
Realistic Modelling of Dynamics at Nanostructured Interfaces Relevant to Heterogeneous Catalysis
by
 , and
, and
Kevin Rossi
1,*, 
Tzonka Mineva
2,
Jean-Sebastien Filhol
2,
Frederik Tielens
3 and
Hazar Guesmi
2,* 1
Ecole Polytechnique Federale de Lausanne, Laboratory of Nanochemistry for Energy, 1950 Sion, Switzerland
2
ICGM, University Montpellier, CNRS, ENSCM, 34090 Montpellier, France
3
General Chemistry (ALGC), Materials Modeling Group, Vrije Universiteit Brussel (Free University Brussels-VUB), Pleinlaan 2, 1050 Brussel, Belgium
*
Authors to whom correspondence should be addressed.
Catalysts 2022, 12(1), 52; https://doi.org/10.3390/catal12010052
Submission received: 5 December 2021
/
Revised: 27 December 2021
/
Accepted: 30 December 2021
/
Published: 4 January 2022
(This article belongs to the Special Issue Reactivity and Structural Dynamics of Catalysts)
{kind=link}
{kind=link}
Abstract
:The focus of this short review is directed towards investigations of the dynamics of nanostructured metallic heterogeneous catalysts and the evolution of interfaces during reaction—namely, the metal–gas, metal–liquid, and metal–support interfaces. Indeed, it is of considerable interest to know how a metal catalyst surface responds to gas or liquid adsorption under reaction conditions, and how its structure and catalytic properties evolve as a function of its interaction with the support. This short review aims to offer the reader a birds-eye view of state-of-the-art methods that enable more realistic simulation of dynamical phenomena at nanostructured interfaces by exploiting resource-efficient methods and/or the development of computational hardware and software.
1. Introduction
In heterogeneous catalysis, atomic and electronic structures of surface atoms and their specific interactions with the local environment (gas, liquid solvent, support) govern the binding and the release of reactant molecules, thus controlling the catalytic reaction mechanisms [1]. In this context, computational models have a long-standing tradition in the study of phenomena at solid–gas and solid–liquid interfaces, which determine the activity, selectivity, and lifetime of a catalyst [2,3]. In fact, numerical simulations can provide a full atomistic and electronic structural insight into the process of interest. Furthermore, numerical simulations are of great aid in the deconvolution and control of input parameters, such as temperature, pressure, chemical composition, and the nature of the species in the system—i.e., the manifold variables that strongly contribute to the complexity of dynamical processes at the interfaces. This is rarely accessible to current characterization measurements—especially for the case of in situ and in operando studies, where the interface structure may undergo frequent changes [4,5], notwithstanding the recent advancements in the field, which have been reviewed in great detail elsewhere [6,7,8]. The purpose of this short review is to survey some significant milestones in the modelling of dynamic phenomena occurring at the interfaces in the heterogeneous catalysts, with an emphasis on works related to metallic or oxide nanoparticle (NP) catalysts. Advancements in the theory-guided understandings of the dynamical organization of three relevant interfaces in heterogeneous catalysis—namely, the catalyst–solvent, catalyst–gas, and catalyst–support interfaces—will be discussed.
2. Catalyst-Solvent Interface
An understanding of the solvent–metal structures is key to rationalizing catalytic processes. Indeed, depending on the reaction of interest, the solvent may act as a relatively innocent spectator, influence activity and selectivity by interacting with adsorbates—e.g., by hydrogen bonding with key intermediates—or serve as a proton donor. Because of the ubiquitous relevance of water in catalytic reactions, water–metal and water–metal oxide interfaces have drawn great interest from the heterogeneous catalysis community. Ab initio density functional theory (DFT) modelling has been adopted, together with molecular-dynamics-based sampling, to probe the structure, dynamics, and acidity of water monolayers, double-layers, and over the surfaces of many late transition metals—Cu [9,10], Ag [10,11], Au [9,11,12], Pt [10,12,13,14], Pd [12,13,15], Rh [13], Ru [16], Ni [17], and their alloys [9]—under a number of different conditions. Similarly, the interface between oxides and water was assessed for systems of different natures—namely, silica [18,19], ZnO [20,21,22], goethite [23], and alumina [24,25]. In the field of electrocatalysis, the dynamics at electrified metal surface–water [26,27,28,29,30,31] and oxide surface–water [32,33] interfaces have also been recently modelled and characterized in detail.
When moving from studies on pristine, flat surfaces to those on nanostructured catalysts, we find that they are lesser in number and more recent. We could rationalize this trend in terms of the computational cost required to achieve accurate and realistic simulations of these systems. While works on pristine surfaces can provide information transferable to surface science experiments or to studies involving large and pristine nanocrystals, we believe that it is only by explicitly considering nanoscale features in the catalyst—e.g., curvature effects, edges, vertices, and step sites unique to nanocatalysts—that insightful and non-trivial inferences on nanostructured water–metal interface properties can be obtained.
The few reports appearing in the literature support this statement. Ferreira de Morais et al. [34] utilized MD with a DFT potential energy landscape (ab initio MD) to characterize the interface between a Pt nanoparticle of 201 atoms and 689 explicitly considered water molecules. They observed a re-organization of water molecules at the water–Pt201 interface. This interface consists of an average of two physisorbed water molecules on terraces, which is twice the number of chemisorbed water molecules on edges and corners. The unique adsorption properties of low-coordinated atoms were also highlighted by Soria et al. [35]. These authors adopted a ReaxFF interaction modelling scheme and molecular dynamics to characterize the hydration shells of spherical TiO2 nanoparticles. Their simulations showed that water molecules adsorb in both molecular and dissociative states on the surface. Depending on water concentration, single water molecules’ dissociation mechanisms (low coverage) or Grotthuss-like mechanisms (high coverage) are observed. Several water molecules can bind to the nanoparticle surface simultaneously, with adsorption being enhanced at low-coordinated Ti atoms that do not find a counterpart in low-energy TiO2 surface terminations. Previously, Fazio et al. [36] focused on the same system, and adopted a multiscale multistep integration scheme to prove that the nanoparticle–water interface affects the water molecules’ dipolar orientation in a solvation shell with a thickness of 5 Å, while water structuring was observed up to a distance of 8 Å from the nanoparticle surface. The hydration shell structures around oxide nanoparticles have been also discussed by Liu et al. [37], who used multiscale quantum mechanics/molecular mechanics (QM/MM) simulations exploiting force fields and tight-binding methods to unravel the short-range and long-range arrangement of water molecules around a spherical Fe3O4 nanoparticle. Almost half of the water molecules in the first hydration shell dissociate and hydroxylate the nanoparticle. Clear signatures in the radial distribution function of water around the nanoparticle enable the detection of a well-structured second and third hydration shell, while water molecules at distances ≥5 Å from the nanoparticle organize in bulk-like arrangements. The immersion of Fe3O4 nanoparticles in water was further found to improve their magnetic properties.
Solvent–catalyst interaction may also induce significant restructuring of the catalyst itself. Thermodynamic models indeed predict rich adsorbate coverage–temperature-phase diagrams, growth increasingly complex when further including eventual external electrochemical potential [38,39]. Zhu et al. [40] developed an environmental model based on the density functional theory, the Wulff construction, and the Langmuir adsorption isotherm to explore the shape evolutions of several transition metal NPs exposed to water vapor pressure at different temperatures. The predicted results show that despite the weak metal–water interactions, the evolution of water coverage on different facets could induce dramatic shape changes, depending on the temperature and the water vapor pressure. More recently, Chan et al. [41] adopted a first-principles approach and a micro-solvation model (one water layer) to draw insight on the stability of different closed-shell Au nanoparticles and determine the nature of the rearrangement mechanisms, which stabilize icosahedral shapes with respect to Ino decahedra. These authors found that the adsorption strength per water molecule following a rearrangement is independent of the nanoparticle size and shape, while the water molecule networks around the nanoparticles consist of H2O molecules linked, by and average of two hydrogen bonds. To sample longer time scales, Delgado et al. [42] utilized an implicit potential to model the attractive interaction between metal atoms and solvent molecules. They showed that such metal–adsorbate interactions diminish the nanoparticles’ thermal stability against solid–solid and solid–liquid transitions. Similarly, Braunwarth et al. [43] observed the long-term stability of Pt cubes solvated in water by means of a ReaxFF potential. The cubes underwent a strong restructuring into concave disordered cuboids after a few ns of simulation.
3. Catalyst-Gas Interface
In gas-phase reactions, the dynamic interaction takes place between the surface of the nanostructured catalyst and the gas molecules, adsorbed intermediates, and products form the catalyst−gas interface, which directly impact the available reaction pathways. In addition, the catalyst−gas interactions are expected to modify the catalyst structure by changing its shape and surface organization [44,45,46], as well as the atomic ordering in the case of alloy catalysts [47,48,49,50,51,52]. As these dynamic structural changes modulate the activity, selectivity, and stability of NP catalysts, probing the dynamic evolution of surface structure and chemical ordering at the atomic scale under reaction conditions is crucial to providing a fundamental understanding of reaction mechanisms.
In spite of the huge number of studies in the literature devoted to metal or metal oxide catalyst–gas interactions, few theoretical studies have been dedicated to the investigation of the dynamics of nanostructured catalysts under reactive conditions. In general, to predict the effects of gas adsorption on metallic nanocatalysts, DFT-based geometric optimization methods are used to calculate and compare the stability of different conformations of small metal clusters interacting with adsorbed gas molecules [53]; however, this method is limited to systems of a few dozen atoms. The prediction of equilibrium shape evolution of larger metallic nanoparticles interacting with gas molecules has been generally resolved by means of Wulff constructions based on DFT calculations instead. This approach, however, does not account for dynamical contributions. In addition, Wulff’s construction theorem is only reliable for NP size > 5 nm, and disregards the possibilities of disordered surfaces and twinning.
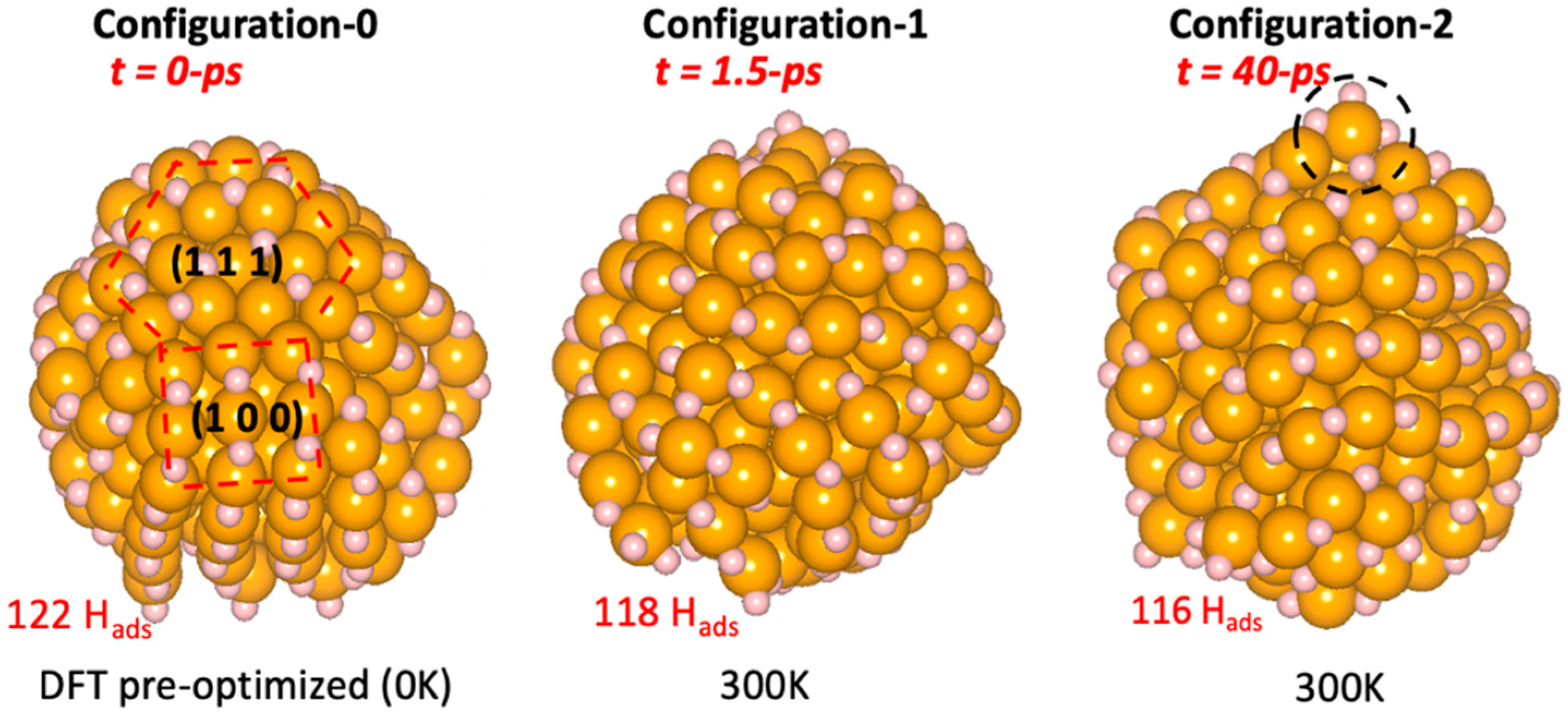
In the last few years, thanks to the strong progress in computational resources, it has been feasible to sample DFT potential energy surfaces via ab initio molecular dynamics (AIMD), exploiting the Born–Oppenheimer (BOMD) or Car–Parrinello (CPMD) formalism. These approaches have emerged as powerful tools to explore the conformational changes of metal NPs when exposed to different pressures of molecules. Very recently, Nassereddine et al. [54] employed BOMD simulations to provide insights on the atomic-scale mechanisms that underpin the experimentally observed size-dependent structural dynamics of Au NPs exposed to atmospheric hydrogen pressure. These authors monitored the structural evolution of hydrogen-covered Au NPs of 2 nm in size for more than 60 ps of simulation time, in order to demonstrate that the strong Au–H interactions may induce a high mobility of gold atoms, which drastically change the shape and the symmetry of the whole NP at room temperature (Figure 1). Similarly, by performing AIMD simulations, He et al. [55] revealed size-dependent order-to-disorder transformation of ultrasmall gold clusters supported on ceria upon exposure to carbon monoxide and oxygen. This transformation was found to lead to the generation of dynamic low-coordinated atoms, which may boost the oxidation reaction of carbon monoxide. Thermodynamic models of gas–catalyst interaction inducing dynamical changes become even more complex when it comes to the case of nanoalloys. Restructuring, reshaping, segregation, and many other processes may occur, and must be taken in account [56].
In addition to the well-known solid-state nanosized alloy catalysts, new emerging classes of materials—namely, single-atom alloy catalysts (SAACs) [50,51] and supported catalytically active liquid metal solutions (SCALMSs)—have been shown to undergo significant changes during reactions. In a recent work, Wang et al. [52] reported the stability phase diagram of single-atom Pt anchored in various metal host NPs as a function of hydrogen pressures and temperatures. This work shows non-intuitive segregation behavior of SAACs where the stability of diluted single atoms is found to depend on either the electronic and geometric ensembles or the affinity of both alloy elements to the reactive gas. Humphrey et al. [57] performed picosecond-long AIMD to show that the diffusion properties of single Pt atoms on a rutile TiO2 (110) support are dictated by the interaction strength with adsorbates and supports, where strong metal–adsorbate interactions determine higher diffusion coefficients. Bauer et al. [58] have recently reported in operando DRIFT and DFT results of GaxPty catalyst systems, showing interesting dynamical behavior of isolated Pt atoms. Running AIMD simulations of Pt atoms evolving in Ga matrixes as a function of time and temperature, these authors showed that the rate of appearance at the surface of a single Pt atom depends on several factors, such as the size of the NP and the mobility of the atoms which, in turn, depend on the temperature and the composition of the catalyst. At temperatures higher than 350 °C, a homogeneous and liquid Ga/Pt metal phase appears, leading to a higher mobility of atoms; thus, the ability of Pt to reach the surface and to interact with the reactant becomes stronger. In addition, the residence time of Pt at the surface is found to be strongly influenced.
4. Catalyst-Support Interface
It is well documented that the arrangements of the catalyst–support interface can have a tremendous impact on the reactivity of the catalyst’s adsorption sites which, in turn, may cause an increase in the catalyst’s activity and/or selectivity due to geometric and/or electronic effects. By the same token, support–catalyst interaction can dramatically affect the stability of the latter with respect to the gas phase. Conversely, the effects of electronic [61], reactive [62], and strong metal–support interactions [63,64] have been strong drivers in the study and analysis of the dynamics of supported nanostructured metal catalysts.
To investigate the dynamics at the support–catalyst interface, ab initio simulations stand as one of the most promising tools. These are key tools to monitor the time- and condition-dependent evolution of the structure of a supported nanocluster. Vila et al. [65] utilized DFT modelling and molecular dynamics to characterize the peculiar negative thermal expansion and large structural disorder of Pt nanoparticles supported on a gamma-Al2O3 surface in terms of a combination of thermal vibrations and low-frequency disorder. Vila et al. [66] previously fitted a hybrid version of the classical Sutton–Chen force field to predict diffusion coefficients and bond-breaking rates as a function of the nanocluster size. By looking at sizes between 10 and 20 atoms, they identified size-dependent dynamical changes in the thermally driven phase change and diffusion of the nanoparticles. Xu et al. [67] adopted AIMD to demonstrate the complexity of the structural evolution of Au–Pd nanoalloys deposited on rutile TiO2, as a function of different in operando conditions. These authors ran simulations for 40–50 ps of the Au–Pd cluster of 38 atoms deposited on a four-layer (7 × 3) rutile TiO2 (110) support. The results show that when simulating reducing conditions (the adsorption of one hydrogen atom), Pt atoms are more likely to lie at the metal–oxide interface. Conversely, under oxidizing conditions (interactions with oxygen), Pd atoms are more likely to be located at the surface of the nanocluster.
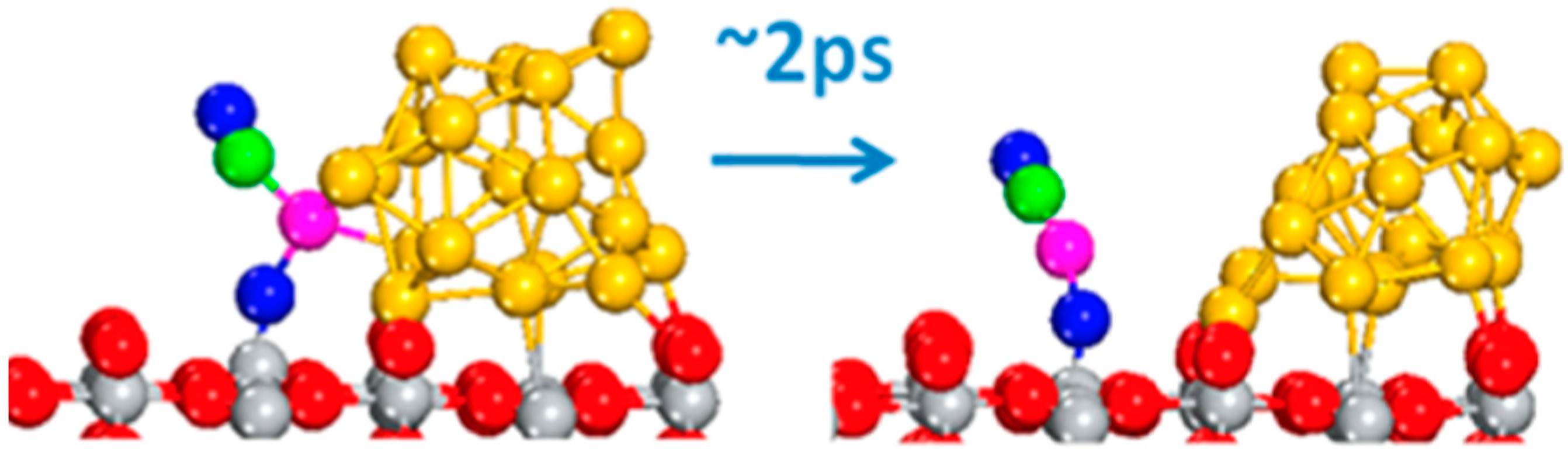
Structural properties and catalytic activity are intimately interlinked. In turn, simulations of the dynamical evolution of the system can capture and reveal non-trivial catalytic reaction pathways. Li et al. [68] utilized ab initio molecular dynamics to monitor the time-dependent evolution of the geometric and electronic structure in a Pt nanocluster supported on CeO2. They identified the dynamic nature of the Pt0−O vacancy, with Ce3+ sites as the key to rationalizing activity trends for water gas shift catalyzed by this system. Similarly, Wang et al. [69] considered Au nanoclusters on ceria, and utilized AIMD to unravel a previously unreported single-atom mechanism through which the system can drive the oxidation of CO. A gold cation can momentarily break away from the gold nanoparticle to catalyze CO oxidation by moving adjacent to the metal–oxide interface (Figure 2), and then re-insert into the nanocluster after the reaction is completed. Additionally, in relation to the dynamical nature of active sites in supported catalysts, Daelman et al. exploited AIMD to show that metal charge of single Pt atoms on CeO2 (100) surfaces evolves over time; they thus rationalized these strong metal–support interactions in terms of the relative position of the Ce 4f levels relative to the ones in Pt, allowing for electron injection and withdrawal between the two [70]. The dynamical modification of the metallic charge further enabled the rationalization of the high reactivity of single Pt atoms for CO oxidation.
Moving from nanoclusters (a few tens of atoms at maximum) to nanoparticles (i.e., systems with at least a few hundred of atoms), several works have focused on the development of accurate interatomic potential parameters to reproduce and predict the effect of the support on the stability of the nanostructured catalyst. Ngajdong et al. [71] fitted an explicit analytical potential to DFT in order to model dynamics at the Ag–SiO2 interface; they demonstrated that the likelihood of observing Ag nanoparticles with twin boundaries decreases when a SiO2 support is considered; furthermore, they observed that melting initiates at the interface between the nanoparticle and the substrate. In the same year, Rossi et al. [72] showed that the support induces a wetting of the nanoparticle, crystalline rather than polycrystalline morphologies, and plays a strong role in stabilizing the nanoparticles against solid–liquid transition. This conclusion was achieved from the analysis of simulations adopting an implicit metal oxide force field parametrized on DFT data.
In addition to oxide supports, carbon also finds an application in devices—often as a substrate where metallic nanocatalysts are deposited. This motivation, together with the accuracy with which carbon–carbon and carbon–metal interactions can be modelled while utilizing efficient approaches, has stimulated a number of investigations on carbon–metal interfaces. Foster et al. [73] utilized an implicit approach where atoms were fixed in contact with the implicit carbon support, in order to show that the substrate tends to increase the stability of the nanocatalysts against solid–liquid transitions. Previously, Morrow et al. [74] and Qiu et al. [75] fitted a semi-empirical force field to DFT data in order to study the stability of metal nanoparticles on carbon nanostructures, which are modelled explicitly. The carbon support nanostructure was identified as a tunable parameter to control nanoparticle size and chemical composition. Later, Cheng et al. [76] also adopted a reactive force field to present an additional proof that the carbon support structure is a free parameter to modify the coordination number distribution of surface sites in a Pt nanoparticle. These changes, in turn, affected the catalytic properties of the system, where the abundance of highly coordinated sites was found to correlate with the activity for CO oxidation.
5. Outlook: Reactive Events at Support/Catalyst/Gas-Liquid Interfaces
The accurate modelling of dynamical processes at the catalyst–solvent, catalyst–gas, and catalyst–support interfaces has thus far benefited greatly not only from the improvement in the hardware performance—which enables faster computations—but also from the advancement in the methods and algorithms with which interatomic interactions are predicted. The investigations we have reported in this short review, in our opinion, represent significant and paradigmatic examples of these trends.
In the future, we believe that the explicit simulation of reactive events—while adopting a non-idealized description of the catalyst, its support, the solvent or the gas environment, and the applied potential for electrified interfaces—is likely to become the state of the art. To date, the investigations have mostly been devoted to pristine surfaces. Example studies of the reconstruction of pristine surfaces in the presence of a solvent have been recently reported by Natarajan et al. [77] and Michalka et al. [78]. The former developed a neural network force field for Cu, H, and O interaction, and utilized it to sample the self-diffusion of adatoms at the interface between a Cu surface and bulk water; they concluded that hopping mechanisms are more favorable than exchange mechanisms, and that Cu–water interaction lowers the barrier associated with self-diffusion. The latter utilized an analytical force field fitted to DFT data to simulate CO-induced restructuring on stepped Pt surfaces, and identified a stochastic edge-doubling nucleation followed by zippering as the most likely reconstruction mechanism.
Notable examples for the case of chemical reactions on pristine surfaces are represented by the studies of Goddard III et al. [79,80]—who utilized ab initio MD and enhanced sampling methods to pinpoint critical intermediates in electrochemical CO2 reduction—and by Vandermause et al. [81], who fitted a Gaussian process of many-body potential to probe reactive events at the H2–Pt interface, and predicted an activation energy for H2 splitting and recombination in striking agreement with the experimental value. Machine learning has also been instrumental in the simulation of reactions in solutions, such as urea decomposition in water [82], proton transfer in Na/water mixtures [83], and CH3SO3H deprotonation in phenol and phenol/H2O2 solvents [84].
Author Contributions
H.G. and K.R. conceived the ideas of this short review. All authors contributed to the preparation of the paper. H.G. and K.R. supervised the project. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
All DFT and MD simulation results depicted in Figure 1 are available from the corresponding author H.G. on request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martinez, T.J. Ab initio reactive computer aided molecular design. Acc. Chem. Res. 2017, 50, 787–795. [Google Scholar] [CrossRef]
- Collinge, G.; Yuk, M.T.; Nguyen, S.F.; Lee, M.S.; Glezakou, V.A.; Rousseau, R. Effect of collective dynamics and anharmonicity on entropy in heterogenous catalysis: Building the case for advanced molecular simulations. ACS Catal. 2020, 10, 9236–9260. [Google Scholar] [CrossRef]
- Grajciar, L.; Heard, C.J.; Bondarenko, A.A.; Polynski, M.V.; Meeprasert, J.; Pidko, E.A.; Nachtigall, P. Towards operando computational modeling in heterogeneous catalysis. Chem. Soc. Rev. 2018, 47, 8307–8348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timoshenko, J.; Cuenya, B.R. In situ/operando electrocatalyst characterization by x-ray absorption spectroscopy. Chem. Rev. 2021, 121, 882–961. [Google Scholar] [CrossRef]
- He, B.; Zhang, Y.; Liu, X.; Chen, L. In-situ transmission electron microscope techniques for heterogeneous catalysis. ChemCatChem 2020, 12, 1853–1872. [Google Scholar] [CrossRef]
- Rupprechter, G. Operando Surface Spectroscopy and Microscopy during Catalytic Reactions: From Clusters via Nanoparticles to Meso-Scale Aggregates. Small 2021, 17, 2004289. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, H.; Yu, Y.; Liu, Z. In Situ Characterization of Catalysis and Electrocatalysis Using APXPS. ACS Catal. 2021, 11, 1464–1484. [Google Scholar] [CrossRef]
- Li, X.; Yang, X.; Zhang, J.; Huang, Y.; Liu, B. In Situ/Operando Techniques for Characterization of Single-Atom Catalysts. ACS Catal. 2019, 9, 2521–2531. [Google Scholar] [CrossRef]
- Clabaut, P.; Fleurat-Lessard, P.; Michel, C.; Steinmann, S.N. Ten facets, one force field: The GAL19 force field for water–noble metal interfaces. J. Chem. Theory Comput. 2020, 16, 4565–4578. [Google Scholar] [CrossRef] [PubMed]
- Heenen, H.H.; Gauthier, J.A.; Kristoffersen, H.H.; Ludwig, T.; Chan, K. Solvation at metal/water interfaces: An ab initio molecular dynamics benchmark of common computational approaches. J. Chem. Phys. 2020, 152, 144703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gim, S.; Cho, K.J.; Lim, H.K.; Kim, H. Structure, dynamics, and wettability of water at metal interfaces. Sci. Rep. 2019, 9, 14805. [Google Scholar] [CrossRef] [Green Version]
- Le, J.; Cuesta, A.; Cheng, J. The structure of metal-water interface at the potential of zero charge from density functional theory-based molecular dynamics. J. Electroanal. Chem. 2018, 819, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Bellarosa, L.; García-Muelas, R.; Revilla-López, G.; López, N. Diversity at the water–metal interface: Metal, Water Thickness, and Confinement Effects. ACS Cent. Sci. 2016, 2, 109–116. [Google Scholar] [CrossRef]
- Lan, J.; Rybkin, V.V.; Iannuzzi, M. Ionization of water as an effect of quantum delocalization at aqueous electrode interfaces. J. Phys. Chem. Lett. 2020, 11, 3724–3730. [Google Scholar] [CrossRef] [PubMed]
- Filhol, J.S.; Neurock, M. Elucidation of the Electrochemical Activation of Water over Pd by First Principles. Angew. Chem. Int. Ed. 2006, 45, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Lespes, N.; Filhol, J.S. Using the electrochemical dimension to build water/Ru(0001) phase diagram. Surf. Sci. 2015, 631, 8–16. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Michel, C.; Schwiedernoch, R.; Filhol, J.S.; Sautet, P.H. Modeling the HCOOH/CO2 electrocatalytic reaction: When details are key. Chem. Phys. Chem. 2015, 16, 2307–2311. [Google Scholar] [CrossRef] [PubMed]
- Sulpizi, M.; Gaigeot, M.P.; Sprik, M. The silica–water interface: How the silanols Determine the surface acidity and modulate the water properties. J. Chem. Theory Comput. 2012, 8, 1037–1047. [Google Scholar] [CrossRef]
- Gierada, M.; De Proft, F.; Sulpizi, M.; Tielens, F. Understanding the Acidic Properties of the Amorphous Hydroxylated Silica Surface. J. Phys. Chem. C 2019, 123, 17343–17352. [Google Scholar] [CrossRef]
- Tocci, G.; Michaelides, A. Solvent-induced proton hopping at a water–oxide interface. J. Phys. Chem. Lett. 2014, 5, 474–480. [Google Scholar] [CrossRef] [Green Version]
- Hellström, M.; Quaranta, V.; Behler, J. One-dimensional vs. two-dimensional proton transport processes at solid–liquid zinc-oxide–water interfaces. Chem. Sci. 2018, 10, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Quaranta, V.; Behler, J.; Hellström, M. Structure and Dynamics of the Liquid–Water/Zinc-Oxide Interface from Machine Learning Potential Simulations. J. Phys. Chem. C 2018, 123, 1293–1304. [Google Scholar] [CrossRef]
- Zarzycki, P.; Rosso, K.M. Surface charge effects on Fe(II) sorption and oxidation at (110) goethite surfaces. J. Phys. Chem. C 2018, 122, 10059–10066. [Google Scholar] [CrossRef]
- Réocreux, R.; Jiang, T.; Iannuzzi, M.; Michel, C.; Sautet, P. Structuration and dynamics of interfacial liquid water at hydrated γ-alumina determined by ab initio molecular simulations: Implications for nanoparticle stability. ACS Appl. Nano Mater. 2017, 1, 191–199. [Google Scholar] [CrossRef]
- Réocreux, R.; Girel, É.; Clabaut, P.; Tuel, A.; Besson, M.; Chaumonnot, A.; Cabiac, A.; Sautet, P.; Michel, C. Reactivity of shape-controlled crystals and metadynamics simulations locate the weak spots of alumina in water. Nat. Comm. 2019, 10, 3139. [Google Scholar] [CrossRef] [PubMed]
- Le, J.; Iannuzzi, M.; Cuesta, A.; Cheng, J. Determining Potentials of Zero Charge of Metal Electrodes versus the Standard Hydrogen Electrode from Density-Functional-Theory-Based Molecular Dynamics. Phys. Rev. Lett. 2017, 119, 016801–016807. [Google Scholar] [CrossRef] [Green Version]
- Le, J.B.; Yang, X.H.; Zhuang, Y.B.; Jia, M.; Cheng, J. Recent progress toward ab Initio modeling of electrocatalysis. J. Phys. Chem. Lett. 2021, 12, 8924–8931. [Google Scholar] [CrossRef] [PubMed]
- Sakong, S.; Groß, A. Water structures on a Pt(111) electrode from ab initio molecular dynamic simulations for a variety of electrochemical conditions. Phys. Chem. Chem. Phys. 2018, 22, 10431–10437. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K.; Sundararaman, R. The electrochemical interface in first-principles calculations. Surf. Sci. Rep. 2020, 75, 100492. [Google Scholar] [CrossRef] [PubMed]
- Le, J.B.; Chen, A.; Li, L.; Xiong, J.F.; Lan, J.; Liu, Y.P.; Iannuzzi, M.; Cheng, J. Modeling electrified Pt(111)-Had/water interfaces from ab Initio molecular dynamics. JACS Au 2021, 1, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Khatib, R.; Kumar, A.; Sanvito, S.; Sulpizi, M.; Cucinotta, C.S. The nanoscale structure of the Pt-water double layer under bias revealed. Electrochim. Acta 2021, 391, 138875. [Google Scholar] [CrossRef]
- Laporte, S.; Finocchi, F.; Paulatto, L.; Blanchard, M.; Balan, E.; Guyot, F.; Saitta, A.M. Strong electric fields at a prototypical oxide/water interface probed by ab initio molecular dynamics: MgO(001). Phys. Chem. Chem. Phys. 2015, 17, 20382–20390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hutter, J.; Sprik, M. Coupling of Surface Chemistry and Electric Double Layer at TiO2 Electrochemical Interfaces. J. Phys. Chem. Lett. 2019, 10, 3871–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, R.F.; Kerber, T.; Calle-Vallejo, F.; Sautet, P.; Loffreda, D. Capturing Solvation Effects at a Liquid/Nanoparticle Interface by Ab Initio Molecular Dynamics: Pt201 Immersed in Water. Small 2016, 12, 5312–5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, F.A.; Di Valentin, D. Reactive molecular dynamics simulations of hydration shells surrounding spherical TiO2 nanoparticles: Implications for proton-transfer reactions. Nanoscale 2021, 13, 4151–4166. [Google Scholar] [CrossRef]
- Fazio, G.; Selli, D.; Ferraro, L.; Seifert, G.; Di Valentin, C. Curved TiO2 Nanoparticles in Water: Short (Chemical) and Long (Physical) Range Interfacial Effects. ACS App. Mat. Int. 2018, 10, 29943–29953. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Siani, P.; Bianchetti, E.; Zhao, J.; Di Valentin, C. Multiscale simulations of the hydration shells surrounding spherical Fe3O4 nanoparticles and effect on magnetic properties. Nanoscale 2021, 13, 9293–9302. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Chem, J.F.; Wang, H.F.; Hu, P.J.; Ma, W.; Long, Y.T. Exploring dynamic interactions of single nanoparticles at interfaces for surface-confined electrochemical behavior and size measurement. Nat. Comm. 2020, 11, 2307. [Google Scholar] [CrossRef]
- Khelfa, A.; Meng, J.; Byun, C.; Wang, G.; Nelayah, J.; Ricolleau, C.; Amara, H.; Guesmi, H.; Alloyeau, D. Selective shortening of gold nanorods, when surface functionalization dictates the reactivity of nanostructures. Nanoscale 2020, 12, 22658–22667. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, Z.; Wang, C.; Gao, Y. Shape evolution of metal nanoparticles in water vapor environment. Nano Lett. 2016, 16, 2628–2632. [Google Scholar] [CrossRef]
- Chan, C.H.; Poignant, F.; Beuve, M.; Dumont, E.; Loffreda, D. A Water Solvation Shell Can Transform Gold Metastable Nanoparticles in the Fluxional Regime. J. Phys. Chem. Lett. 2019, 10, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Callico, L.; Rossi, K.; Pinto-Miles, R.; Salzbrenner, P.; Baletto, F. A universal signature in the melting of metallic nanoparticles. Nanoscale 2021, 13, 1172–1180. [Google Scholar] [CrossRef]
- Braunwarth, L.; Jung, C.; Jacob, T. Exploring the Structure–Activity Relationship on Platinum Nanoparticles. Top. Catal. 2020, 63, 1647–1657. [Google Scholar] [CrossRef]
- Hansen, P.L.; Wagner, J.B.; Helveg, S.; Rostrup-Nielsen, J.R.; Clausen, B.S.; Topsøe, H. Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science 2002, 295, 2053–2055. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Lee, S.; Sell, K.; Barke, I.; Kleibert, A.; von Oeynhausen, V.; Meiwes-Broer, K.H.; Rodríguez, A.F.; Elam, J.W.; Pellin, M.M.; et al. Combined temperature-programmed reaction and in situ X-ray scattering studies of size-selected silver clusters under realistic reaction conditions in the epoxidation of propene. J. Chem. Phys. 2009, 131, 121104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, A.I.; Meng, J.; Zhu, B.; Gao, Y.; Guesmi, H.; Prunier, H.; Alloyeau, D.; Wang, G.; Louis, C.; Delannoy, L.; et al. Reshaping Dynamics of Gold Nanoparticles under H2 and O2 at Atmospheric Pressure. ACS Nano 2019, 13, 2024–2033. [Google Scholar] [CrossRef]
- Guesmi, H. Theoretical insights on the effect of reactive gas on the chemical ordering of gold-based alloys. Gold Bull. 2013, 46, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Dhifallah, M.; Dhouib, A.; Aldulaijan, S.; DI Renzo, F.; Guesmi, H. First-principles study of Au–Cu alloy surface changes induced by gas adsorption of CO, NO, or O2. J. Chem. Phys. 2016, 145, 024701. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Zepeda, F.; Gatalo, M.; Pavlišič, A.; Dražić, G.; Jovanovič, P.; Bele, M.; Gaberšček, M.; Hodnik, N. Atomically Resolved Anisotropic Electrochemical Shaping of Nano-electrocatalyst. Nano Lett. 2019, 19, 4919–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Walsh, A.G.; Yu, J.; Zhang, P. Single-atom alloy catalysts: Structural analysis, electronic properties and catalytic activities. Chem. Soc. Rev. 2021, 50, 569–588. [Google Scholar] [CrossRef] [PubMed]
- Kiani, D.; Sourav, S.; Taifan, W.; Calatayud, M.; Tielens, F.; Wachs, I.E.; Baltrusaitis, J. Existence and Properties of Isolated Catalytic Sites on the Surface of β-Cristobalite-Supported, Doped Tungsten Oxide Catalysts (WOx/β-SiO2, Na-WOx/β-SiO2, Mn-WOx/β-SiO2) for Oxidative Coupling of Methane (OCM): A Combined Periodic DFT and Experimental Study. ACS Catal. 2020, 10, 4580–4592. [Google Scholar]
- Wang, Q.; Zhu, B.; Tielens, F.; Tichit, D.; Guesmi, H. Mapping surface segregation of single-atom Pt dispersed in M surfaces (M = Cu, Ag, Au, Ni, Pd, Co, Rh and Ir) under hydrogen pressure at various temperatures. Appl. Surf. Sci. 2021, 548, 149217. [Google Scholar] [CrossRef]
- Sun, G.; Sautet, P. Metastable Structures in Cluster Catalysis from First-Principles: Structural Ensemble in Reaction Conditions and Metastability Triggered Reactivity. J. Am. Chem. Soc. 2018, 140, 2812–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassereddine, A.; Wang, Q.; Loffreda, D.; Ricolleau, C.; Alloyeau, D.; Louis, C.; Delannoy, L.; Nelayah, J.; Guesmi, H. Revealing Size Dependent Structural Transitions in Supported Gold Nanoparticles in Hydrogen at Atmospheric Pressure. Small 2021, 17, 2104571. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, J.C.; Luo, L.; Wang, Y.G.; Zhu, J.; Du, Y.; Li, J.; Mao, S.X.; Wang, C. Size-dependent dynamic structures of supported gold nanoparticles in CO oxidation reaction condition. Proc. Natl. Acad. Sci. USA 2018, 115, 7700–7705. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.F.; Chee, S.W.; Baraissoc, Z.; Jin, H.; Tan, T.L.; Mirsaidov, U. Real-Time Imaging of Nanoscale Redox Reactions over Bimetallic Nanoparticles. Adv. Funct. Mater. 2019, 29, 1903242. [Google Scholar] [CrossRef]
- Humphrey, N.; Bac, S.; Sharada, S.M. Adsorbate-assisted migration of the metal atom in atomically dispersed catalysts: An ab initio molecular dynamics study. J. Chem. Phys. 2021, 154, 234709. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.; Maisel, S.; Blaumeiser, D.; Vecchietti, J.; Taccardi, N.; Wasserscheid, P.; Bonivardi, A.; Görling, A.; Libuda, J. Operando DRIFTS and DFT Study of Propane Dehydrogenation over Solid- and Liquid-Supported GaxPty Catalysts. ACS Catal. 2019, 9, 2842–2853. [Google Scholar] [CrossRef] [PubMed]
- Vecchietti, J.; Baltanás, M.A.; Gervais, C.; Collins, S.E.; Blanco, G.; Matz, O.; Calatayud, M.; Bonivardi, A. Insights on hydride formation over cerium-gallium mixed oxides: A mechanistic study for efficient H2 dissociation. J. Catal. 2017, 345, 258–269. [Google Scholar] [CrossRef]
- Lewandowska, A.E.; Calatayud, M.; Lozano-Diza, E.; Minot, C.; Bañares, M.A. Combining theoretical description with experimental in situ studies on the effect of alkali additives on the structure and reactivity of vanadium oxide supported catalysts. Catal. Today 2008, 139, 209–213. [Google Scholar] [CrossRef]
- Shi, Y.; Ma, Z.R.; Xiao, Y.-Y.; Yin, Y.-C.; Huang, W.-M.; Huang, Z.-C.; Zheng, Y.-Z.; Mu, F.-Y.; Huang, R.; Shi, G.-Y.; et al. Electronic metal–support interaction modulates single-atom platinum catalysis for hydrogen evolution reaction. Nat. Comm. 2021, 12, 3021. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cui, Y.; Wu, Z.; Milligan, C.; Zhou, L.; Mitchell, G.; Xu, B.; Shi, E.; Miller, J.T.; Ribeiro, F.H.; et al. Reactive metal–support interactions at moderate temperature in two-dimensional niobium-carbide-supported platinum catalysts. Nat. Catal. 2018, 1, 349–355. [Google Scholar] [CrossRef]
- Wu, P.; Tan, S.; Moon, J.; Yan, Z.; Fung, V.; Li, N.; Yang, S.-Z.; Cheng, Y.; Abney, C.-W.; Wu, Z.; et al. Harnessing strong metal–support interactions via a reverse route. Nature Comm. 2020, 11, 3042. [Google Scholar] [CrossRef] [PubMed]
- van Deelen, T.W.; Mejía, C.H.; de Jong, K.P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Vila, F.D.; Rehr, J.J.; Nuzzo, R.G.; Frenkel, A. Anomalous Structural Disorder in Supported Pt Nanoparticles. J. Phys. Chem. Lett. 2017, 8, 3284–3288. [Google Scholar] [CrossRef] [PubMed]
- Vila, F.D.; Hayashi, S.T.; Moore, J.M.; Rehr, J.J. Molecular dynamics simulations of supported pt nanoparticles with a hybrid Sutton-Chen potential. J. Phys. Chem. C 2016, 120, 14883–14891. [Google Scholar] [CrossRef]
- Xu, C.Q.; Lee, M.S.; Wang, Y.G.; Cantu, D.C.; Li, J.; Glezakou, V.A.; Rousseau, R. Structural Rearrangement of Au-Pd Nanoparticles under Reaction Conditions: An ab Initio Molecular Dynamics Study. ACS Nano 2017, 11, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kottwitz, M.; Vincent, J.L.; Enright, M.J.; Liu, Z.; Zhang, L.; Huang, J.; Senanayake, S.D.; Yang, W.C.D.; Crozier, P.A.; et al. Dynamic structure of active sites in ceria-supported Pt catalysts for the water gas shift reaction. Nat. Commun. 2021, 12, 914. [Google Scholar] [CrossRef]
- Wang, Y.G.; Cantu, D.C.; Lee, M.S.; Li, J.; Glezakou, V.A.; Rousseau, R. CO Oxidation on Au/TiO2: Condition-Dependent Active Sites and Mechanistic Pathways. J. Am. Chem. Soc. 2017, 138, 10467–10476. [Google Scholar] [CrossRef]
- Daelman, N.; Capdevila-Cortada, M.; López, N. Dynamic charge and oxidation state of Pt/CeO2 single-atom catalysts. Nat. Mat. 2019, 18, 1215–1221. [Google Scholar] [CrossRef] [Green Version]
- Ngandjong, A.C.; Mottet, C.; Puibasset, J. Freezing and melting of silver nanoparticles on silica substrate using a simple interatomic potential for Ag–SiO2 interaction on the basis of ab initio calculations and experimental Data. J. Phys. Chem. C 2017, 121, 3615–3622. [Google Scholar] [CrossRef]
- Rossi, K.; Ellaby, T.; Paz-Borbón, L.O.; Atanasov, I.; Pavan, L.; Baletto, F. Melting of large Pt@MgO(1 0 0) icosahedra. J. Phys. Condens Matter. 2017, 29, 145402. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.M.; Pavloudis, T.; Kioseoglou, J.; Palmer, R.E. Atomic-resolution imaging of surface and core melting in individual size-selected Au nanoclusters on carbon. Nat. Comm. 2019, 10, 2583. [Google Scholar] [CrossRef] [Green Version]
- Morrow, B.H.; Striolo, A. Platinum nanoparticles on carbonaceous materials: The effect of support geometry on nanoparticle mobility, morphology, and melting. Nanotechnology 2008, 19, 195711. [Google Scholar] [CrossRef]
- Qiu, C.; Zhao, C.; Sun, X.; Deng, S.; Zhuang, G.; Zhong, X.; Wei, Z.; Yao, Z.; Wang, J.G. Multiscale Simulation of Morphology Evolution of Supported Pt Nanoparticles via Interfacial Control. Langmuir 2019, 35, 6393–6402. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Kvande, I.; Zhu, Y.A.; Hammer, N.; Rønning, M.; Walmsley, J.C.; Li, P.; Qi, Z.; Zhou, X.G.; Chen, D. Decoding Atomic-Level Structures of the Interface between Pt Sub-nanocrystals and Nanostructured Carbon. J. Phys. Chem. C 2018, 122, 7166–7178. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Behler, J. Self-Diffusion of Surface Defects at Copper–Water Interfaces. J. Phys. Chem. C 2017, 121, 4368–4383. [Google Scholar] [CrossRef]
- Michalka, J.R.; Latham, A.P.; Gezelter, J.D. CO-Induced Restructuring on Stepped Pt Surfaces: A Molecular Dynamics Study. J. Phys. Chem. C 2016, 120, 18180–18190. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.; Fortunelli, A.; Goddard, W.A. Reaction intermediates during operando electrocatalysis identified from full solvent quantum mechanics molecular dynamics. Proc. Natl. Acad. Sci. USA 2019, 116, 7718–7722. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Negreiros, F.R.; Sun, Q.; Xie, M.; Sementa, L.; Stener, M.; Ye, Y.; Fortunelli, A.; Goddard, W.A., III; Cheng, T. Predictions of Chemical Shifts for Reactive Intermediates in CO2 Reduction under Operando Conditions. ACS Appl. Mater. Interfaces 2021, 13, 31554–31560. [Google Scholar] [CrossRef] [PubMed]
- Vandermause, J.; Xie, Y.; Lim, J.S.; Owen, C.J.; Kozinsky, B. Active learning of reactive Bayesian force fields: Application to heterogeneous hydrogen-platinum catalysis dynamics. arXiv 2021, arXiv:2106.01949. [Google Scholar]
- Yang, M.; Bonati, L.; Polino, D.; Parrinello, M. Using metadynamics to build neural network potentials for reactive events: The case of urea decomposition in water. Catal. Today, 2021; in press. [Google Scholar] [CrossRef]
- Hellström, M.; Ceriotti, M.; Behler, J. Nuclear Quantum Effects in Sodium Hydroxide Solutions from Neural Network Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 10158–10171. [Google Scholar] [CrossRef] [PubMed]
- Rossi, K.; Jurásková, V.; Wischert, R.; Garel, L.; Corminbœuf, C.; Ceriotti, M. Simulating Solvation and Acidity in Complex Mixtures with First-Principles Accuracy: The Case of CH3SO3H and H2O2 in Phenol. J. Chem. Theory Comput. 2020, 16, 5139–5149. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of the drastic changes in Au NPs exposed to hydrogen. Configurations extracted from AIMD simulation at 300 K, at simulation times of 0 ps (Configuration-0) where truncated octahedron initial structure was pre-optimized by DFT, 1.5 ps (Configuration-1), and 40 ps (Configuration-2). Color coding of atoms: gold in yellow and hydrogen in white. The number of adsorbed hydrogen Hads is indicated in red.
Figure 1.
Illustration of the drastic changes in Au NPs exposed to hydrogen. Configurations extracted from AIMD simulation at 300 K, at simulation times of 0 ps (Configuration-0) where truncated octahedron initial structure was pre-optimized by DFT, 1.5 ps (Configuration-1), and 40 ps (Configuration-2). Color coding of atoms: gold in yellow and hydrogen in white. The number of adsorbed hydrogen Hads is indicated in red.

Figure 2.
Initial (left) and successive (right) configurations sampled after 2ps during an AIMD simulation at 700 K comprising a supported Au nanocluster, one CO, and one Oad. Color coding: C: green, O: blue and red, Ti: gray, and Au: gold and pink. The pink Au atom labels the cation. Reprinted with permission from [69]. Copyright 2016 American Chemical Society.
Figure 2.
Initial (left) and successive (right) configurations sampled after 2ps during an AIMD simulation at 700 K comprising a supported Au nanocluster, one CO, and one Oad. Color coding: C: green, O: blue and red, Ti: gray, and Au: gold and pink. The pink Au atom labels the cation. Reprinted with permission from [69]. Copyright 2016 American Chemical Society.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rossi, K.; Mineva, T.; Filhol, J.-S.; Tielens, F.; Guesmi, H. Realistic Modelling of Dynamics at Nanostructured Interfaces Relevant to Heterogeneous Catalysis. Catalysts 2022, 12, 52. https://doi.org/10.3390/catal12010052
AMA Style
Rossi K, Mineva T, Filhol J-S, Tielens F, Guesmi H. Realistic Modelling of Dynamics at Nanostructured Interfaces Relevant to Heterogeneous Catalysis. Catalysts. 2022; 12(1):52. https://doi.org/10.3390/catal12010052
Chicago/Turabian StyleRossi, Kevin, Tzonka Mineva, Jean-Sebastien Filhol, Frederik Tielens, and Hazar Guesmi. 2022. "Realistic Modelling of Dynamics at Nanostructured Interfaces Relevant to Heterogeneous Catalysis" Catalysts 12, no. 1: 52. https://doi.org/10.3390/catal12010052
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.