KBr pellets; wavenumbers are in cm
−1; abbreviations: vs, very strong; s, strong; m, medium; w, weak.
1H and
13C NMR spectra were run on a Varian Unity 400 spectrometer (Varian, USA) at ambient temperature. TG-DTA data were collected with a Perkin Elmer STA6000 Thermal Analyzer (Perkin-Elmer, Waltham, MA, USA) at a heating rate of 10 K min
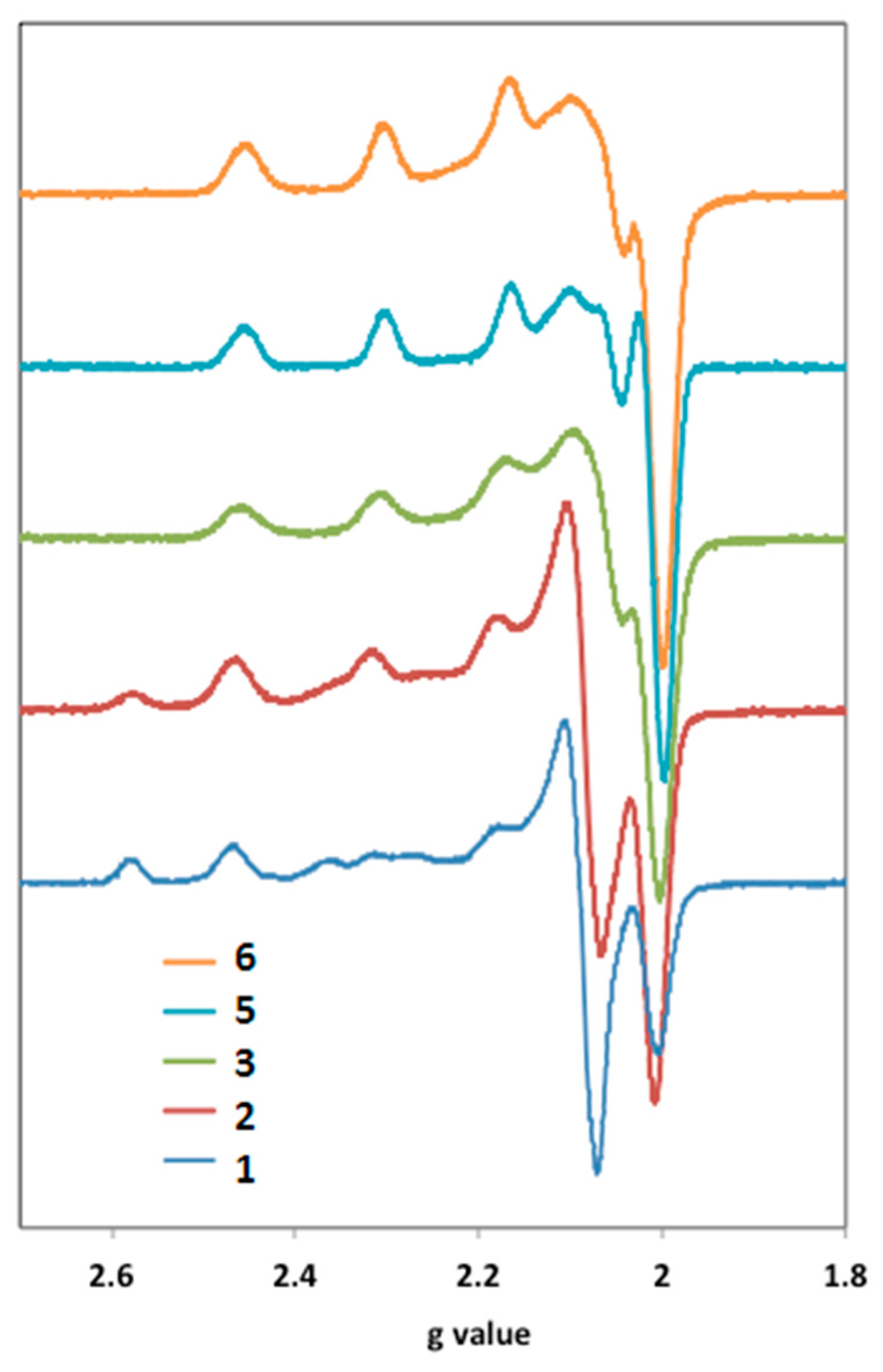
−1 under an air atmosphere. The EPR spectra were obtained at 100 K on a CW X-band Bruker ESP 300E Spectrometer (Bruker, Bremen, Germany). The X-band EPR spectra were analyzed using a program developed by Rockenbauer and Korecz [
61]. The EPR spectra for compounds
1 and
2 were simulated by a superposition of two component curves. The fitting was achieved assuming either axial or rhombic
g and
A tensors. The electrical conductivity measurements (Λ
M, reported as S cm
2 mol
−1) of solutions of the copper compounds
1–
6 (ca. 1.5 × 10
−3 M) were taken with a WTW Multi 340 conductometer at 15 °C. The UV–Vis absorption spectra of dichloromethane/methanol or DMF solutions of
1–
6 (ca. 1.5 × 10
−3 M) in 1.00 cm quartz cells were recorded at room temperature on a Lambda 35 UV–Vis spectrophotometer (Perkin-Elmer) by scanning the 200–800 nm region at a rate of 240 nm min
−1. Reactions under microwave irradiation were performed by using a focused Anton Paar Monowave 300 reactor (Anton Paar GmbH, Graz, Austria).
3.3. Catalytic Studies
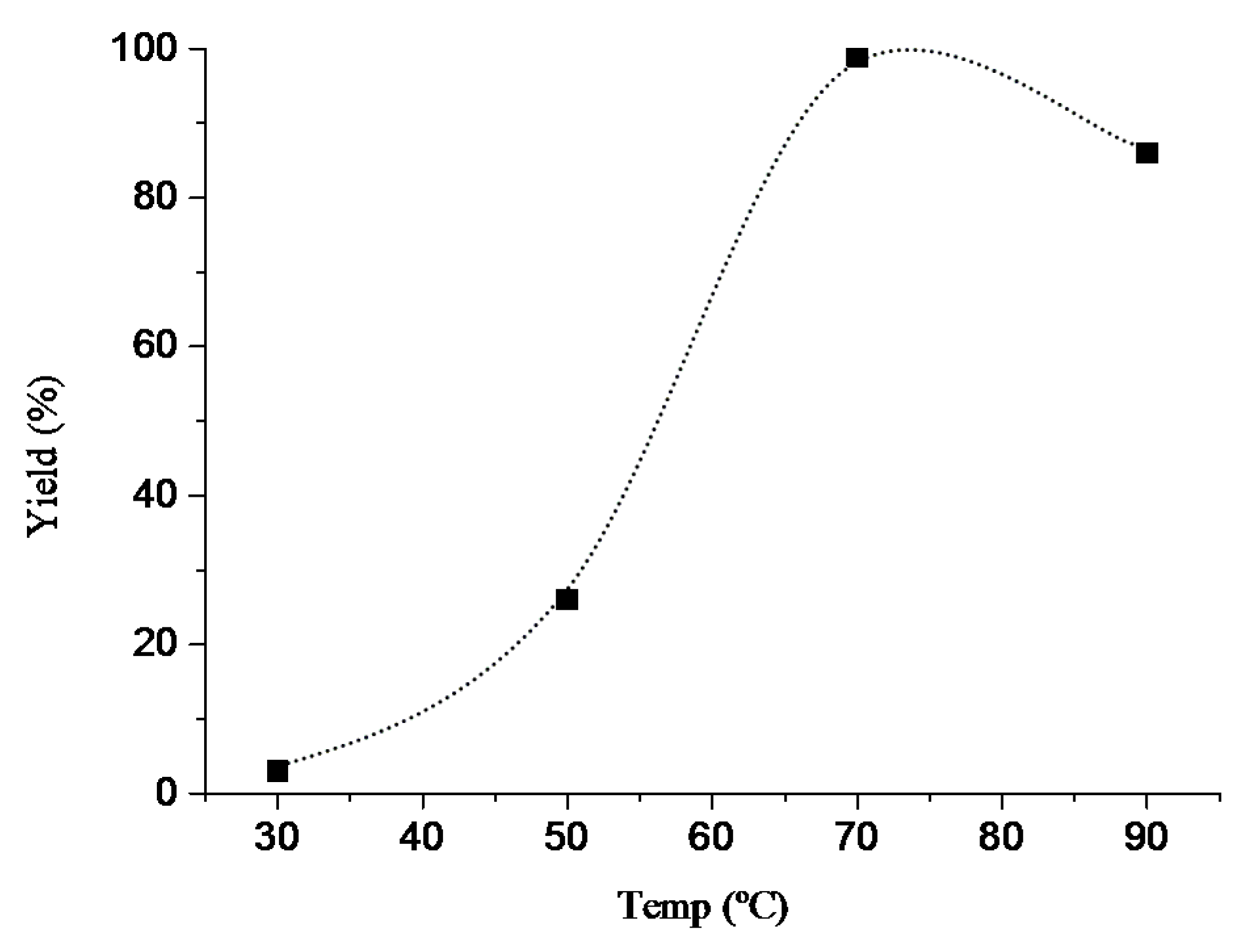
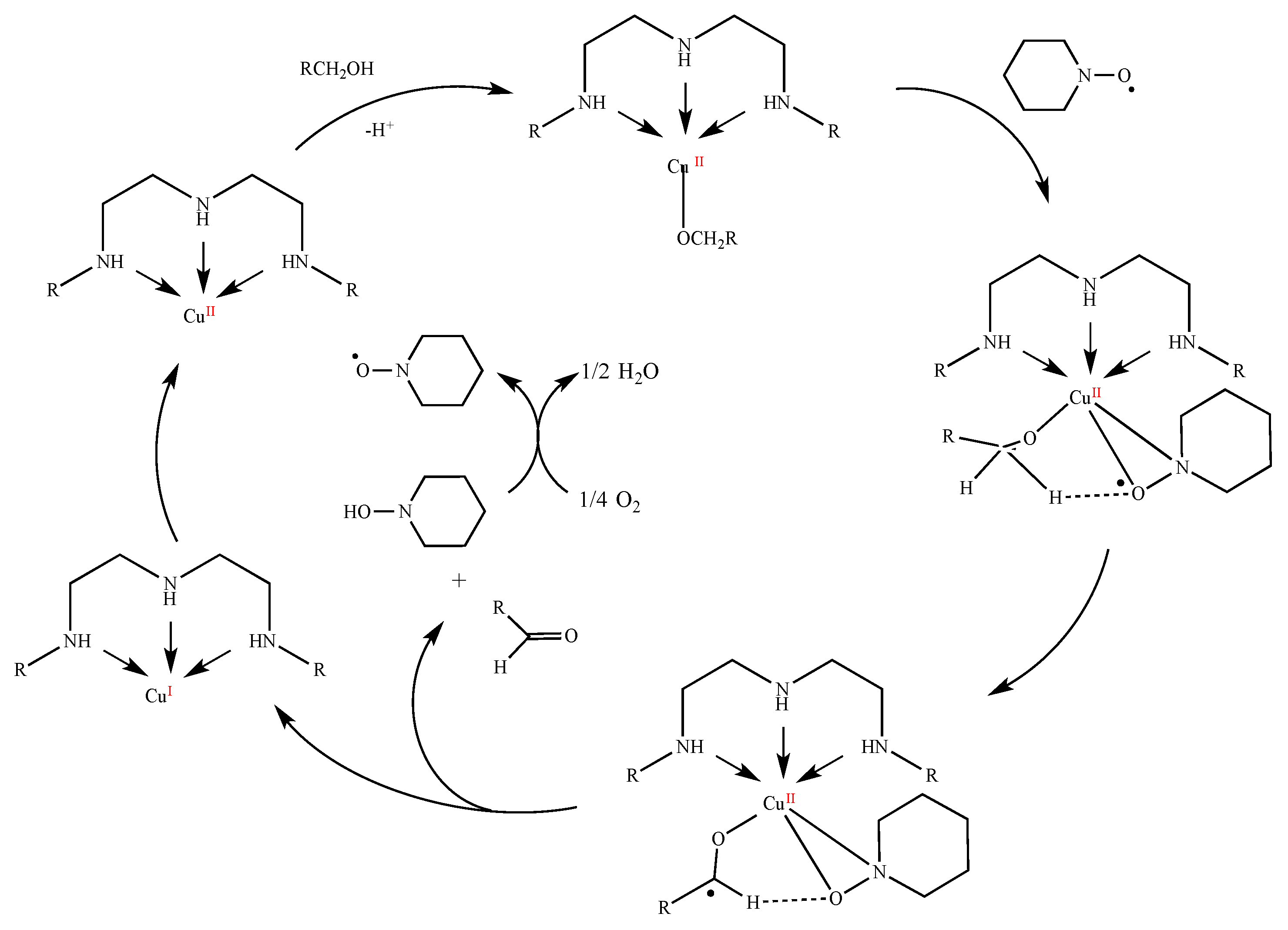
The aerobic oxidations of benzylic alcohols, cinnamyl alcohol, and 1-phenylethanol were typically performed in flasks fitted with water circulating condensers and under air at atmospheric pressure. 1.5 mmol of the substrate, 0.0075 mmol (0.5 mol% based on substrate) of Cu catalyst 1–6, 0.075 mmol (5 mol%) of TEMPO and 5 mL of 1 M K2CO3 aqueous solution were added, and the reaction mixture was stirred for 20 h at 70 °C after which the additions of 0.5 mL of HCl 1 M (for neutralization) and 5 mL of EtOAc (for the extraction of the substrate and of the organic products from the reaction mixture) were accomplished. Then, 120 μL of cycloheptanone was added as internal standard, and the products were analyzed and quantified by GC (the internal standard method).
The MW-assisted solvent-free peroxidative oxidation experiments of 1-phenyethanol were performed using a 10 mL capacity reaction tube (13 mm Ø), fitted with a rotational system and an IR temperature detector. The alcohol (2.5 mmol), 5 µmol of Cu catalyst 1–6 (0.2 mol% based on substrate) and a 70% aqueous solution of t-BuOOH (5 mmol) were introduced in a sealed cylindrical Pyrex tube. The system was left under stirring and under irradiation of 5 W at 80 °C or 15 W at 120 °C for 0.5 h. After cooling to room temperature, 150 µL of benzaldehyde (internal standard) and 2.5 mL of MeCN (for substrate and organic products extraction) were added. After stirring for 10 min, a sample (1 µL) was taken from the organic phase and analyzed by GC.
Oxidation products were analyzed by gas chromatography using a FISONS Instruments GC 8000 series gas chromatograph with a flame ionization detector (FID) and a capillary column (DB-WAX, column length: 30 m; Ø: 0.32 mm) (He as the carrier gas) and the Jasco-Borwin v.1.50 software. The temperature of injection was 240 °C. The initial temperature of the column was kept at 120 °C for 1 min, elevated 10 °C/min up to 200 °C, and finally held at this temperature for 1 min. The attribution of peaks was made by comparison with commercial samples (GC) and, in some cases, by GC-MS analyses using a Perkin Elmer Clarus 600 C instrument (He as the carrier gas), equipped with a 30 m × 0.22 mm × 25 μm BPX5 (SGE) capillary column.
Synthesis of L. Compound L was synthesized by hydrogenation, with KBH
4, of the Schiff-base 1,4,19,22,25,40-hexaaza-10,13,31,34-tetraoxa-6,14,27,35(1,4)-tetrabenzenacyclotria contaphane-5,18,26,39-tetraene [
68]. Then, 1.0 g (1.5 mmol) of this precursor and 400 mL of methanol were mixed in a 1000 mL flask, and a white emulsion was obtained. Next, 2.0 g (37 mmol) of KBH
4 were added in portions of 400 mg every 20 min. The colorless solution thus obtained was stirred for half an hour, filtered, and the solution taken to dryness. Upon the addition of 200 mL of water, it was left stirring for 1 h. Compound L was obtained as a white solid, which was isolated by filtration, recrystallized from methanol, and dried under vacuum (0.90 g, yield 89%), m.p. 177–179 °C. Anal. calcd for C
40H
54N
6O
4·CH
3OH·2.5H
2O (C
41H
63N
6O
7.5;
mw 759.97) C, 64.80; H, 8.36; N, 11.06. Found C, 64.87; H, 8.47; N, 10.82;
1H NMR (400 MHz, CD
3OD: CDCl
3 = 1:4),
δ 2.55 (s, 16H, NH–C
H2), 3.50 (s, 8H, C
H2 Ar), 4.09 (s, 8H, O–C
H2), 6.69 (d,
J = 8.4 Hz, 8H, aryl), 7.00 (d,
J = 8.4 Hz, 8H, aryl).
13C NMR (100 MHz, CD
3OD: CDCl
3 = 1:4),
δ 47.59 (
CH
2-N), 47.68 (
CH
2N), 52.59 (CH
2CH
2N), 66.41 (O-
CH
2); 114.32 (aryl), 129.24 (aryl), 131.86 (aryl), 157.61 (aryl). IR (KBr disc) (cm
−1): 3420 (vs, ν
O-H and ν
N-H), 2932 (s), 2832 (s), 1611 (s, ν
aryl-H), 1515 (s), 1453 (s, ν
aryl-H), 1380 (w), 1299 (m), 1243 (vs), 1180 (m), 1113 (m), 1068 (s), 939 (s), 810 (s), 770 (m), 616 (w), 522 (w).
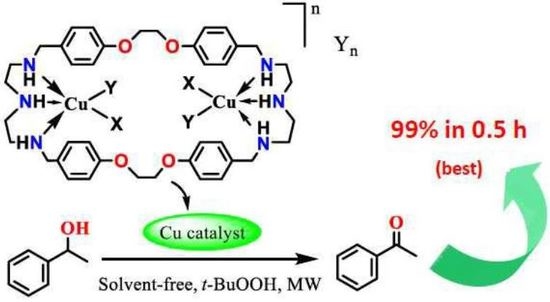
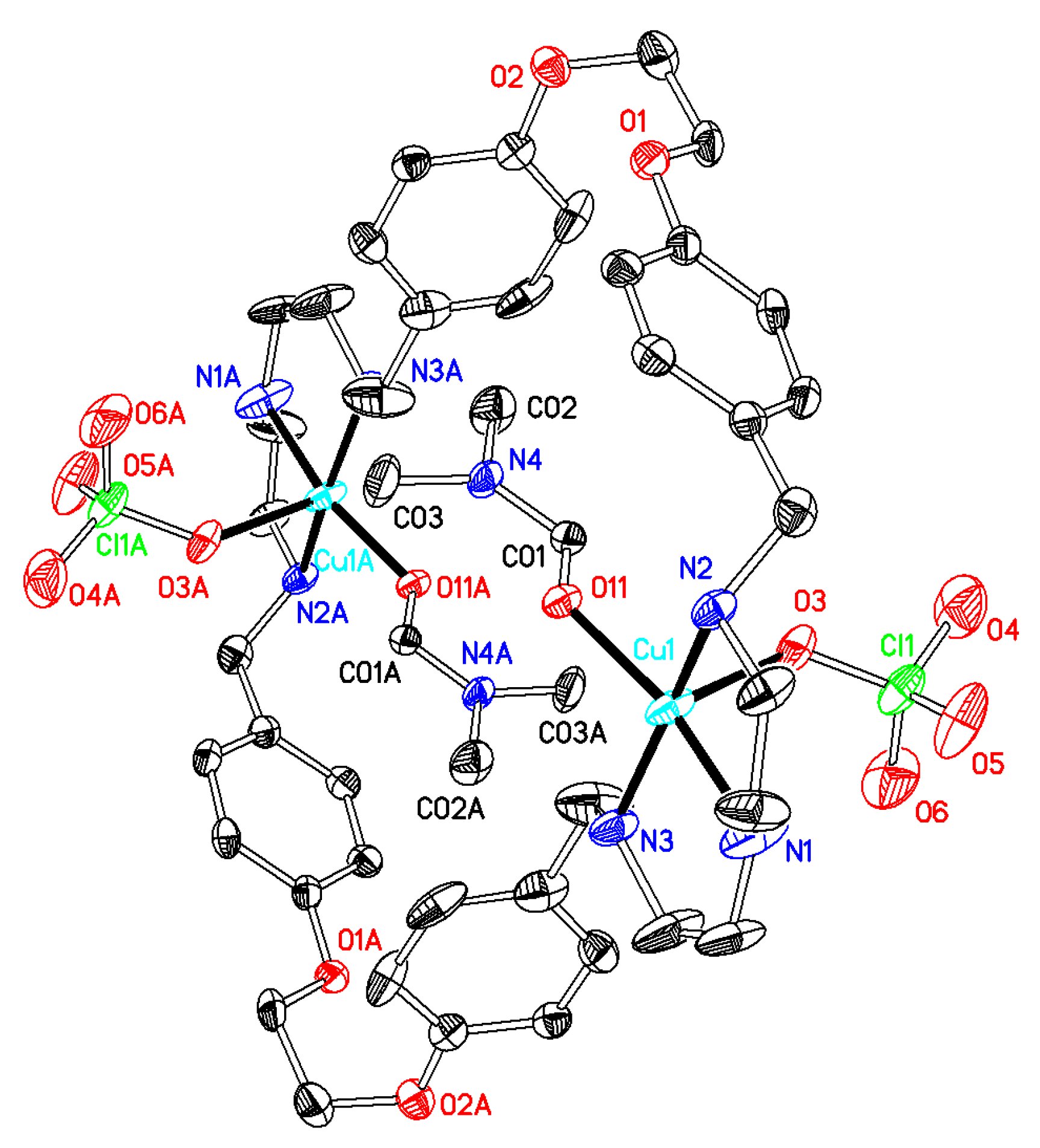
Synthesis of [Cu2(OSO2CF3)2L(DMF)2](SO3CF3)2 (compound 1) Cu(SO3CF3)2·2H2O (0.29 g, 0.72 mmol) was dissolved in 10 mL of methanol, a solution of L·CH3OH·2.5H2O (0.25 g, 0.33 mmol) in 20 mL mixture of methanol and dichloromethane (1:1) was added, and the system was stirred for 24 h. After filtration, the blue filtrate was taken to dryness, and 20 mL of diethyl ether was added, thus affording a blue solid. TG-DTA: CuO% = 11.8, weight% (for loss of solvents) = 1.9. Calcd with the formula of C44H54Cu2F12N6O16S4∙1.5H2O: CuO% = 11.1, weight% (for loss of H2O) = 1.9. After dissolution of the compound in 20 mL of DMF and diffusion of diethyl ether through this solution, dark blue crystals were obtained, suitable for X-ray diffraction. Different batches of the compound were used for microanalysis and single-crystal X-ray diffraction. Total amount obtained: 0.20 g (39% yield based on L). Anal. calcd for C44H54Cu2F12N6O16S4·2C3H7NO·4H2O (C50H76Cu2F12N8O22S4; mw 1624.52): C 36.97; H 4.72; N 6.90. Found: C 37.10; H 4.51; N 6.62. IR (KBr disc) (cm−1): 3466 (vs), 3223 (s), 2946 (m), 2908 (s), 1658 (vs), 1612 (s, νaryl-H), 1586 (w), 1513 (s, νaryl-H), 1444 (s), 1384 (s), 1252 (vs), 1166 (vs), 1110 (s), 1100 (m), 1049 (vs), 926 (m), 821 (m), 765 (m), 701 (w), 652 (s), 578 (m), 521 (m). UV–Vis max in DCM/MeOH, nm (ε, M−1cm−1): 616 (35), 273 (401). ΛM in DCM/MeOH: 110 Scm2mol−1.
Synthesis of [Cu2(OSO2C6H4Me)2L(DMF)2](SO3C6H4Me)2 (compound 2) Cu(SO3C6H4Me)2·4H2O·CH3OH (0.15 g, 0.30 mmol) and L·CH3OH·2.5H2O (0.10 g, 0.13 mmol) were dissolved in 20 mL DMF and the solution stirred for 24 h. After filtration, the filtrate was concentrated under vacuum, and 20 mL of diethyl ether was then added to give a blue solid. Different batches of the compound were used for microanalysis, TG, and single crystal X-ray diffraction. Total amount obtained: 0.21 g (79% yield based on L). Anal. calcd for C68H82Cu2N6O16S4·4C3H7NO·2H2O (C80H114Cu2N10O22S4; mw 1823.17): C 52.70; H 6.30; N 7.68. Found: C 52.42; H 6.22; N 7.52. TG-DTA: CuO% = 10.3, weight% (for loss of H2O and DMF) = 17.7. Calc with the formula of C80H114Cu2N10O22S4: CuO% = 8.7, weight% (for loss of H2O and DMF) = 18.0. IR (KBr disc) (cm−1): 3463 (vs), 3189 (m), 2932 (m), 1652 (vs), 1612 (m), 1513 (s, νaryl-H), 1454 (m), 1386 (m), 1183 (vs), 1122 (s), 1035 (s), 1009 (s), 816 (m), 683 (s), 568 (s). UV–Vismax in DMF, nm (ε, M−1cm−1): 619 (39), 285 (884). ΛM in DMF: 132 Scm2mol−1. Suitable crystals for X-ray diffraction analyses were obtained upon slow diffusion of diethyl ether into the above DMF solution of the compound.
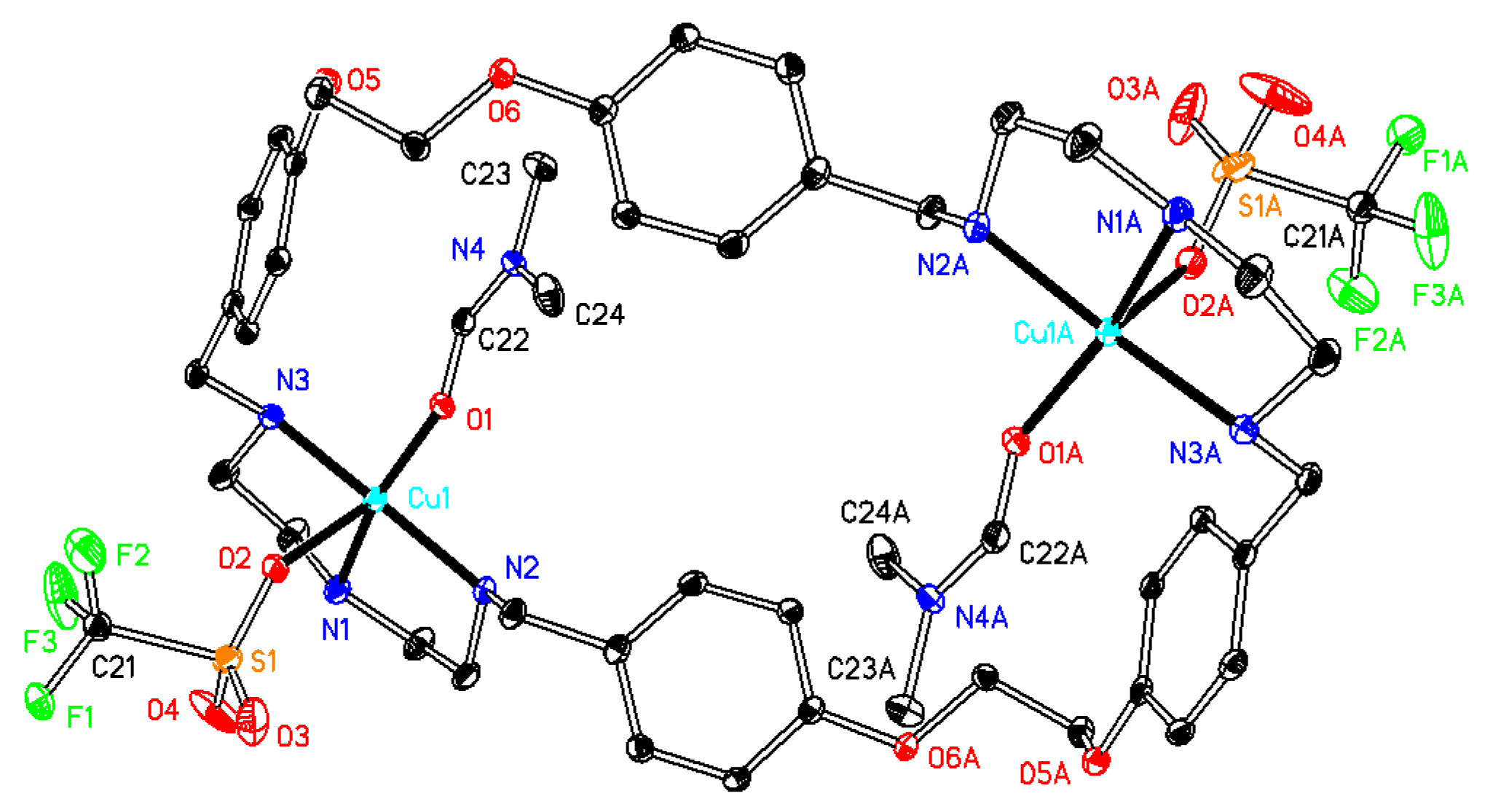
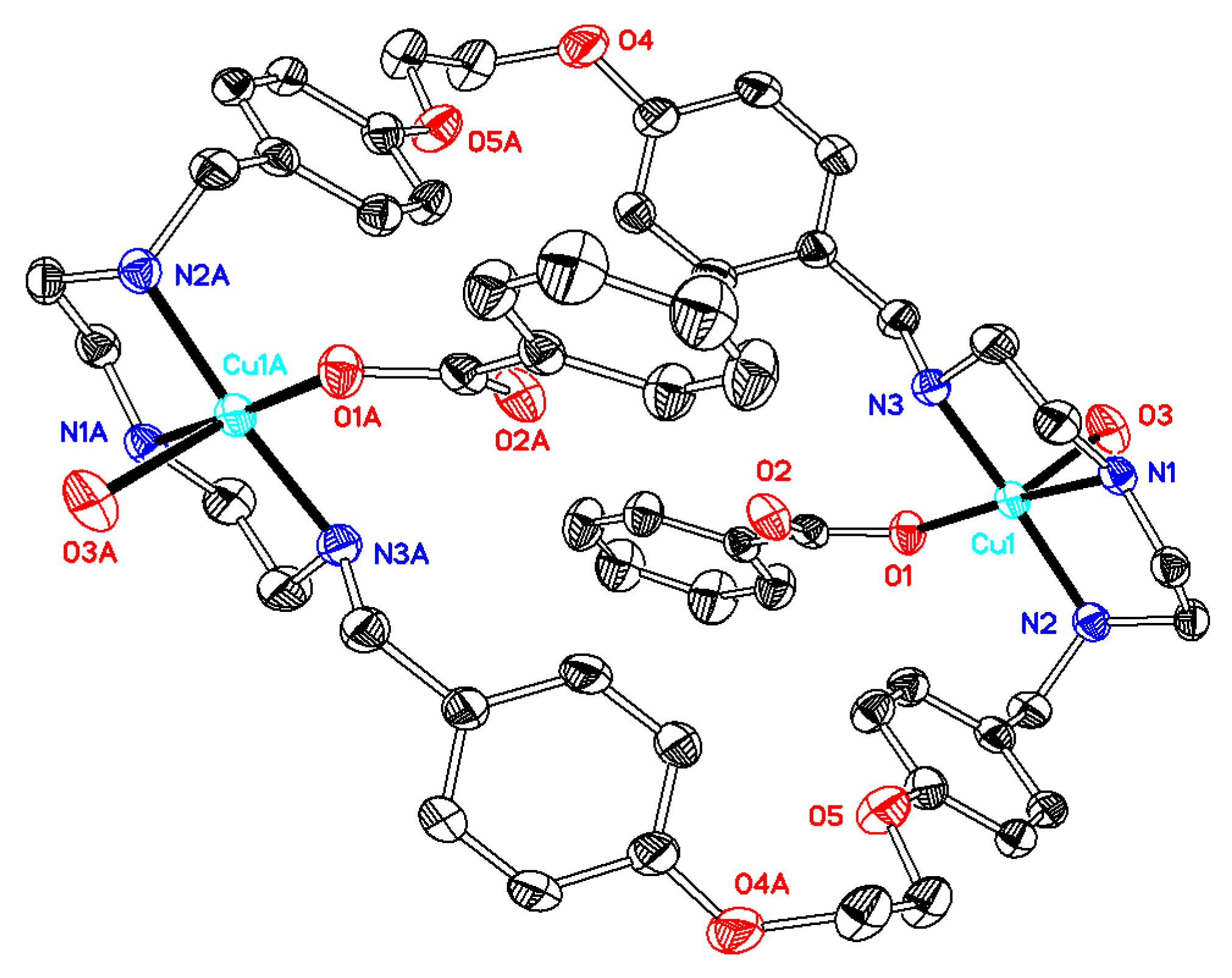
Synthesis of[Cu2(ONO2)2L(DMF)2](NO3)2(compound 3). The starting materials Cu(NO3)2·2.5H2O (0.14 g, 0.60 mmol) and L·CH3OH·2.5H2O (0.21 g, 0.28 mmol) were mixed in 20 mL of DMF, and the system was stirred for 24 h. After filtration, slow diffusion of diethyl ether into the filtrate solution led to the formation of blue crystals, which were suitable for X-ray analysis. Different batches of the compound were used for microanalysis, TG, and single crystal X-ray diffraction. Total amount obtained: 0.17 g (49% based on L). C40H54Cu2N10O16·2C3H7NO·3.5H2O (C46H75Cu2N12O21.5; mw 1267.25): C 43.60; H 5.97; N 13.26. Found: C 43.58; H 6.21; N 13.06. TG-DTA: CuO% = 12.8, weight% (for loss of solvents) = 19.6. Calc with the formula of C40H54Cu2N10O16·3C3H7NO·3H2O (C49H81Cu2N13O22): CuO% = 12.0, weight% (for loss of H2O and DMF) = 20.5. IR (KBr disc) (cm−1): 3429 (vs), 3200 (m), 2933 (m), 2878 (m), 1652 (vs), 1611 (m), 1585 (s), 1513 (s, νaryl-H), 1384 (vs), 1307 (m), 1248 (s), 1224 (s), 1180 (m), 1054 (m), 1031 (m), 918 (w), 826 (m), 775 (m), 693 (w), 618 (w), 518 (w). UV–Vismax in DMF, nm (ε, M−1cm−1): 619 (38), 282 (862). ΛM in DMF: 166 Scm2mol−1.
Synthesis of[Cu2(OClO3)2L(DMF)2](ClO4)2(compound 4). The starting materials Cu(ClO4)2·6H2O (0.12 g, 0.32 mmol) and L·CH3OH·2.5H2O (0.10 g, 0.13 mmol), as well as 20 mL of methanol were mixed in a 50 mL flask, and the system was stirred for 24 h. After filtration, the filtrate was concentrated, and diethyl ether was added. A blue solid was obtained (0.15 g). Suitable crystals for X-ray diffraction analyses were obtained upon slow diffusion of diethyl ether into a DMF solution of the compound. (0.090 g, yield 44% based on L). Anal. calcd for C40H54Cu2Cl4N6O20·2C3H7NO·4.5H2O (C46H77Cu2Cl4N8O26.5; mw 1435.04): C 38.50; H 5.41; N 7.81. Found: C 38.38; H 5.38; N 7.97. IR (KBr disc) (cm−1): 3438 (vs), 3240 (m), 2931 (m), 2876 (m), 1656 (vs), 1613 (m), 1513 (s, νaryl-H), 1455 (m), 1415 (w), 1385 (m), 1306 (m), 1250 (s), 1226 (s), 1117 (vs), 1084 (vs), 922 (w), 834 (s), 702 (m), 626 (s). UV–Vismax in DMF, nm (ε, M−1 cm−1): 618 (37), 279 (859). ΛM in DMF: 141 Scm2mol−1.
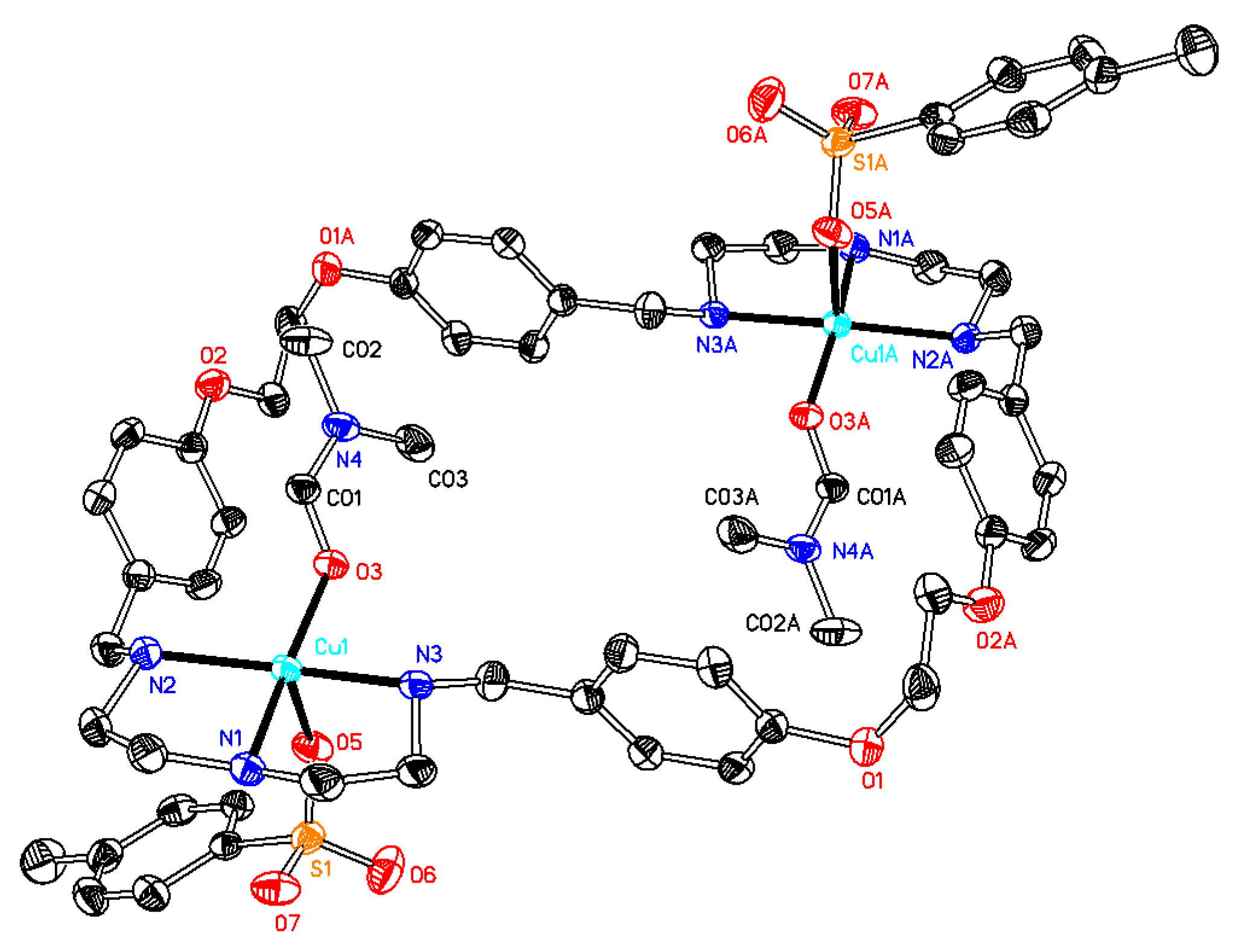
Synthesis of[Cu2(OCOPh)2L(H2O)2](CO2Ph)2 (compound 5). The starting materials Cu(CO2Ph)2 (0.15 g, 0.50 mmol) and L·CH3OH·2.5H2O (0.17 g, 0.22 mmol) were dissolved in 20 mL of a 1:1 mixture of methanol and dichloromethane, and the system was stirred for 24 h. After filtration, the blue filtrate was taken to dryness and recrystallized from DMF and diethyl ether to give a blue solid (0.26 g, yield 71% based on L), which was isolated by filtration and dried under vacuum. The mother solution was then mixed with 1,4-dioxane in a 2:1 ratio followed by slow evaporation, which led to the formation of X-ray quality dark blue crystals. Anal. calcd for C68H74Cu2N6O12·2C3H7NO·2H2O·CH2Cl2 (C75H94Cl2Cu2N8O16; mw 1561.59): C 57.68; H 6.07; N 7.18. Found: C 57.97; H 6.01; N 7.05. TG-DTA: CuO% = 13.1, weight% (loss of solvents) = 6.6. Calc with the formula of [Cu2(CO2Ph)4L(H2O)2]·0.5DMF (C69.5H81.5N6.5O14.5Cu2; mw 1367.01): CuO% = 11.6, weight% (loss of solvents) = 5.3. IR (KBr disc) (cm−1): 3430 (vs), 3219 (m), 2931 (m), 2878 (m), 1665 (m), 1598 (s), 1551 (s, νaryl-H), 1512 (s), 1456 (m), 1384 (vs), 1304 (w), 1244 (s), 1179 (m), 1067 (m), 1024 (w), 934 (w), 840 (m), 723 (m), 681 (w), 618 (w). UV–Vismax in H2O or DCM/MeOH, nm (ε, M−1 cm−1): 616 (45), 271 (704) and 226 (1,931), or 611 (43) and 273 (521). ΛM in H2O or DCM/MeOH: 226 or 131 Scm2mol−1.
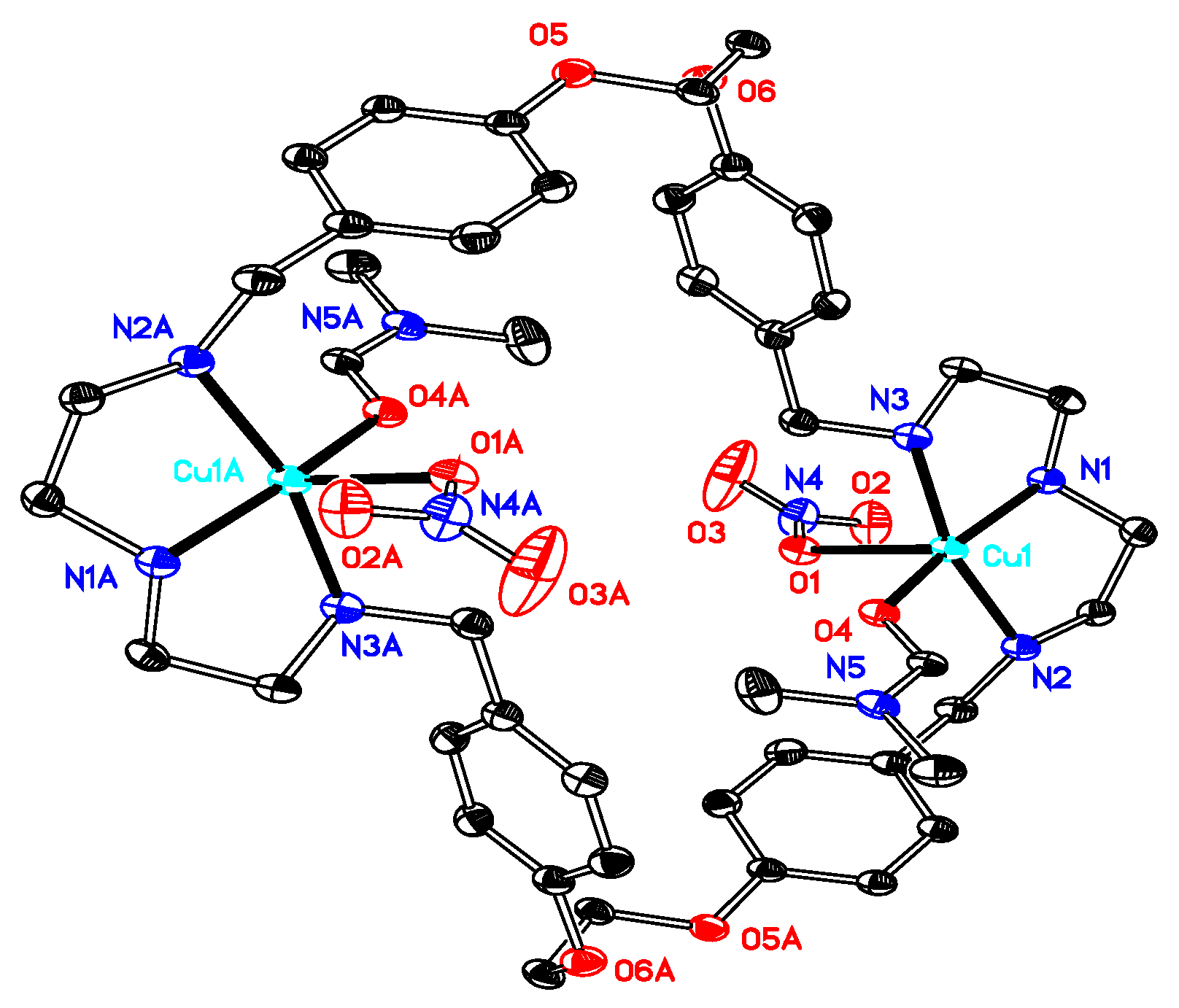
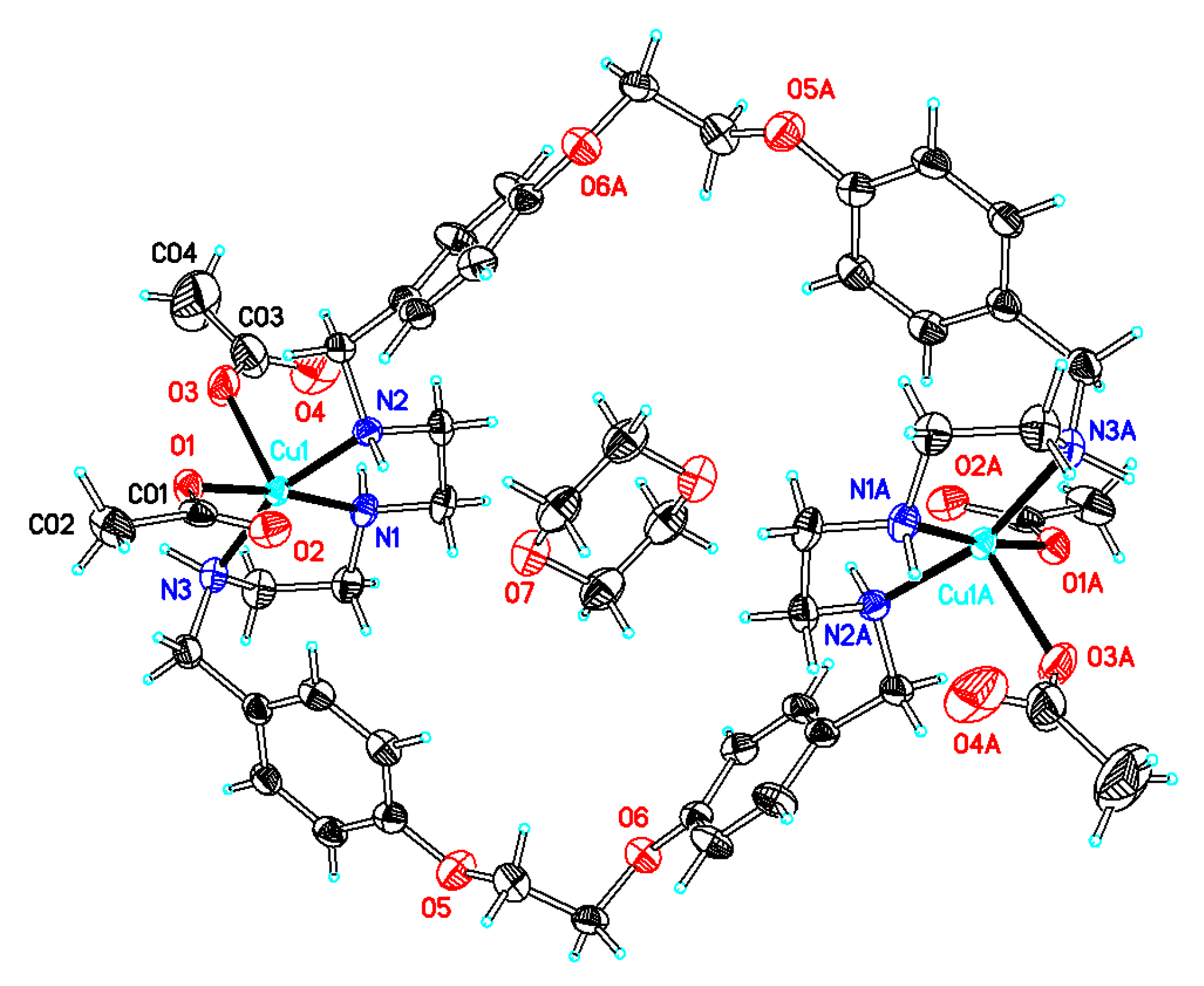
Synthesis of[Cu2(OCOMe)4L] (compound 6). The starting materials Cu(CO2Me)2·H2O (0.059 g, 0.30 mmol) and L·CH3OH·2.5H2O (0.10 g, 0.13 mmol) were dissolved in 20 mL methanol and stirred for 2 h. After filtration, the filtrate was concentrated under vacuum, and diethyl ether was added. A dark blue solid was obtained (0.15 g). Dark blue crystals of compound 6 suitable for X-ray diffraction analyses were obtained upon dissolution of 0.10 g of the compound in 15 mL of a 2:1 mixture of methanol and 1,4-dioxane followed by slow evaporation at room temperature (0.067 g, yield 67% based on L). Anal. calcd for C48H66Cu2N6O12·C4H8O2·5H2O (C52H84Cu2N6O19; mw 1224.34): C 51.01; H 6.92; N 6.86. Found: C 50.75; H 6.96; N 6.80. TG-DTA: CuO% = 13.9, weight% (lost for solvents) = 6.9. Calc with the formula of [Cu2(CO2CH3)4L]·0.5C4H10O2·2.5H2O (C50H76Cu2N6O15.5; mw 1136.3): CuO% = 14.0, weight% (lost for the solvents) = 7.9. IR (KBr disc) (cm−1): 3433 (vs), 3239 (m), 2931 (s), 2881 (m), 1614 (m), 1570 (vs), 1514 (s, νaryl-H), 1409 (s), 1335 (m), 1306 (m), 1251 (s), 1181 (m), 1117 (m), 1053 (s), 985 (m), 919 (m), 869 (w), 847 (m), 767 (w), 666 (m), 618 (m); UV–Vismax in H2O or DCM/MeOH, nm (ε, M−1 cm−1): 603 (45), 270 (252) and 222 (877), or 612 (33) and 271 (527). ΛM in H2O or DCM/MeOH: 28 or 10 Scm2mol–1.
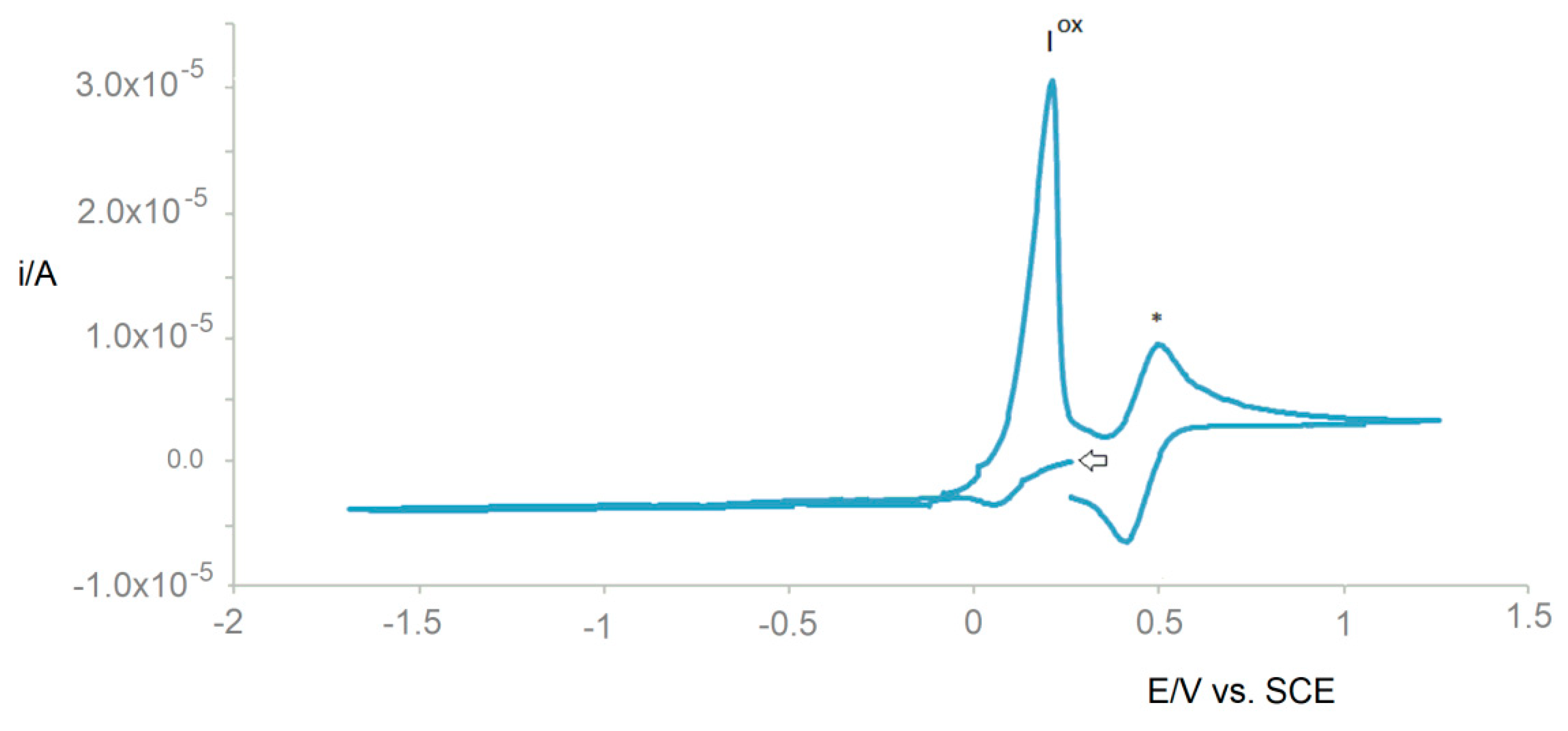
In 1 M K2CO3 aqueous solution, after 20 h at 70 °C: UV–Vismax in H2O or DCM/MeOH, nm (ε, M−1 cm−1): 606 (8), 268 (1771), and 221 (2056) or 610 (<8) and 270 (476). ΛM in H2O or DCM/MeOH: 248 Scm2mol−1.

 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
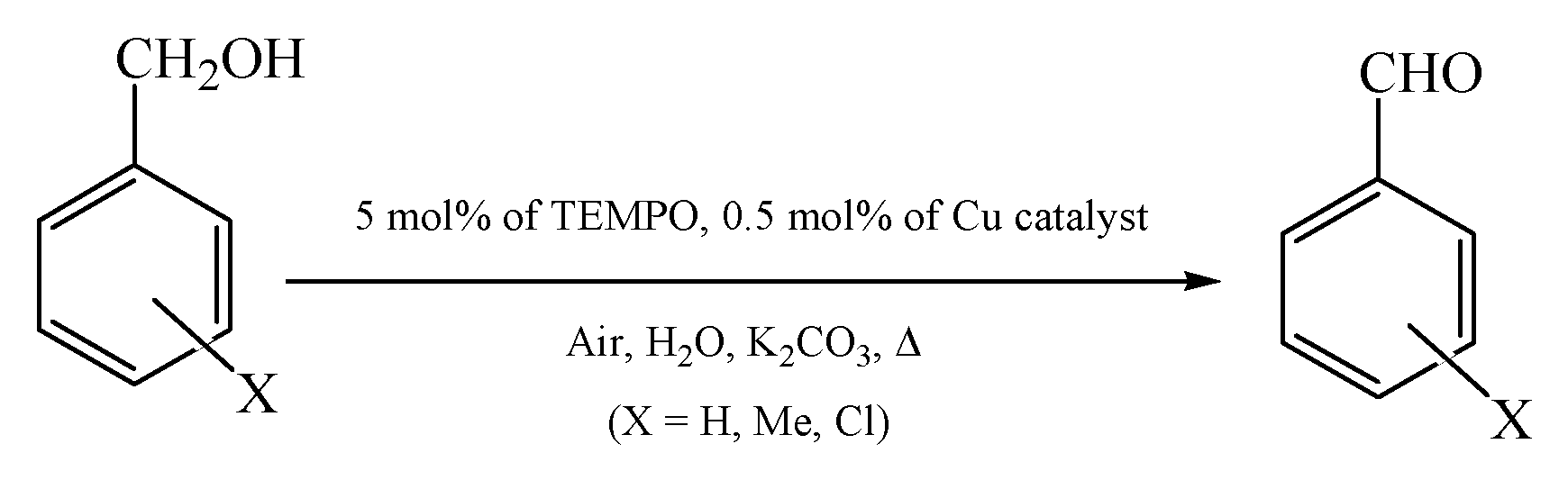{kind=link}
{kind=link}
{kind=link}