Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies

Research and Development Engineering Center “Syntez”, Moscow 119571, Russia
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(2), 216; https://doi.org/10.3390/catal10020216
Submission received: 24 December 2019
/
Revised: 19 January 2020
/
Accepted: 23 January 2020
/
Published: 11 February 2020
(This article belongs to the Special Issue Progress in Catalytic Hydrodechlorination)
Abstract
:The commercial implementation of hydrodechlorination processes will be an essential step in resolving the problem of environmentally sound organochlorine wastes processing. By now, there is a number of fundamental, applied, and process elaborations of such processes, in which chlorine is almost completely removed from wastes. The review article contains a significant number of results including basic regularities of thermal hydrodechlorination, comprehensive and selective catalytic hydrodechlorination. It discusses thermodynamics, kinetics, and catalysts of gas and liquid phase processes. Considerable attention is paid to hydrodechlorination of vinyl chloride production wastes and utilization of tetrachloromethane, which is the ozone-depleting substance. It also discusses hydrodechlorination of mono- and (poly)chlorobenzenes. The important examples of liquid phase data include hydrogenation using complex hydrides of elements. It also includes several flow sheets of hydrodechlorination processes.
1. Introduction
The reduction of organochlorine compounds is essential to the organic synthesis and, for example, is applied for aldehydes synthesis from carboxylic acid chlorides or for obtaining by this method compounds containing a lower number of chlorine atoms compared to the initial ones.
Of great importance is knowledge of the C–Cl bonds behavior under reduction conditions of other functional groups in the same molecule. The reduction of C–Cl bond to C–H bond is also used to prove the structure and analysis of organochlorine compounds [1].
In principle, any C–Cl bond can be converted to a C–H bond; however, the reduction ease and selectivity strongly depend on the structure of the chlorine-containing functional group on presence and location of other functional groups in the molecule, and on the nature of the reducing agent. So, for example, chlorine bound to a vinyl carbon atom is usually replaced with harder hydrogen, and that to an allyl easier than in saturated chlorohydrocarbons. The accumulation of chlorine atoms or other electron-withdrawing groups on a carbon atom bound to chlorine facilitates the reduction. A chlorine atom located in the alpha position to the electron-withdrawing group is replaced more easily than more distant chlorine atoms in the same molecule.
The reactivity series of organochlorine compounds in the reducing reactions can, however, vary significantly depending on the reduction mechanism. So, for example, in heterolytic reduction, primary chlorine derivatives are more reactive than secondary ones, and in homolytic reduction the ratio is opposite [1].
Various methods for C–Cl bonds reduction in non-cyclic organochlorine compounds can be divided into the following groups:
- Electrochemical reduction.
- Radiolysis of organochlorine compounds.
- Hydrogenation with hydrogen—thermal and catalytic.
- Reduction with metal hydrides and complex hydrides of elements.
- Metal reduction in the presence of hydrogen donors.
- Reduction with metal salts of lower valency.
Of these methods, the catalytic reduction with hydrogen and reduction with complex hydrides of elements are the most widely used and studied [1,2,3].
Hydrodechlorination processes also have a pronounced applied value. Hydrodechlorination end products are hydrocarbons and HCl.
In general, the reaction can be written as
R1R2HmCln + ℓH2 → R1R2H(m+2ℓ-n) + nHCl
Given the fact that hydrodechlorination makes it possible quantitatively remove chlorine with the formation of environmentally sound hydrocarbons, one can explain the growing interest in these processes from the point of view of their perspective as methods for industrial wastes processing at the most chemical industries [4,5,6,7]. The data in Figure 1 indicate the growth of publications on hydrodechlorination problems in the last few decades.
The amount of information on hydrodechlorination processes is very significant and includes materials on kinetics and mechanism, as well as flow charts of these processes.
In order to identify the general patterns of hydrodechlorination processes, attempts have repeatedly been made to generalize and systematize data published in [2,3,8,9]. Of the summarizing materials, one should note the reviews [4,10] published in the 1990s. It is noted that catalytic methods are more widely used for hydrodechlorination.
2. Electrochemical Reduction
Electrochemical reduction is a complex process in which the speed and direction of reactions depend on many factors. It is characterized by the following features. Two electrons are spent for breaking the C–Cl bond; the half-wave potential in most cases does not depend on the pH of the medium; in general, the electrode reaction is irreversible [1]. There is an approximate linear relationship between the half-wave potential of the reactions
and RX bond dissociation energy, where X = Cl, Br, I [11].
RX + 2e + H+ ➔ RH + X−
In [12], a scheme was proposed describing the mechanism of electrochemical reduction of the C–Cl bond
where [R.] are [R-] are the electrode complexes.
RCl + e ➔ [R.] + Cl−
[R.] ➔ R.
2R. ➔ R–R
[R.] + e ➔ [R−]
[R−] + solvent ➔ RH,
However, there is another point of view, according to which there is a simultaneous transfer of a pair of electrons, similar to that for nucleophilic substitution reactions [1].
Electrochemical reduction is carried out on cathodes of copper, zinc, amalgamated zinc, palladium, cadmium, mercury, and lead [13]. The cathodic dechlorination proceeds in an alkaline environment more efficiently than in an acidic one; however, in most cases, acidic electrolytes are used to avoid such side reactions as saponification [13,14]. The recovery is usually carried out in inert solvents, in particular in alcohols. In [13], the influence of the reduction conditions—the cathode material, current density, the nature of electrolyte and solvent, the concentration of the reducible substance, and temperature—on the course of the reaction is described.
The recovery of monochlorohydrocarbons is difficult and results mainly in the replacement of chlorine by hydrogen in accordance with the overall equation
RCl + 2e + H+ ➔ RH + Cl−
In some cases, dimeric compounds (R-R) are formed according to the scheme
2RCl + 2e ➔ R–R + 2Cl−
The recovery of vinyl chlorine is difficult, and that of the allyl one is facilitated in comparison with chlorine in saturated compounds, while the double bond is preserved. The triple bond is also remained intact at polarographic reduction of the compounds similar to RC≡C–CH2Cl [1].
Polychlorinated derivatives with the vicinal position of chlorine are dechlorinated to form the corresponding unsaturated compounds [15,16] according to the scheme
R2CClCClR2 + 2e ➔ R2C = CR2 + 2Cl−
In heme-polychlorinated derivatives, chlorine is sequentially replaced by hydrogen.
It was noted that an increase in the number of chlorine atoms or other electronegative groups on the same carbon atom results in easier recovery (half-wave potential shifting to the positive direction). Such a regularity, as indicated in [1], was shown on the examples of the reduction of mono-, di-, and trichloroacetic acids, chlorinated methanes, and alpha-chloronitroalkanes.
Electrochemical reduction of organochlorine compounds is used for analytical and preparative purposes; this method has no applied value.
3. Radiolytic Reduction and Dechlorination
Water-soluble fatty organochlorine compounds react with the water radiolysis products—hydrated electron, hydrogen atom, and hydroxyl radical—to produce chlorine ions and organic radicals. As a rule, the hydrated electron (eaq) is a stronger reducing agent than the hydrogen atom [17]. Often, the H and eaq give the same reaction products. However, H atoms, as opposed to eaq, have a dehydrogenating effect in reactions with unsaturated compounds and, therefore, the nature of the reducing agents can be determined from the final reaction products. The reactions of the hydrated electron are quite diverse; below, we will briefly discuss some dissociative addition reactions
eaq + ClCH2COOH ➔ [ClCH2COOH]− ➔ Cl− + C.H2COOH
Using the chloroacetate ion as an example, it was shown that its interaction with the hydrated electron results in a quantitative separation of the chlorine ion [18]. In contrast, the hydroxyl radical detaches hydrogen rather than chlorine atom from the chlorine derivatives [1,19]. The reaction of the chlorine derivatives with the hydrogen atom flows in two directions: with separation of the chlorine ion (1) and the hydrogen atom (2)
ClCH2COO− + H ➔ C.H2COO− + Cl− + H+
ClCH2COO− + H ➔ ClC.HCOO− + H2
Radiolysis of tetrachloromethane leads to removal of chlorine and formation of hexachloroethane [20] according to the scheme
CCl4 ➔ C.Cl3 + Cl.
2Cl. ➔ Cl2
2C. Cl3 ➔ C2Cl6
Radiolysis of chloroform [20] results in formation of hydrogen chloride and higher boiling compounds than chloroform, as well as small amounts of methylene chloride and tetrachloromethane. The formation of hydrogen chloride can be described by the following Equations (3)–(6)
CHCl3 ➔ C.HCl2 + Cl.
CHCl3 ➔ C.Cl3 + H.
Cl. + CHCl3 ➔ HCl + C.Cl3
H. + CHCl3 ➔ HCl + C.HCl2
Dichloromethyl and trichloromethyl radicals formed in these reactions recombine to form higher products. The appearance of small amounts of CH2Cl2 and CCl4 indicates [1,20] as well the course of the reaction according to the Equations (7), (8) or (9), (10)
or
C.HCl2 + CHCl3 ➔ CH2Cl2 + C.Cl3
C.Cl3 + CHCl3 ➔ CCl4 + C.HCl2
C.HCl2 + HCl ➔ CH2Cl2 + Cl.
C.Cl3 + HCl ➔ CCl4 + H.
Radiolysis of chloroform does not produce hydrogen and chlorine; this indicates that the reactions (11) and (12) below do not compete with the reactions (5) and (6) [20]
Cl. + CHCl3 ➔ Cl2 + C.HCl2
H. + CHCl3 ➔ H2 + C.Cl3
The radiochemical stability of lower perchlorolefins and dienes declines in the series [1]
C5Cl8 > C4Cl6 > C3Cl6 > C2Cl4
Moreover, the destruction processes are of relatively small importance, the formation of more complex molecules predominates.
The radiolytic reduction process also has no applied value and is used for preparative or analytical purposes.
4. Hydrodechlorination Using Hydrogen
The process of hydrodechlorination using hydrogen can be carried out both on catalysts and under direct temperature exposure to chlorohydrocarbons in a reducing medium (thermal process). The thermal process occurs at temperatures of 600–800 °C and, as a rule, results in total dechlorination of the initial compounds and in formation of a mixture of organic products and hydrogen chloride. This fact determines its undoubted good perspectives for implementation in an industrial environment.
Catalytic hydrodechlorination allows adjusting the degree of dechlorination and directing the process to obtain specific organic products. Such an approach is the most relevant when solving the problem of the competent disposal of organochlorine wastes from such large-scale industries as vinyl chloride, dichloroethane, chloroform, perchloroethene, epichlorohydrin, and monochlorobenzene.
Based on the foregoing and the desirability of obtaining marketable products from wastes, one can propose the following classification of the processes for organochlorine compounds hydrodechlorination:
- thermal hydrodechlorination resulting in formation of a mixture of hydrocarbons and hydrogen chloride
- exhaustive catalytic hydrodechlorination also resulting in formation of hydrocarbons and hydrogen chloride
- selective catalytic hydrodechlorination resulting in formation of target organic products (hydrocarbons and chlorohydrocarbons) and hydrogen chloride
The common advantages of the hydrodechlorination method include disposal of both the chlorine and organic components of the wastes, as well as the elimination of the possibility of dioxins formation in a reducing environment.
The thermal hydrodechlorination is carried out at a sufficiently high (600–800 °C) temperature, which complicates its hardware design. As a hydrogenating agent, hydrogen is mostly used. The advantage of the process is the possibility of feeding the wastes into the process in the unclarified form.
In the thermal process, a complex mixture of organic products with a low chlorine content is formed because of hydrodechlorination. It is rather difficult to get marketable organic products in this process. By tightening the process conditions, it is possible to achieve complete dechlorination of wastes organochlorine components. The resulting mixture of hydrocarbons (after extracting HCl from it) can be used as fuel [21,22].
The application of catalysts allows significantly reduce the temperature of the process (up to 100–400 °C) and simplify its hardware design. However, a serious problem of ensuring the operation stability of the used catalytic systems in these processes occurs. Moreover, the wastes should be fed into the reactor in a clarified form, which somewhat reduces their level of disposal. Nevertheless, the current level of process developments allows feeding more than 80% of wastes into the reactor. Application of unclarified wastes is almost impossible due to the deactivation of the catalysts.
In the catalytic hydrodechlorination process, both hydrogen and other reducing agents can be used. For implementation of the industrial waste disposal, it is preferable to use hydrogen.
However, in the presence of heterogeneous and homogeneous catalysts, not only molecular hydrogen is used as hydrogen donors. Organic compounds (formic acid, formates, etc.) [2,3], as well as hydrazine hydrides and complex metal hydrides can also be used.
Let us consider, for example, the hydrodechlorination of chlorinated hydrocarbons of C2 fraction, which form the majority of the wastes in production of the vinyl chloride, the most important large-scale product of the organochlorine synthesis.
The catalytic hydrodechlorination of chlorohydrocarbons of C2 fraction can be carried out to produce chlorohydrocarbons with a low chlorine content (for example, vinyl chloride) or hydrocarbons (ethane, ethylene, methane).

The selective hydrodechlorination process is aimed at obtaining target products that can be used in the organic synthesis. The best option is to obtain from wastes organic products—vinyl chloride and ethylene—that can be used in the main production [23,24,25]. This makes possible to regenerate the raw materials spent for waste formation.
The preferred option is the selective hydrodechlorination of the wastes to produce ethylene. As will be shown in the description below, the developed catalytic systems allow carrying out the process with an ethylene formation selectivity of 90% and higher. In this case, a mixture of ethylene and HCl leaving the waste hydrodechlorination unit can be used to produce dichloroethane in the ethylene oxychlorination step.
C2H4 + 2HCl + 1/2O2 → C2H4Cl2 + H2O
Figure 2 shows a process flowchart for vinyl chloride production from ethylene according to a non-waste, chlorine-balanced scheme with a stage of selective hydrodechlorination of wastes.
The hydrodechlorination process, in which vinyl chloride along with ethylene is formed from wastes, can also be introduced into the vinyl chloride production, but it will differ by the presence of a hydrodechlorination products condensation and separation scheme for their return to production.
In the case of the selective hydrodechlorination process, all waste components of the C2 fraction are used to produce marketable products. Due to this, more than 70% of ethylene and chlorine (or HCl) spent on waste generation is returned to the vinyl chloride production. In this case, a decrease in the consumption of ethylene and chlorine for vinyl chloride production is achieved.
The catalytic hydrodechlorination product can also be a mixture of hydrocarbons, containing mainly ethane and hydrogen chloride. As explained above, the obtained hydrocarbons can be used as fuel (after HCl separation) [26].
Impurities of C1, C4, C6 compounds contained in the wastes complicates the separation and use of the waste hydrodechlorination products.
Small amounts of chlorohydrocarbons with four carbon atoms contained in the wastes are converted to butenes and butanes, those having one carbon atom—to methane, and chlorobenzenes—to benzene.
CCl4 + 2H2 → CH4 + 4HCl
C4H8Cl2 + H2 → C4H8 + 2HCl
C6H5Cl + H2 → C6H6 + HCl
Benzene, butanes, and butenes are separated from the products by condensation (or absorption) and a mixture of ethylene (with a small content of methane and ethane) and hydrogen chloride is fed to oxychlorination.
Catalytic hydrodechlorination processes are carried out mainly in the gas phase. A number of publications are devoted to the liquid phase hydrodechlorination process using heterogeneous or homogeneous catalysts. Such processes have their own specifics, but can also be used to carry out both non-selective (exhaustive) and selective hydrodechlorination [27,28]. Chemistry and technology of these processes are of considerable interest and will be considered separately. The results obtained in them can be used, inter alia, to interpret the process mechanism on heterogeneous catalysts and in the gas phase.
Hydrodechlorination of chlorohydrocarbon components contained in the vinyl chloride production wastes is accompanied by heat generation. Table 1 shows the standard heats of formation (∆H298) of the waste components and their conversion products, as well as the thermal effect of hydrodechlorination reactions. As can be seen from these data, the thermal effect of hydrodechlorination reactions is not very large, especially in conditions of the selective ethylene production. An increase in the chlorine atoms content in the C2 chlorohydrocarbon molecules and production of ethane as a final product result in a noticeable increase in the thermal effect.
The average thermal effect of clarified wastes processing from the vinyl chloride production (with ethylene (90%) and ethane (10%) obtaining from C2 chlorohydrocarbons and selective hydrogenation of chloromethanes to methane, chlorobutenes to butenes (90%), chlorobenzenes to benzene) calculated based on these data was 104.5 kJ/mol (or 947.2 kJ/kg) of wastes.
The hydrodechlorination thermal effect was 8–10 times lower compared with the oxidation process, but it can be used to reduce the energy costs for the production.
The small thermal effect of the process makes it possible to use reactors with either fluidized or fixed catalyst bed. The possibility of using reactors of various types will be discussed in the description below.
4.1. Thermal Hydrodechlorination
The process of thermal hydrodechlorination of organochlorine compounds, including those being the components of vinyl chloride production waste, is described in a number of papers [21,22,26,27,28,29,30,31]. The obtained positive results made it possible to determine the scheme of the industrial waste disposal process using this method [30].
The thermal process occurs, as a rule, at a temperature of 600–1000 °C, residence time 1–10 sec and in presence of molecular hydrogen as a hydrogenating agent. Along with hydrogen, hydrocarbons (methane, butane, etc.) can be fed into the process as hydrogen donor [30]. Use of hydrocarbons can significantly reduce the cost of waste processing.
In the thermal process, C–Cl bonds hydrogenolysis in the used organochlorine raw materials and the formation of less chlorinated chlorohydrocarbons or hydrocarbons occur.
R–Cl + H2 → RH + HC
The process rate largely depends on the type of chlorohydrocarbon being processed and the number of chlorine atoms in its molecule.
The process proceeds mainly by a radical-chain mechanism and is also accompanied by breaking of C–C bonds and the formation of condensation products. In view of this, a wide range of organic products obtained is a characteristic feature of the thermal hydrodechlorination process. When describing hydrodechlorination processes, as a rule, conditions are specified that ensure complete dechlorination of organochlorine compounds.
Table 2 shows the studies results of the thermal hydrodechlorination of chloroaromatic hydrocarbons and chloromethanes, which is stipulated by the relevance of the process for these compounds processing and the possibility of hydrodechlorination simulation using the examples of C1 and C6 chlorohydrocarbons. The possibility of achieving high (up to 100%) conversions of the studied chlorohydrocarbons is shown. One should note the low conversion of 1,2-dichloroethane at a temperature of 580 °C. However, based on the data on the hydrodechlorination of less reactive dichlorobenzene (conversion at 600 °C is about 1%), almost 100% conversion of 1,2-dichloroethane at 950 °C should be expected.
Figure 3 summarizes data [22] on reactivity of a number of chlorohydrocarbons in the thermal hydrodechlorination process carried out with a 2–5-fold excess of hydrogen with respect to chlorine. As illustrated in Figure 3, chloroform and tetrachloromethane react most readily under hydrodechlorination conditions. Total conversion of these compounds can be achieved at 500–550 °C. Methyl chloride and chlorobenzene hydrodechlorination rates are quite low, and for their total conversion a temperature exceeding 800 °C is necessary. According to [22], chlorobenzene hydrodechlorination rate slightly decreases with an increase in the chlorine content in the Ph-Cl molecule.
Methyl chloride and C2 chlorohydrocarbons occupy an intermediate position between the aforementioned groups of chlorohydrocarbons. The authors of [4] argue that the reactivity of di-, tri-, and tetrachloroethenes is at vinyl chloride level. The reactivity of the other C2 chlorohydrocarbons remains obviously at the same level.
The basic product of chlorobenzenes processing is benzene.
The basic products of hydrodechlorination during processing of chlorohydrocarbons C2 fraction include ethane and ethylene. Significant amounts of methane, acetylene, C3–C4 hydrocarbons, and aromatic hydrocarbons (benzene) were also recorded.
The products of the thermal process include soot as well. However, most authors argue that with a sufficiently high excess of hydrogen, soot is not produced or is produced in minimal amounts. This contrasts with the behavior of these compounds during pyrolysis, where the formation of acetylene and soot is significant.
There are plenty of publications devoted to the mechanism and kinetics of the thermal process. The main studies of hydrodechlorination of organochlorine compounds were carried out for the processing of chloromethanes, C2 chloroolefins, and chloroarenes. Generalized materials on the kinetics and mechanism of these compounds conversion during hydrodechlorination see in [26,28,29]. Despite the extensive information provided, the interpretation of the mechanism raises a number of questions.
A few data on the thermal hydrodechlorination of chloroethanes with two or more chlorine atoms, which constitute the basis of the vinyl chloride production wastes, show that their conversion is of stepwise character. The initial stage of the process is the thermal dehydrochlorination of polychloroethanes followed by the chloralkenes involvement into the hydrogenolysis reaction.
An analysis of [33,34] made it possible to give a reasonable interpretation of the chloroethenes hydrodechlorination process. A probable scheme of reactions for vinyl chloride conversion, where HX is any compound with a mobile hydrogen atom, can be represented as follows:

As is clear from the scheme under consideration, the implemented process includes competition between two radical reactions and monomolecular release of HCl, whilst reaction 7 performs regeneration of hydrogen atoms. A high value of the rate constant for this reaction [35] and a large excess of hydrogen cause a much higher stationary concentration of hydrogen atoms in comparison with the concentration of chlorine atoms. In the process of thermal hydrodechlorination, the concentration of [H●] is nearly equilibrium. Ultimately, this determines the predominant path of radical reactions 1–2 and 3–4 in comparison with monomolecular decomposition 6 and reactions of condensation products formation:
Regardless of the mechanism being implemented, the authors of [26,29] suggest the presence of various intermediates in the hydrodechlorination process.
The virtual absence of acetylene in the hydrodechlorination products can be explained by low rate of its formation and rapid expenditure, although this has not been experimentally proven.
Secondary exhaustive dechlorination reactions, such as thermal hydrodechlorination, are hydrogenation of ethylene to ethane and destructive hydrogenation of ethane to methane.
A radical mechanism has been proposed for hydrodechlorination of chlorarenes [4]; it is confirmed by the following kinetic equation with one-half-order in hydrogen
Using this equation, it is easy to calculate the constant in the presence of data obtained for different reaction times (t).
The above equation satisfactorily describes the hydrodechlorination rates for a number of chlorohydrocarbons. Figure 4 shows the temperature dependences of the calculated values for the reaction involving chloromethanes, a number of chloralkenes, and chlorarenes.
From the calculated preexponential factor, energy, and activation entropy (Table 3) the authors of [4] made a number of conclusions clarifying the mechanism of the process.
It can be seen that the activation energy for the thermal hydrodechlorination of the studied compounds varies in the range of 150 to 260 kJ/mol. The exceptions are dichloroethenes.
Negative values of the activation entropy can be explained by the formation of a transition state with a stable structure. Positive value of entropy apparently is consistent with the molecular cleavage of the C–Cl bond at the limiting stage, followed by the formation of free radicals as intermediates.
Thermal Hydrodechlorination Process
The implementation of the thermal hydrodechlorination process is associated with a number of difficulties caused by maintaining the required temperature regime in the reaction space. The heat released during hydrodechlorination (see Table 1) cannot provide the necessary temperature (up to 1000 °C). This problem can be solved by additional heat introducing by burning fuel or by simultaneously carrying out hydrodechlorination and a more exothermic process in the same reaction unit.
Figure 5 shows a schematic diagram of an option for thermal hydrodechlorination of organochlorine products [5,30]. The diagram includes a hydrodechlorination reactor and units (stages) for reaction gas cooling and HCl absorption. To ensure total dechlorination of the wastes, a temperature of 950–1000 °C is maintained in the reactor (1). Hydrogen is fed in a threefold excess (mol) compared with the stoichiometric amount.
The reactor (1) was created as a cracking unit with a fluidized heat carrier bed (sand). The use of a heat carrier provides a fairly uniform temperature profile along the height of the reactor. Sand is continuously withdrawn into the regenerator (2), into which air and fuel are supplied. The soot formed during hydrodechlorination burns out in a hot air stream. The regenerated sand heated to the required temperature is returned to the reactor. The conveyor lines between the reactor and the regenerator are provided with a gas shutter to avoid any contact between the hydrogen-rich reactor medium and the regenerator medium.
A gas mixture of wastes with hydrogen is introduced into the fluidized bed of hot sand, where the rapid direct heat exchange between the sand and gas occurs. The soot formed in the process is deposited on the sand. Hot reaction gases representing a mixture of hydrocarbons, hydrogen, and HCl, leave the reactor through a cyclone (4) and are fed to a two-stage cooling system consisting of a quenching column (5) and a condenser (6).
In the quenching column (5), the gas stream is rapidly cooled by evaporation of the atomized liquid in direct contact with the reaction gas. Water or aromatic compounds are used as a quenching agent.
Figure 5 shows the scheme for the water use conditions. In the condenser (6), the quenching liquid is cooled and organic compounds and HCl are stripped from it.
The cooled stream of hydrocarbons, HCl and excess hydrogen is fed to the absorber (7), where HCl absorption by water occurs. Gas separated from HCl is used as fuel.
The operating conditions in the condenser and the absorber are selected so that the effluent streams from both units form suitable for use concentrated waste hydrochloric acid.
However, the authors of [26,28] consider such an implementation of the hydrodechlorination process to be unpromising. They propose to solve the problem of heat supply by combining the hydrodechlorination process and other chemical processes, which are the sources of the necessary additional amount of heat, in one reaction unit. The processes of fractional methane combustion [36] and coal gasification [37] can be combined with the hydrogenolysis. The significance of these processes is not limited to heat generation; each of them can act as a hydrogen donor for the hydrogenolysis. Therefore, methane can directly act as a hydrogenating agent, and the gasification process produces necessary for this purpose hydrogen. Another solution involves the implementation of high-pressure thermal hydrogenolysis [38], which provides high hydrogenolysis rates, the ability to increase specific heat generation, and ensures maintenance of the required temperature regime.
High temperatures result in low selectivity of the hydrodechlorination and soot formation. It is possible to lower the temperature of the hydrodechlorination (by about 200 °C) using activated carbon, which results in an increase in the process rate and makes it possible to lower the temperature and reduce soot formation. This is because the condensation (sooting) reaction has a higher activation energy compared to the hydrogenolysis one with C–Cl bond cleavage.
Activated carbon, obviously, partially plays the role of a process catalyst and the results obtained in its presence cannot be considered as the data for a purely thermal process.
4.2. Catalytic Hydrodechlorination
A significant number of review articles on the problems of catalytic hydrodechlorination published over the past 25 years [4,9,10,26,28] point to the serious role of this direction in the development of chemistry and organochlorine synthesis, as well as to the great attention that the authors give to the solution of the environmental issues. This is also confirmed by the extensive data on the processing of organochlorine wastes by various methods (oxidation, oxychlorination, pyrolysis, chlorination, electrocracking), among which the catalytic hydrodechlorination plays a significant role [5,9].
These articles discuss the basic issues related to use of catalytic systems, and mechanism and kinetics of the process and contain the limited specific application information on the hydrodechlorination of a number of compounds being the components of organochlorine wastes of industrial productions.
Much attention in the review [10] was given to the publications in the field of biocatalysis with biodehalogenation of various halohydrocarbons using miscellaneous microorganisms and enzymes, as well as to the process implementation in the presence of phase transfer catalysts.
An organized material on the hydrodechlorination of tetrachloromethane, the proper processing of which plays an important role in the implementation of the 1987 Montreal Protocol decisions on the ozone layer protection, is given in the review [28].
A significant part of the cited materials on the catalytic effect of the systems used, the mechanism and kinetics of the process can also be used to interpret the hydrodechlorination of C2 chlorohydrocarbons—the basic components of vinyl chloride production wastes.
The process is carried out both in liquid and in gas phases. However, the basic direction in the processing of organochlorine production wastes is the gas phase process.
4.2.1. Catalytic Systems
The choice of catalytic systems for hydrodechlorination is quite wide. The vast majority of the reactions are carried out on metals of IB, VB, VIB, VIIB, and VIII groups of the periodic table. The most frequently used metals include Pt, Pd, Rb, Ru, Ir, Os, Ni, Co, Fe, Re, Mn, Mo, W, Cr, V, Cu, Ag, and Au [4].
Properties of metal catalysts: fusion with other metals, introduction of promoters, and variation of carriers can significantly change activity, selectivity, service life, etc. Catalytic systems based on the bimetallic alloys Rh-Ir, Cu-Rh, Co-Mo [33], Ni-W, Ni-Mo [39], Pd-Pt, Pd-Ga, Pd-Al, Pd-Ru [40], Pd-Sn, Pd-Pb, Pd-Ge, Pd-K, Pd-Fe, Pd-Co, and Pd-Ag [41,42] were proposed. The promoting additives to metal catalysts include Pd, Pt, Ru, Ir, Fe, Co, Ni, Cu, Cd, Ag, Au, V, Cr, Mo, W, Bi, Al, Hg, In, Sn, Te, as well as Si, P, As, Sb [40,43], salts and oxides of aluminum, boron and titanium, phosphines and phosphates, salts, hydroxides and oxides of alkali and alkaline earth metals, phosphonium halides.
Systems containing salts of copper, palladium, ruthenium, platinum, rhodium, nickel, and molybdenum on various carriers have been described as catalysts [21]. The oxides of chromium, nickel, and molybdenum are widely used in the process. These catalysts are subjected to thermal treatment in a reducing environment before being used in the hydrodechlorination process. Under these conditions, a metal phase forms on the surface of the catalyst, as a result of which the behavior of the salt and metal catalysts becomes much closer.
The range of catalyst carriers is rather wide; it includes activated carbon [41], diatomites, alumina [44,45], chromium oxide [44], silicon oxide, iron oxide, aluminosilicates, calcium carbonate, zeolites, barium sulfate, etc. [21].
The hydrodechlorination process is carried out in a wide temperature range, as a rule, 100–400 °C, the process temperature depends on the type of processed chlorohydrocarbons and the used catalyst system. As a rule, the process is carried out at low pressure. However, in some cases, a pressure of up to 10 MPa is used, as a result of which the hydrodechlorination rate increases and it becomes possible to lower the process temperature.
Despite the diversity of the proposed catalysts, the main systems used are the catalysts based on Pt, Pd, Ni, and Cr. Table 4 summarizes some of the data obtained by several authors for hydrodechlorination of the major types of organochlorine compounds, in particular, components of vinyl chloride production wastes. Data on products obtained on systems with different carriers and promoted by a number of metals are also provided.
As can be seen from Table 4, the use of Pd, Pt, and Rh catalysts allows performing partial or complete dechlorination of C2 and C3 chlorohydrocarbons. The process products can be both saturated and unsaturated chlorohydrocarbons and hydrocarbons. Changes in the catalysts selectivity and the selective hydrodechlorination process management will be discussed below. As a rule, these catalysts are used for the complete dechlorination of chlorohydrocarbons. When processing CCl4, various chloromethanes, methane and C2+ hydrocarbons are produced. A similar pattern is observed for the nickel catalysts.
The main components of vinyl chloride production wastes (di-, trichloroethane), as well as other chlorohydrocarbons are hydrogenated on nickel and chromium, as well as modified catalysts to produce a number of products. The main products of C2 chlorohydrocarbons hydrodechlorination, as when using catalysts with noble metals, are ethane, ethylene and chlorohydrocarbons with a low chlorine content.
Despite the various conditions for carrying out the hydrodechlorination on noble metals and nickel and chromium based systems, including modified ones, the activity of catalysts with noble metals is somewhat higher and lower temperatures (50–100 °C) can be used to carry out this process.
The catalytic activity of a number of metals was studied using hydrodehalogenation reactions of chloro- and bromoethyl as an example [67]. The relative activity of metals measured in a stationary process, is presented by a series:
Pt > Pd > Rh >> W > Ni > Fe
Despite the fact that platinum catalysts are usually more active, most researchers prefer palladium systems, apparently, as more affordable ones. In some cases, preference is given to cheaper catalysts based on nickel and chromium.
For hydrodechlorination of tetrachloromethane, a series of noble metal activities looks somewhat different:
Pd > Pt > Rh > Ru
The highest activity of Pd, according to [68], correlates with a number of vacant state density values. This is consistent with the most favorable CCl4 sorption conditions on Pd-active sites, since chlorine atoms in tetrachloromethane exhibit weak donor properties [28].
The oxidative state of the active metal is one of the key factors determining activity of the catalysts [9]. In this a case the ratio Me(0) to Me(n+) is important. This fact was described in the publications of L.M. Gomes-Sainero et al. [68,69], who showed that during CCl4 hydrodechlorination Pd0:Pdn+ ratio of 1:1 ensures high catalyst activity due to balance of hydrogen activation on Pd0 and C-Cl bond activation on Pdn+.
The observed dependences of the metals activity on their position in the periodic table can also be explained by the fact that in the extremum region the most favorable geometric conformity is achieved between the length of the chemisorption bond and the crystal lattice unit cell size of the catalyst [4]. This point of view based on the multiplet theory of catalysis, is confirmed by the dominant contribution of the entropy factor to the change in the specific activity of individual atoms during the transition from one metal to another.
An important approach resulting in some cases in improving of the hydrodechlorination catalysts performance is their modification. Both the active component [70,71,72] and the carrier [73,74,75] can be subject to modification.
The promotion and modification of the hydrodechlorination catalysts bear mainly a descriptive character. Some scientific approaches to the modification and promotion of catalysts are described mainly on the example of systems used in the hydrodehalogenation of organochlorofluorine compounds [10].
Modification of the Pd or Rh catalyst with copper allows achieving high hydrodehalogenation selectivities. Thus, in the presence of supported on carbon catalyst containing 10% copper, 0.044% rhodium, and 6.5% methyltriphenylphosphonium chloride, chloroform is formed from CCl4 with a yield of 52.8% at 100% selectivity [76].
During hydrodechlorination, the formation and preservation of double-bonded compounds (in particular, ethylene, and vinyl chloride) are especially significant. Choosing a modifier system of the Pd/Al2O3, the authors of [43] put an objective to prevent further reduction of double bonds and obtain unsaturated compounds. Metals of IB-IIIB groups (Ag, Bi, Cd, Cu, Hg, In, Pb, Sn, Tl) had a positive effect on the selectivity of olefins formation. On the obtained catalysts 1,1,2-trichlorotrifluoroethane was sequentially dechlorinated into trifluorochloro—and then to trifluoroethene.
As can be seen from [10], the maximum selectivity on the sum of olefins is achieved in the presence of Bi-Pd/SiO2 and Tl-Pd/C, whereas a change in the thallium content in the catalyst results in a drastic change in the selectivity of the alternative olefins formation and predominance of one of the products.
To clarify the nature of the modification effect, R.Ohnishi et al. [43] carried out a thermodynamic analysis of the catalytic cycle. Presented by the authors dependence of the equilibrium constant logarithm for the HCl conversion (13) on the equilibrium constant logarithm for the decomposition of the chloride of the corresponding metal (14) showed that all the effective promoters fall into one category:
MeFn + nHCl = MeCln + nHF
MeCln + n/2 H2 = Me + nHCl
According to [43], these data indicate that the role of the modifier is to suppress the hydrogenating activity of a platinum group metal and its activity in the hydrodechlorination reaction. Using a number of physical and chemical methods for studying the catalysts it was shown that the activating effect of the metal-modifier is achieved by destroying the ensembles of palladium atoms: for example, modification with thallium results in shifting of the broad palladium peak in the X-ray diffraction pattern in the Tl-Pd/C system to lower angles 2θ, which prove the formation of a thallium–palladium alloy or intermetallide.
Interesting results on modification were obtained by studying the effect of 11 additives [77]. The maximum selectivity for the formation of (fluorine) olefins is observed in the presence of catalysts modified with Bi and Tl. More active were the catalysts prepared by supporting of Pd-Bi on SiO2: with Pd content of 2% and a ratio of Bi:Pd = 1:2 selectivity of olefin formation exceeds 90%.
Chlorofluoroethane hydrodechlorination over Ni/TiO2 and MnTiO3 and Ni and Co powders at 300 °C also proceeds with the formation of olefins, while in the presence of Pd/TiO2 significant alkane formation is observed.
In the presence of a Ni/TiO2 catalyst prepared by decomposition of Ti-Ni-H hydride systems with mineral acids, the basic product of chlorofluoroethane (Freon 113) hydrodechlorination at 200–400 °C was fluoroethylene, and the activity and selectivity of the catalysts strongly depended on the initial hydride composition [10]. A catalyst containing 8.3% Ni prepared from TiNiH0,6 hydride had the maximum activity: at the temperatures of 300–350 °C the conversion degree was 98% and the selectivity was 95%. The authors explain the significant differences in the activities of the catalysts prepared from intermetallic hydrides with various ratios of Ti and Ni by a strong interaction of a metal with a carrier in the system, and this approach seems promising for explaining the strong influence of metals-modifiers on the hydrodechlorination selectivity in the presence of supported metal-containing catalysts.
E. Lokteva et al. [9] indicate a frequently encountered synergistic effect when using bimetallic hydrodechlorination catalysts. In particular, the authors of [78] observed that during hydrodechlorination of chlorobenzene on a PdNi/C catalyst, hydrodechlorination of dichlorodifluoromethane and 2-chlorophenol on an AuPd/Sibunit catalyst [79,80]. The synergistic effect, according to [9], is explained by formation of bimetallic alloys, prevention of Pd carbide formation, and by a significant slowdown in the reaction of Pd transition to an oxidized form.
The authors of [74,81,82,83] showed that the addition of Cu to Pd-, Pt-, Ni-containing catalysts results in the predominant formation of unsaturated hydrocarbons during the chloralkanes hydrodechlorination. The reason for this, according to [9,84], is the enrichment of the catalyst surface with copper while reducing the copper content on the surface during adsorption of chloralkanes (1,2-dichloroethane, 1,1,2-trichloroethane). In particular, this effect is observed when modifying the Pd catalysts with copper.
The catalytic hydrodechlorination is structurally sensitive, which is exhibited in the dependence of the specific activity of the catalyst on the active sites availability and structure [85]. The efficient hydrodechlorination requires an interaction between the active site of the catalyst and the substrate: an optimal combination of their structural properties is required, which is best seen in the presence of weak electron-donating substituents. The structural sensitivity of the hydrodechlorination reactions is confirmed by numerous examples of the dependence of the specific activity of catalysts on their dispersion [40,86]. An important factor is also the particle size of the catalyst, which was shown in [87] for Pd/C catalyst and [88] for Pt/Al2O3 catalyst. According to [89], the Pd/C catalyst with a particle size of 4–15 mm is more active than a catalyst with a particle size of 2–5 mm. This parameter also affects the selectivity of the process. Thus, it was shown in [88] that for the Pd/C catalyst, the tetrachloromethane hydrodechlorination proceeds predominantly to methane with particle size of less than 4 mm, while an increase in this size leads to formation of chloroform as the basic product. This indicates a change in the adsorption properties of the catalyst with a change in the size of its particles [9,88]. B. Cuenya made similar conclusions in the review [90].
This implies an important aspect of the modification and methods used for preparation of the hydrodechlorination catalysts. The activity and selectivity of the catalyst largely depend on the temperature regime of the catalysts treatment with an inert gas and reducing agents, type of a reducing agent, as well as on the catalysts treatment with gaseous reagents such as HCl, Cl2, air, H2O, F2, H2S, etc. [4]. The significance of the catalyst preparation method for targeted regulation of its activity is illustrated by the results of [91], where the possibility of “chemical grafting” of palladium ions on Al2O3 surface with varying ionic nature of the metal is shown. A strong correlation between the ionic nature of palladium and its activity was revealed.
E. Lokteva et al. [9] emphasize the high efficiency of the laser electrodispersion (LED) method for preparation of catalysts, which consists of metal nanoparticles deposition from colloidal dispersions on a carrier. It was shown, in particular, that the Pd/Sibunit catalyst prepared in this way showed a high resistance to deactivation by hydrogen chloride. The authors of [92] came to a similar conclusion when studying the hydrodechlorination of chlorobenzene on a Ni–Al2O3 catalyst.
The nature of the carrier also plays a significant role in the selection of the hydrodechlorination catalysts. According to [93,94,95], carbon carriers are most suitable for these purposes due to their inertness with respect to hydrogen chloride as one of the process products. Nevertheless, there are a large number of publications indicating the possibility of using oxide mesoporous carriers, zeolites, and polymers [70,74,83,96,97,98]. From the point of view of applied significance, it seems that the most promising are Pd/C systems that are quite active, selective, and stable in the processes of hydrodechlorination of various classes of organochlorine compounds, for example, 1,2-dichloroethane, 1,1,2-trichloroethane, tetrachloromethane [5], 2,4-dichlorophenol, monochlorobenzene, and 4-chloro-2-methylphenoxyacetic acid [99].
4.2.2. Comparative Data of Hydrodechlorination of Chlorohydrocarbons
Hydrodechlorination of chlorohydrocarbons proceeds at different rates above the catalytic systems used in the process. The direction of the process also varies significantly depending on the type of catalyst and the process conditions. In this regard, the assessment of comparative reactivity of chlorohydrocarbons in the process is a rather difficult task. Data in this area is limited and inconsistent. They are fragmented, but the obtained results suggest that the reactivity of chlorohydrocarbons is determined by thermodynamic, polar, steric and adsorption factors, as well as the specificity of the catalytic systems. The certain results of hydrodechlorination of various chlorohydrocarbons will be discussed in detail below.
In some cases, the observed antibasis of change in the rate of C-Cl bonds hydrogenolysis in the processed compounds and the energy of these bonds indicate that the thermodynamic factor can have a decisive influence on the reactivity of the compounds and the selectivity of the process. Thus, a decrease in the chloralkanes reactivity above a Pd catalyst in the series below is accompanied by an increase in the dissociation energy of C–Cl bonds in the same direction [100].
tri-C4H9Cl >> i-C3H7Cl > n-C3H7Cl > C2H5Cl
The above dependence is typical for the platinum group metals, which do not exhibit high specificity with respect to the adsorption of compounds of various nature.
The effect of the steric factor can be stipulated either by screening of reaction sites with volume substituents, or by the dependence of the catalyst active sites accessibility for being adsorbing particles on the size of the latter. Thus, the inhibitory effect of ortho substituents [4] on the hydrogenolysis rate of C–Cl bonds in chlorobenzenes and chlorophenols is explained by the blocking effect of these substituents.
The role of the polar factor is confirmed by the correlation between the rates of C–Cl bond hydrogenolysis and the electron-donating properties of substituents in the nucleus during hydrodechlorination of chlorobenzenes in the liquid-phase process above Pd/C and its antibasis for the gas-phase hydrodechlorination [4].
The composition of the products formed can also be controlled by the adsorption capacity of the reactants or their functional groups.
The data provided in [4] that, during the tetrachloromethane hydrodechlorination above, a number of systems methane can be predominantly formed, indicate the determining effect of the adsorption factor on the products composition. The inhibitory effect of hydrogen chloride on hydrogenolysis revealed in a number of studies is usually associated with the adsorption of this component on the surface and blocking of the active sites of the catalyst.
The tetrachloromethane hydrodechlorination is one of the research priorities of hydrogenolysis of organochlorine compounds. This issue was considered, for example, in the reviews [4,9,101]. According to [102], the products of CCl4 conversion above the Pt/Al2O3 catalyst include methane, chloroform (trichloromethane), and hydrogen chloride, whereas trichloromethane is inactive in the hydrogenation reaction, even when it is added to the initial mixture. The authors proposed a radical reaction mechanism, in which hydrogen and tetrachloromethane dissociate on platinum, and then two parallel reactions proceed: C.Cl3 interacts with H2 or absorbs H to give trichloromethane. Alternatively, C.Cl3 re-adsorbs on platinum and does not desorb until it turns into C.H3 and three moles of hydrogen chloride. The radical C.H3 interacting with hydrogen gives methane.
At the interaction of tetrachloromethane with hydrogen on membrane palladium catalysts the formation of perchloroethene and hexachlorobutadiene-1,3 was revealed [42].
When studying the interaction of tetrachloromethane with hydrogen by a pulsed compression technique at typical process times of 10−3 sec, two reaction regions were found. At the temperature of 1150K (conversion rate up to 20%), the process proceeds relatively slowly. By adjusting the composition of the initial mixture and the temperature of the process, it is possible to obtain trichloromethane yield of 16% with the selectivity close to 100%. In a certain temperature range under conditions of autoignition of the mixture, the reaction can be directed to the predominant formation of perchloroethene.
The tetrachloromethane hydrodechlorination in the gas phase over the palladium catalyst, according to [46], has two features: first, the reaction does not stop at the intermediate stage, but proceeds to the formation of methane, and secondly, the catalyst is rapidly deactivated.
To maintain the stable operation of the catalyst (Pt/Al2O3) during hydrodechlorination of trichloromethane to dichloromethane, it is recommended to add water to the initial mixture, which, according to [103], stimulates chlorine removal from alumina, which prevents excessive acidity and the associated rapid carbonization.
E. Golubina et al. [104] indicate that upon the tetrachloromethane hydrodechlorination above the Pd/C catalyst at 150–220 °C, CCl4 is completely converted, while Cl2, which is formed as an intermediate at dissociative adsorption of CCl4, oxidizes palladium and this contributes to the generation of active sites of oligomerization and catalyst deactivation. Carbon-containing deposits can be removed by feeding argon into the system.
M. Bonarowska et al. [105] propose to use Pt catalyst in the process in interaction with carbon nanotubes. It is argued that catalysts of this type are quite stable and show a high selectivity for the formation of chloroform (trichloromethane). Catalysts based on Pt supported on inorganic carriers (Al2O3, MgO, CeO2) behave similarly and chloroform is also the basic product [106]. In contrast, Pd-based catalysts accelerate alkylation reactions to form C1-C7 hydrocarbons according to the Schultz–Flory mechanism [106].
Also note some patents devoted to this problem. It was shown in [107] that for the selective production of chloroform by the tetrachloromethane hydrodechlorination, it is most preferable to use the Pt/Al2O3 catalyst with a uniform distribution of platinum over the entire mass of the microspherical alumina particle. For process in the fluidized bed of the catalyst, the CCl4 conversion exceeds 90% with stable operation of the catalyst for at least 200 h.
A. Avetisov et al. [108] indicate that pretreatment of the Pt/Pt/Al2O3 catalyst with water vapor increases the selectivity of chloroform formation from 65 to 82% at tetrachloromethane hydrodechlorination at 80–120 °C. However, the conversion of tetrachloromethane under these conditions is low and does not exceed 60%.
Within the development of an integrated technology for production of lower olefins from natural gas through the intermediate synthesis of methyl chloride, the authors of [109,110] suggested to process the methane oxychlorination by-products containing CH2Cl2, CHCl3, and CCl4 by hydrodechlorination with the predominant production of methyl chloride or methane and return them to the technological cycle. The process is carried out on a Pd or Ni-Mo catalyst at 150–300 °C with high selectivity of the target products formation.
The specificity of the catalysts affects the relative contribution of various reactions occurring under hydrodechlorination conditions, and especially on the selectivity of the process. The noted role of the adsorption factor is to some extent stipulated by the specificity of the catalyst active sites with respect to the adsorption of reagents. However, the specificity nature of the catalytic systems is not limited only to adsorption interactions and in general it is necessary to take into account the structural features of both the catalyst active site and the reagent. This was shown in [45], where dichloroethenes hydrodechlorination above Pt/Al2O3 was studied. According to the authors, the higher reactivity of the C–Cl bond in dichloroethenes compared to that in chloralkanes is due to stabilization of the dichloroethene quasitautomeric form, which acts as a surface intermediate in the hydrogenolysis reaction, by the active site of the catalyst.

A similar approach is considered by the authors [4] on the example of hydrogenolysis of aromatic compounds.
4.2.3. Mechanism and Kinetics of the Process
The theoretical framework of the catalytic hydrogenolysis of organohalogen compounds are not well studied, and the kinetic data are fragmented and contradictory [4]. The absence in some works of data on the reaction pathways makes the series of catalytic activity and reactivity given in the previous section rather conditional.
A number of studies are dedicated to creating the formal mathematical models of processes. However, such models being useful for designing of reactors and optimizing the process conditions, provide little for the interpretation of the catalytic hydrogenolysis mechanism [4].
For hydrodechlorination, the catalytic systems are used, pathway above which may be different. Most authors of articles in this field believe that the mechanisms of the process for the noble metals based and nickel-chromium systems are significantly different, although they have some common aspects.
In some cases, the process speed equations of the same type are used to describe the processes with reagents that differ in adsorption capacity, which can result in change in the process limiting stage and the kinetic equation form.
The inconsistency of data on the hydrodechlorination kinetics and the catalytic activity series reflects the complexity of the studied objects stipulated by specificity of each of the catalytic systems and the ambiguity of the influence of various factors on the course of chemical processes [4]. It should be noted that most of the studies on the kinetics and mechanism of the process were carried out using chloroaromatic compounds and tetrachloromethane. Therefore, some of the reaction mechanisms proposed below are of limited importance and cannot always be extended to other hydrodechlorination objects. Nevertheless, a discussion of the hydrodechlorination mechanisms described in the scientific literature seems to be useful.
Two main groups of mechanisms can be distinguished.
The first group assumes adsorption of the reacting components on the catalytic surface, interaction of the adsorbed particles, and subsequent desorption of products from the surface (Langmuir–Hinshelwood mechanism) [41,111,112]. This approach was used to interpret the kinetics of the hydrogenolysis of n-chloronitrobenzene to n-chloroaniline on ruthenium and platinum catalysts [40,112]. A feature of the proposed mechanism is the continuation of hydrogen adsorption on the catalytic surface even after its saturation with an organic reagent.
According to the authors, the physical prerequisites for this are free sites on the surface due to the strong dipole–dipole repulsion of the polar nitrogroups of the adsorbed substrates and the small size of the hydrogen molecules. Such a mechanism was proposed, in particular, for hydrodechlorination of a number of components of vinyl chloride production wastes.
In the process of tetrachloromethane hydrodechlorination above a Pt/η-Al2O3 catalyst, a zero order reaction was observed on chlorohydrocarbon and half order on H2. The authors [28,102] substantiate this by the following hydrogen dissociation scheme:
 This scheme assumes predominant adsorption of chlorohydrocarbons on the surface of the catalyst (metal).
This scheme assumes predominant adsorption of chlorohydrocarbons on the surface of the catalyst (metal).
It should be noted that in the above scheme the formation of products is assumed to be in the gas phase. Nevertheless, the interaction of the adsorbed particles is more probable with the products release into the gas phase. However, this should not affect the form of the catalytic equation.
A slight increase in the reaction orders on the reactants with increasing of temperature may indicate that the kinetics of the process are consistent with the Langmuir–Hinshelwood mechanism.
Similar kinetic data were obtained when studying the hydrodechlorination of cis- and trans-dichloroethenes on Pt/Al2O3 [45].
It can be assumed that in this case as well, the observed orders on dichloroethene indicate adsorption saturation of the catalytic surface with dichloroethenes, and the half order of the reaction on hydrogen corresponds to the dissociative nature of its adsorption.
In the hydrodechlorination reaction of 1,2-dichloroethane [48] and dichloroethenes [23] on a nickel-chromium catalyst, the absence of the process rate dependence on the concentration of chlorohydrocarbons and first order on hydrogen concentration were also observed. This indicates hydrogen sorption on a surface saturated with chlorohydrocarbons. It should be noted that the above reactions were observed for catalysts of various types.
Another group of catalytic hydrodechlorination mechanisms involves alternating chlorination and reduction of a catalyst that acts as a carrier of hydrogen and chlorine between reactants and products. This approach is based on the mechanism proposed for the hydrogenolysis of chlorobenzenes on palladium-rhodium and palladium-tin catalysts, which includes the following stages:

However, the authors of [4] show that the experimental data obtained for the chlorobenzene hydrodechlorination are not totally consistent with the kinetic equation derived from this scheme by the method of stationary concentrations
where , , and are the current partial pressures of chlorobenzene, hydrogen, and hydrogen chloride. The equation for the reaction rate obtained in practice is
As a result, the authors of [4] come to the conclusion that the Langmuir–Hinshelwood model is preferable. This conclusion, first of all, relates to the chlorobenzene hydrodechlorination, but, obviously, it can be extended to other chlorohydrocarbons.
Of fundamental importance is the conclusion about the dissociative nature of hydrogen adsorption on the surface of metals, arising from the proportionality of the reaction rate to . The relatively low activation energies of the gross catalytic hydrodechlorination processes of a number of chlorohydrocarbons (Table 5) is an additional argument in favor of the dissociative adsorption of hydrogen on noble metal catalysts.
The fruitfulness of the Langmuir–Hinshelwood model is confirmed by the fact that it allows predicting the inhibitory effect of reactants and reaction products on the process and evaluate the adsorption capacity of the components. The low sensitivity of the hydrodechlorination reactions to the acidic active sites in the catalyst is apparently associated with the high adsorption capacity of hydrogen chloride: the adsorbed hydrogen chloride molecules act as such sites and the contribution of the natural active sites of the catalyst to its catalytic effect becomes insignificant. This may explain the failure of made in [4] efforts to modify the hydrodechlorination catalysts with alkaline agents. However, this statement is doubtful. As will be shown below, a number of authors managed to increase the catalysts stability by introducing alkali metals into them.
The products composition analysis and special studies of the mechanism of these reactions assist in clarifying the nature of the elementary stages of halogenated hydrocarbons hydrogenolysis. Obviously, the substitution of halogen for hydrogen requires the preliminary dissociation of the carbon-halogen bonds. The preferred dissociation of these very bonds is due to thermodynamic factors resulting from the relatively low C–halogen bond energies [115] and high metal–halogen bond energies, which is the driving force for the halogenated hydrocarbons chemisorption. The nature of the C–Cl bonds cleavage on the catalytic surface was studied in [113], which was devoted to the hydrogenolysis of chloromethane and dichloromethane on titanium, palladium, and other metal films. The use of 13C and 35Cl radioisotopes made it possible to establish that the cleavage of C–Cl bonds is irreversible. In the same work, it was shown that the deuterium exchange rate in chloralkanes is negligible compared to the rate of hydrogenolysis. The composition of the products formed during the hydrogenolysis of the halogenated hydrocarbons shows that the dissociation of the C–Cl bond is not accompanied by its synchronous attack by an adsorbed hydrogen atom or other surface intermediate, but is an independent elementary stage resulting in the formation of surface radicals. The latter being kinetically independent particles are involved in further interactions, forming different products depending on the conditions and type of the catalytic system. This is confirmed by the formation of polymerization products along with the products of direct halogen replacement with hydrogen.
Possible types of interactions involving surface radicals formed as a result of the primary dissociation of carbon-halogen bonds were analyzed in [4].
Reactions of chlorine radical cleavage. For reactions of chlorine radical cleavage, one of the way for turning a surface radical into a radical with a lower chlorine content can be its dechlorination with the chlorine radical cleavage followed by the interaction of the adsorbed fragment with hydrogen. This point of view [42] allows explaining the formation of products with a low content of chlorine or hydrocarbons at low conversion degrees under the hydrodechlorination conditions. In the study of the dichloroethane hydrodechlorination on a number of catalysts [5], it was shown that at dichloroethane conversion, 5–10% ethane and ethylene are the basic products (>90%) of the process. The type of catalyst used and the process conditions determines the predominant formation of ethylene and ethane.
The specificity of the catalysts used in relation to the adsorption and reactivity of the surface radicals makes it possible to vary the results of the hydrodechlorination process. Thus, at the tetrachloromethane hydrodechlorination both methane and a mixture of methane and chloromethanes can be obtained. Interesting data were obtained in [5], where it was shown that the tetrachloromethane hydrodechlorination above Pt/η-Al2O3 results in the formation of chloroform and methane exclusively with a constant ratio of [CHCl3]/[CH4] in the entire range of conversion degrees.
Hydrogenation of radicals. Hydrogenation of surface radicals results in the formation of hydrodechlorination products. This was shown in [41,113], which established an increase in the fraction of hydrogenation products with increasing hydrogen concentration. The participation of surface radicals is directly confirmed by the study of chloralkanes deuteration [67,100,113]. On the one hand, the isotopic composition of the being formed alkane indicates the structure of the surface radical involved in the hydrogenation, and on the other hand, indicates that this radical is indeed hydrogenated by hydrogen (deuterium).
Recombination of radicals. The possibility of such interactions is confirmed by the formation of hexachloroethane as one of the products of the tetrachloromethane hydrogenolysis on nickel-zeolite catalysts and alloyed metal membranes Pd-Pt, Pd-Ga, Pd-Al, and Pd-Ru [5,40]. The obtaining of the hydrogenolysis products containing twice the number of carbon atoms compared to the initial organic reagent can be considered as a result of the surface radicals recombination. The mismatch between the degree of substitution of carbon atoms in the resulting primary fragments and in the recombination products is a consequence of the primary fragments transformation into new surface radicals with the same number of carbon atoms. The observed formation of 1,1,1,2-tetrachloroethane on the Ni-zeolite catalysts [5] as one of the tetrachloromethane hydrodechlorination products should be considered as a result of [CCl3•]ads and [CH2Cl•]ads surface radicals recombination, of which the latter is a product of the [CHCl3•]ads radical transformation.
The observed effect of oligomerization on supported Pd-containing catalysts is related to the proximity of adsorbed carbon-containing particles on the catalyst surface—monoradicals and carbenes [68,116]. Obviously, the probability of such reactions progressively decreases with increasing the catalyst specific surface area determined by the carrier properties. This is clearly illustrated by an increase in the yield of the oligomerization products at transition from Pd/C to palladium deposited on inorganic carriers (from 4 to 80–94%) [68], as well as with an increase in the carbon surface for the Pd/C system (from 4 to 70%) [68,116]. An additional factor contributing the oligomerization accelerating is the irregularity of the metal distribution across the carrier surface [68]. X-ray photoelectron spectroscopy showed that Pd/C has a uniform distribution of active sites over the entire surface [68], which results in a decrease in the oligomerization rate.
Polimerization. The occurrence of such the reactions is confirmed by the formation of polymer products in some processes of catalytic hydrogenolysis. Thus, when studying the hydrogenolysis of CHnCl4-n compounds above Ni-, Co-, and Fe-catalysts [117], the formation of a wide range of products from C1 to C4 was observed. The formation of C1–C5 hydrocarbons during the tetrachloromethane hydrodechlorination above the palladium catalysts supported on a carbon carrier was observed [116].
The formation mechanism of low molecular weight polymers is poorly understood. It can be assumed that this reaction is the interaction of the surface carbene radicals with monoradicals. The acceleration of C1–C4 hydrocarbons formation from CO and H2 on Ni-, Co-, and Fe-catalysts when adding chloromethanes to the reaction mixture indicates that the formation mechanism of low molecular weight polymers under conditions of the catalytic hydrodechlorination is similar to that of the Fischer–Tropsch synthesis.
Despite the progress achieved, many aspects of the catalytic hydrodehalogenation mechanisms are still not clear. Depending on the conditions and catalysts, anionic, radical, and anion-radical hydrodehalogenation mechanisms have been proposed. Given the fundamental differences in the reaction conditions (for example, hydrogen source can be either hydrogen gas or organic hydrogen-containing donor molecules; the presence or absence of electron carriers, counter-ions in the case of complex catalysis, etc.), the mechanisms in each the specific case may also be different [4].
In addition to the general kinetics aspects and mechanism of hydrodehalogenation, it seems appropriate to provide some additional data on the chlorohydrocarbons hydrodechlorination, including wastes from the vinyl chloride production.
In the reaction scheme proposed for 1,2-dichloroethane hydrodechlorination on a rhodium catalyst [55], ethyl chloride is considered as an intermediate compound, which is then hydrogenated to ethane:
 where *H is adsorbed hydrogen; *CH2CH3 – adsorbed ethyl.
where *H is adsorbed hydrogen; *CH2CH3 – adsorbed ethyl.The authors assume that the adsorbed ethyl *CH2CH3 can serve as an intermediate in the formation of ethane, on the one hand, and of ethylene, on the other hand. The observed formation of ethylene at elevated temperatures within the framework of the proposed scheme is explained by the dehydrogenation of adsorbed ethyl *CH2CH3, which occurs due to the additional energy obtained.
The interpretation of the obtained experimental data on the 1,2-dichloroethane hydrodechlorination is of great interest, as it would allow clarifying the scheme and mechanism of the proceeding reactions [53]. According to the authors, 1,2-dichloroethane hydrodechlorination in the presence of a catalyst (1,5% Pd) at 200–300 °C, also proceeds through the intermediate formation of ethyl chloride, which can then be hydrodechlorinated to ethane. An increase in temperature to 350 °C results in ethylene appearance in the reaction products. The reason for this may be the ongoing reaction
CH3–CH2Cl → C2H4 + HCl
Since a decrease in the ethyl chloride formation selectivity in the temperature range of 300–350 °C always exactly corresponds to an increase in the ethylene formation selectivity, the authors note that the observed changes in the 1,2-dichloroethane hydrodechlorination selectivity are consistent with the ethyl chloride hydrodechlorination scheme on the palladium films [67].
Of undoubted interest are the data on the chlorinated olefins hydrodechlorination mechanism.
In the above-mentioned paper [45] devoted to the hydrodechlorination of cis-, trans-dichloroethenes, a reaction scheme and mechanism are proposed. From the reaction system the authors distinguish the reactions of ‘hydrogenation’ and ‘hydrodechlorination’. Hydrogenation of dichloroethenes resulting in dichloroethane and ethyl chloride formation are characterized by zero order on dichloroethenes and by first order on hydrogen. Hydrodechlorination reactions resulting in ethane formation in the conversion range up to 1% are characterized by zero order on dichloroethene and half order on hydrogen. The authors justify such distinguishing of the reactions by the fact that the initial dichloroethenes, as well as the intermediate product, vinyl chloride, are adsorbed on the catalyst surface in two tautomeric forms.

The hydrodechlorination mechanism was significantly refined by the results of [64,118]. It was previously established [118] that the hydrodechlorination of dichlorinated aromatic compounds occurs mainly through the sequential removal of chlorine atoms with the desorption of partially dechlorinated intermediates. The mechanism for chlorinated olefins hydrodechlorination proposed by the authors of [64] includes the stages of both sequential removal of chlorine atoms and multiplet chlorine separation. Comparison of the calculated rate constants of the corresponding stages and determination of their contribution to the chlorinated olefin consumption shows that the multiplet chlorine removal prevails. According to the authors, the multiple removal of chlorine atoms occurs without desorption of partially dechlorinated intermediates.
A decrease in the selectivity of less chlorinated hydrocarbon formation in the trichloroethene hydrodechlorination with an increase in its conversion allows suggesting these compounds being the primary reaction product, which can further be hydrodechlorinated to ethane [55] according to the sequence transformation scheme:
CHCl=CCl2 + H2 → CH2=CCl2 + HCl
CH2=CCl2 + H2 → CH3–CHCl2
CH3–CHCl2 + H2 → CH3–CH2Cl + HCl
CH3–CH2Cl + H2 → CH3–CH3 + HCl
Due to the lack of data on the adsorbed intermediates structure, the authors do not discuss the details of the reaction mechanism. The authors explain the presence of saturated less chlorinated hydrocarbons in the reaction products by the fact that these compounds are more stable on the surface of rhodium catalysts. This is consistent with the lower reactivity of 1,2-dichloroethane under the same conditions.
Some additional information on the hydrodechlorination mechanism is provided by the chloropropenes conversion data.
From the proposed reaction scheme of 1,3-dichloropropene hydrodechlorination [66], it is clear that the authors consider all three variants of interaction with hydrogen:
- (a)
- addition of hydrogen across a double bond in the molecule of 1,3-dichloropropene;
- (b)
- sequential removal of chlorine atoms;
- (c)
- multiplet removal of chlorine atoms.

The possible mechanism of chlorine atoms removal during 1,3-dichloropropene hydrodechlorination in the presence of platinum and nickel-based catalysts is not discussed in the paper. The authors only note the tendency of platinum to saturate double bonds, providing correspondingly higher yields of chloropropane, propane, and the ability of nickel to hydrodechlorinate selectively 1,3-dichloropropene to propene. However, when comparing the hydrodechlorination reactions of 1,3-dichloropropene [66] and perchloroethene [64] in the presence of nickel, the authors confine themselves to comparing certain values of the activation energies and rate constants of the initial reagents decrease, which in both cases obey the pseudo-first order, suggesting the similarity of chlorine atom removal mechanisms from dichloropropene and perchloroethene molecules, i.e., the priority of multiple separation.
Of interest are the data on the tetrachloromethane hydrodechlorination, which in the presence of nickel on zeolites results in the formation of chlorine-substituted methane, ethane, propane, and butane [65]. Based on the data obtained, the authors substantiate the formation of all the observed products by a free radical mechanism, including the initial dissociation of hydrogen on nickel.
CCl4 + ●H → ●CCl3 + HCl
●CCl3 + H2 → CHCl3 + ●H
CHCl3 + ●H → CHCl2 + HCl
Thermal reactions between the free radicals adsorbed give the oligomerization products:
●CCl3 + ●CCl3 → Cl3CCCl3
●CCl3 + ●CHCl2 → Cl3C–CHCl2
●CCl3 + ●CH2Cl → Cl3C–CH2Cl
According to the authors, the resulting C2 compounds can undergo hydrodechlorination with the formation of the corresponding free radicals. The latter, in turn, can also undergo oligomerization and then be hydrodechlorinated to the observed C3 and C4 compounds.
4.2.4. Catalyst Deactivation and Stability
The maintaining of catalytic activity over time is one of the main problems for the hydrodechlorination process. This problem exists for all the catalytic processes as well.
In a number of articles on catalytic hydrodechlorination, after a certain period of stability a significant deactivation of catalysts was noted [56,64,65].
The reasons for the deactivation and stabilization of the hydrodechlorination catalysts are reflected in the review articles [28,86]. Partial poisoning of the catalyst can even result in a change in the kinetics and mechanism of the process.
The reasons for deactivation of hydrodechlorination catalysts may be different.
The decrease in the activity of metal catalysts is usually explained by partially blocking of the catalyst surface by the obtaining hydrogen chloride and by the deposition of high molecular weight products and coke [56,58,64,65,67,119,120].
It is believed that deactivation by polymer compounds on the catalyst surface is most typical at elevated temperatures, when the oligomerization and polymerization processes proceed at a higher rate. High molecular weight compounds or coke can be formed both inside the carrier cells and at the active sites of the catalyst. The contribution of various factors to the catalyst deactivation is difficult to assess.
Analysis of samples of spent and partially deactivated Rh catalyst [55] for the chlorine content shows that after 10 h of operation the amount of chlorine in the catalyst increased from 0.11 wt % to 0.47 wt %. According to the authors, the increase in chlorine content may be one of the basic factors resulting in the Rh/SiO2 catalyst deactivation. Treatment of the catalyst with hydrogen at elevated temperature (300 °C) resulted in more than 80% recovery of the initial activity of the catalyst. Analysis of the catalyst showed that its chlorine content decreased from 0.47 wt % to 0,17 wt %. Subsequent hydrodechlorination on such a regenerated catalyst results in a decrease in the catalyst activity.
As experimental data show, it is not possible to restore its activity by treatment with hydrogen. Obviously, there is an irreversible modification of the catalyst surface, which leads to a constant loss of the catalytic activity. The authors do not exclude that agglomeration and coke formation are the possible reasons for the decrease in the catalyst activity. However, the agglomeration absence is indirectly confirmed by X-ray analysis of the catalyst samples before and after the reaction, showing that the Rh particle size remains unchanged [55].
The coke formation in the form of fibers on the outer catalyst surface is confirmed by scanning electron microscopy and spectroscopy [60]. Decontamination caused by the coking or polymerization products formation is apparently more inherent in carbon [66] and zeolite (ZSM-5) [60,65,66] catalysts having small pore sizes. In view of this, to ensure the stable operation of the hydrodechlorination catalysts, carriers and process conditions are selected so that ensure the absence of or minimum polymerization and coking rates on the catalyst.
A number of references have confirmed that the probable cause of the catalysts poisoning is the oligomerization occurring on their surfaces followed by the oligomers coking at the active sites [120,121,122,123]. The arguments in favor of this point of view are the correlation between the amount of condensation products and the rate of the catalyst deactivation, the correspondence between the amount of pyrolytic carbon in the catalyst and a decrease in its activity, the regenerating effect of oxygen on the catalyst with restoration of its initial activity, the presence of carbon dioxide in the regeneration products [120,122]. Obviously, the oxidative regeneration successfully used for inorganic-supported catalysts cannot be expanded to the metal/activated carbon systems, and the reduction method, as shown, is ineffective.
A similar disadvantage resulted in the unstable operation of the catalysts was revealed during the catalytic tetrachloromethane hydrodechlorination in the gas and liquid phases [116,124,125]. Special studies of the Pd/Sibunit catalysts stability demonstrated a monotonic decrease in their productivity over time [124,125]. So, the ICT-3-31 catalyst (0.5% Pd) lost 93% of its original activity over 140 h of operation. Attempts to regenerate the catalyst were unsuccessful: when it was treated with hydrogen at 400 °C, its activity was restored, but only for a while.
Factors contributing to catalyst deactivation: low H2:chlorohydrocarbon ratio, temperature increase and acidic sites on the carrier [65,119,120,121,122,123]. An excess of hydrogen promotes the hydrogenation of surface intermediates (CH2)ads responsible for condensation processes [42,113,117,126]. With an increase in surface temperature, pyrolysis and associated coke formation proceed, resulting in blocking of active sites [122]. Therefore, it is necessary to identify the optimal temperature for each catalytic system, combining the catalysts stability with a sufficient process rate. The absence of the acidic sites on the catalyst is the problem of carrier choice. It was shown that the catalyst deactivation rate is related to the carrier acidity [120]. In this regard, catalysts on zeolite carriers are of little promise [5.65]. It makes sense to focus researchers’ attention on basic carriers, such as MgO, or neutral carriers with minor acidic impurities. Undesirable condensation processes can be caused by Al, Fe, and Cr impurities present in activated carbon, which act as Friedel–Crafts catalysts [121].
An effective way to stabilize catalysts is to modify them with weakly basic impurities, such as alkali and alkaline earth metal chlorides, which neutralize the acidic centers [126]. An interesting example of a stable catalytic system is associated with the use of porous glass as a platinum group metal carrier, which has sufficient indifference to the oligomerization processes [127].
The catalyst stability, its activity and selectivity of the products formation substantially depend on the conditions of the catalyst preparation and pre-treatment, including various activation and passivation methods.
The proposed methods of preparation are diverse and often contradict each other. A special role the treatment of the catalyst with hydrogen [65] and hydrodechlorination products (hydrogen chloride), as well as chlorine plays.
When studying the hydrodechlorination process, all the tested catalysts (Pt, Ni on various carriers) were pre-treated using a separate method for each [109]. Catalysts containing platinum were mainly subjected to hydrogen activation at a temperature of 225–400 °C for several hours. In some cases, these techniques allow significantly increasing the time of stable operation of the catalysts, which opens the way to the industrial implementation of the hydrodechlorination process and minimizing the number of regeneration cycles.
According to the authors of [56], rhodium catalysts pretreated with hydrochloric acid or gaseous hydrogen chloride are obviously preferable for use in hydrodechlorination. It is also recommended to pre-treat the catalytic carriers with hydrochloric acid or gaseous hydrogen chloride, alkali or alkaline earth metal hydroxides or their salts before rhodium is supported. Before the direct use of the catalysts treated in such a way, the activation is carried out with hydrogen at 100–500 °C.
Pretreatment of the noble metal-based catalysts with gaseous chlorine at elevated temperatures increases the efficiency of their use in the hydrodechlorination process [128,129]. Obviously, this is primarily not due to their activity, but to their stability. We can assume the partial blocking (poisoning) of the active sites of the catalyst with chlorine and the stable operation of the major body of the catalyst over time.
The data on the effectiveness of the catalysts’ and carriers’ pretreatment with hydrogen chloride are contradictory.
It is noted that the supported platinum catalyst pretreated with a chlorine-containing substance, could be successfully used in the tetrachloromethane hydrodechlorination to produce chloroform and methylene chloride [130]. The proposed pretreatment included the following steps:
- drying of the catalyst in an atmosphere of diluent gas (nitrogen) for 2–6 h at an elevated temperature (100–500 °C);
- catalyst treatment with hydrochloric acid for 2–4 h at an elevated temperature (150–300 °C);
- catalyst recovery with a reducing agent (hydrogen, hydrazine, formaldehyde) for 2–24 h; at elevated temperature (150–500 °C);
- catalyst reprocessing with hydrochloric acid from 15 min to 2 h at a temperature of 80–150 °C.
The authors of this article were not able to stabilize the noble metal-based catalysts, as well as nickel systems by pretreatment with hydrogen chloride. The hydrodechlorination process on such systems was studied using di- and trichloroethane as an example. In these processes, the catalyst pretreated with HCl at t <350 °C did not show high activity and stability over time.
According to some reports, an increase in the service life of the hydrodechlorination catalyst is achieved by treating a noble metal (Pt, Pd) supported on an oxide carrier with chlorinated hydrocarbons in the range of 100–500 °C. Pretreatment time may vary from 15 min to 24 h, depending on the desired increase in the catalyst life time [126].
In order to increase the catalyst life time, it is also proposed to pretreat the porous catalytic support with an aqueous solution of ammonium chloride before applying platinum and/or palladium [131].
Many organic wastes contain sulfide impurities. In view of this, a number of articles are devoted to the study of such impurities effect on the catalyst and hydrodechlorination indices, including the catalyst stability The effect of catalyst sulfiding is usually studied by preliminary introducing small amounts of sulfur (mainly hydrogen sulfide) into it.
In particular, the effect of catalyst sulfiding on o-dichlorobenzene hydrodechlorination in the gas phase at 300 °C and a pressure of 2 MPa over reduced and sulfidied Ni, Mo, and Ni-Mo catalysts supported on Al2O3 was studied [132]. On all the catalysts, the reaction proceeds sequentially, with the intermediate formation of chlorobenzene. Sulfiding affects the activity of catalysts in different ways: for Ni/Al2O3, Mo/Al2O3, and Ni- Mo/Al2O3 the relative activities of the sulfided and non-sulfided forms are 0.3; 1.4; 2.2 respectively.
The proposed method for a Ni-Mo/Al2O3 catalyst treating differs from the previously described ones in that the catalyst undergoes a preliminary three-hour sulfiding at 400 °C in a stream of a mixture consisting of 10% hydrogen sulfide with hydrogen [64]. According to the method validation results, the catalyst further remains active without additional makeup with hydrogen sulfide [118].
Sulfiding of the catalyst significantly affects its stability. In the most studies an improvement in the stability of such catalysts were noted. The mechanism of this phenomenon is practically not studied.
The authors of this article also note a significant increase in the time of stable operation of the catalyst in the di- and trichloroethane hydrodechlorination when using sulfided systems with noble metals.
The preliminary sulfiding of the carrier also positively affects the stability of the catalyst supported on it. Most industrial catalysts used in hydrogenation processes are made using sulfided or pre-sulfided carriers. As the experiments showed, the optimal sulfur content in the catalyst is 0.1–0.3 wt %.
The inhibitory effect of hydrogen chloride on hydrogenolysis revealed in a number of studies is usually associated with the adsorption of this component on the surface and blocking of the active sites of the catalyst. In order to prevent blocking of the active sites of the catalyst with hydrogen chloride, some processes are carried out in the presence of alkalies, bases, or buffer mixtures (NaOH, KOH, NH4OH, NaOOCCH3-N(C2H5)3, CH3COONH4-CH3COOH, etc.) [133,134,135,136,137]. In this respect, the hydrogenation of o-chloronitrobenzene is especially indicative [137]: in the presence of NaOH the hydrogenolysis of the C-Cl bond prevails, while in its absence the nitrogroup recovery takes precedence. These and other data suggest that there are active sites of various types on the catalytic surface, each of which is specific for a particular reaction [33,39].
The use of a number of bimetallic and modified catalysts can also significantly affect their stability.
It is noted that when using a Pd-Ag bimetallic catalyst [53], with an increase in the silver content in the alloy, the activity of the catalyst decreases and the deactivation rate becomes lower.
The authors of this article showed that the most stable catalytic systems for hydrodechlorination of the main components of vinyl chloride production wastes are Ni-Cr, Cr-Cu, and Ni-Pd catalysts [5].
4.2.5. Selective Catalytic Hydrodechlorination
From the literature on catalytic hydrodechlorination, it can be seen that the selectivity of product formation depends on a number of factors. The composition of the products obtained is primarily determined by the used catalytic system and the process conditions. The conditions of preliminary preparation (thermal treatment) of the catalyst and that of its commissioning have a significant impact. In view of this, the results obtained using similar catalytic systems can vary significantly. By varying the conditions for the preparation of the catalyst, its composition and hydrodechlorination conditions, it is possible to achieve sufficiently selective obtaining of the target organic products in the process.
The possibility of introducing the hydrodechlorination of organochlorine wastes of vinyl chloride production is determined by the stability of the used catalytic systems. The data are largely contradictory, and the most articles do not contain any results obtained during the long-term operation of the studied catalysts. In view of this, this section mainly deals with materials on hydrodechlorination obtained by the authors of the article on catalytic systems characterized by a fairly high stability [5,138]. The possibility of selective hydrodechlorination of vinyl chloride production wastes is determined by the processing of the basic waste components: di- and trichloroethane.
It is most often proposed to use catalysts containing palladium and nickel for hydrodechlorination, as well as basing modified systems on them.
Hydrodechlorination on Noble Metal Based Catalysts
Catalysts based on palladium (and other noble metals), which stipulate high rate of the exhaustive hydrodechlorination process, are hardly suitable for the selective ethylene production in the processing of vinyl chloride production wastes.
An increase in the metal content in the catalyst and the concentration of hydrogen in the reaction mixture contributes to an increase in ethane yield during the processing of the C2 chlorohydrocarbons. In view of this, the efforts of selective hydrodechlorination were made under conditions of using a reduced ratio hydrogen:C2 chlorohydrocarbon.
The most consistent the use of Pd catalysts in the hydrodechlorination of vinyl chloride production wastes (di-, tri-, and tetrachloroethanes and dichloroethenes) is described in [46,47]. Table 6 and Table 7 show the results of the hydrodechlorination of di- and trichloroethanes above the Pd catalyst containing ~0.5% metal on γ-Al2O3 [47].
As can be seen from the Table 6, the basic product of 1,2-dichloroethane conversion even at H2:DCE = 1:1 is ethane. The formation of ethylene and intermediates is negligible. An increase in the process temperature from 200 to 350 °C results in an increase in the selectivity of ethylene formation, but ethane remains the basic product of the process.
The authors suggest that a decrease in the ethane yield and an increase in the ethylene yield are associated with a decrease in the rate of ethylene hydrogenation.
The selectivity of product formation significantly depends on the reagent ratio. The decrease in the H2:DCE ratio in the reaction mixture results in an increase in the selectivity of ethylene formation (up to 40–50%). However, the hydrodechlorination at a low H2:C2 ratio cannot be realized due to low catalyst stability under these conditions.
Varying 1,1,2-trichloroethane hydrodechlorination conditions (Table 7) also did not result in the selective production of ethylene.
Somewhat different results of hydrodechlorination of the same chlorohydrocarbons on Pd catalyst are given in [48,49]. The selectivity of the ethylene formation during dichloroethane hydrodechlorination with an increase in temperature up to 350 °C achieves 62% (Table 8).
Selectivity of the ethylene formation on a catalyst containing a reduced (0.25%) amount of Pd is slightly higher.
For 1,1,2-trichloroethane hydrodechlorination the authors of [49] noted an increased yield of vinyl chloride and an increase in the selectivity of the ethylene formation with temperature increasing (Table 9).
The results similar to stated above were obtained using rhodium (Rh/SiO2) [55] and platinum (Pt/Al2O3) [45,51] catalysts during 1,2-dichloroethane hydrodechlorination. An increase in temperature results in an increase in the selectivity of the ethylene formation also reported [53].
Despite these trends, it is not possible to achieve the selective production of ethylene and vinyl chloride on catalysts containing only the compounds of the platinum group noble metals. The ethane formation on all of these catalysts occurs at high rate. Obviously, the use of in the process of other metals with variable valency, as well as various binary and complex catalytic systems, including those supported on porous materials, is more promising. In this case, the platinum group metals can serve as an efficient modifier of the hydrodechlorination catalyst, on which the selective production of ethylene and vinyl chloride is carried out.
Catalysts Based on Nickel, Chromium, and other Metals of Variable Valency, Binary, and Complex Catalytic Systems
During hydrodechlorination the catalysts containing various metals of variable valency: nickel, chromium, cobalt, molybdenum, copper, manganese, bismuth, etc., as well as various combinations including these metals and platinum group metals are used. The ability to selectively produce ethylene and vinyl chloride from C2 polychlorides via hydrodechlorination on these systems, as shown, largely depends on the catalyst composition and the process conditions.
The comparative data on individual catalytic activity of a number of the most frequently used metals in the hydrodechlorination process are of interest. Table 10 shows the data on 1,2-dichloroethane hydrodechlorination at the residence time of ~4 s and the molar ratio of H2:C2H4Cl2 = 2:1 [5].
The study was carried out on a fluidized-bed unit with a catalyst containing compounds of Ni, Cr, Cu, Mn, Cd, Mo, Bi, and V (mainly in the form of chlorides). Active alumina (microsphere, specific surface area ~150 m2/g) was used as a carrier. The metal content in the catalyst was ~4 wt %. The catalyst was pretreated with hydrogen at the temperature of 400 °C. To better identify the catalytic activity of metals, the study was carried out under conditions providing a relatively low yield of hydrodechlorination products.
Under the conditions of using the catalysts with the studied metals, the formation of ethane in the hydrodechlorination process was practically not observed. This indicates the fundamental possibility of creating catalytic systems providing selective production of ethylene during hydrodechlorination. During 1,2-dichloroethane processing, along with ethylene a significant amount of vinyl chloride is obtained, which can also be considered as the target product.
As the special studies showed, the vinyl chloride is formed as a result of the dehydrochlorination occurring both on the carrier and on the metal component of the catalyst with the participation of adsorbed hydrogen. The yield of vinyl chloride is determined by the composition of the catalytic system (primarily, by the metal content) and the process conditions.
An increase in temperature from 250 to 350 °C results in a significant increase in 1,2-dichloroethane conversion and ethylene yield of on all the studied catalysts (Table 10). The main products of the process at elevated temperatures are also ethylene and vinyl chloride, and the formation of ethane was not observed.
As can be seen from the Table 10, the ethylene production on catalysts containing Mn, Cd, Bi proceeds at a low speed with a low selectivity of its formation. In view of this, these metals do not seem promising as catalysts for the selective hydrodechlorination.
The highest selectivity for the ethylene formation is observed on a nickel containing catalyst. It was found that the content of this metal in the catalyst significantly affects the results of 1,2-dichloroethane hydrodechlorination. Catalysts based on microspheric γ-Al2O3 with a nickel concentration of 4–12 wt % were used. The process was carried out on a fluidized-bed unit at 250 and 350 °C, the ratio H2:DCE = 2:1 (mol.), residence time: ~ 8 s. [5].
At a hydrodechlorination temperature of 250 °C, an increase in the nickel concentration in the catalyst from 4 to 8% results in an increase in the catalyst activity. At the same time, the selectivity of ethylene formation (from 63 to 96%) and its yield increase. A further increase in the nickel concentration of results in a slight decrease in the catalyst activity.
The most comprehensive picture of the nickel concentration effect at a temperature of 350 °C is shown in Figure 6. The 1,2-dichloroethane conversion achieves 100% at a nickel concentration of 8–10 wt %. At this concentration, vinyl chloride in the products is absent and ethylene is predominantly formed. As shown in Figure 6, an increase in nickel concentration to 8% results in an increase in the selectivity and yield of ethylene.
The selectivity of ethylene formation on the Mo catalyst is slightly lower than that on the Ni/Al2O3 system, but its yield is higher. Obviously, the presence of Mo may be desirable in a selective hydrodechlorination catalyst system.
The Cr-containing catalyst provedes an increased conversion of 1,2-dichloroethane and a reduced selectivity of the ethylene formation (as compared to a nickel-based catalyst). Chromium, obviously, can be considered as a modifier of the hydrodechlorination catalyst [5].
The process on a copper-containing catalyst does not differ in the high speed and selectivity of the ethylene formation. However, when combining with nickel, a known synergistic effect is possible, when the activity of the binary system can significantly exceed the sum of the activities of each separate catalyst. In view of this, the Ni-Cu/carrier system may be of interest for the hydrodechlorination process. A similar effect can occur when using other binary systems (Ni-Cr or Ni-Mo).
Thus, the selective production of ethylene during hydrodechlorination can be carried out on a catalyst containing nickel or binary systems containing it.
Modification of the nickel catalyst containing 3–6 wt % of metal by introducing a restricted amount (~ 0.1%) of molybdenum, chromium, copper into it does not practically affect the hydrodechlorination performance, including the selectivity of the ethylene formation and its yield.
Binary systems containing comparable amounts of nickel, chromium, molybdenum, and copper are of much greater interest.
The results of 1,2-dichloroethane hydrodechlorination on these systems are shown in Table 11 and Table 12.
As can be seen from the Table 11, the use of binary systems containing comparable amounts of nickel and molybdenum, as well as chromium and molybdenum during hydrodechlorination results in an increase in the ethylene yield compared to nickel and chromium catalysts with the virtual absence of ethane. However, the selectivity of the ethylene production using these systems under the studied conditions is not high enough (~60%) and vinyl chloride remains one of the basic products of the process.
The study of the process on the Ni-Mo or Cr-Mo systems showed that it is possible to achieve significantly more selective (above 90%) ethylene production during 1,2-dichloroethane hydrodechlorination on both systems by varying the process conditions (Table 12 and Table 13).
As can be seen from Table 12 and Table 13, increasing the process temperature increases the conversion of 1,2-dichloroethane and slightly reduces the selectivity of the ethylene formation. A distinctive feature of the process on these binary catalysts is the high (90%) selectivity of the ethylene formation under hydrogen excess conditions. The need to increase the hydrogen concentration in the reaction medium for the selective ethylene production complicates the use of the obtained ethylene at the oxychlorination stage and does not allow recommending modified with molybdenum systems for implementation of the selective hydrodechlorination process.
The hydrodechlorination on binary systems containing commensurate amounts of copper and chromium results in an almost selective formation of ethylene (Table 11) and a significant increase in its yield. This system is of significant practical interest.
During 1,2-dichloroethane hydrodechlorination on the binary Ni-Cr system, an increase in the ethylene formation selectivity to almost 80% was observed, which also makes possible to consider it as a promising one for this process [5]. Moreover, an increase in the metal content in the catalyst results in an increase in 1,2-dichloroethane conversion and an increase in the ethylene formation selectivity (up to 90%) [5].
According to [53,139], 1,2-dichloroethane hydrodechlorination in the presence of a Pd-Ag bimetallic catalyst proceeds with the selective ethylene formation. It was shown that pure silver is completely inert during 1,2-dichloroethane hydrodechlorination. The authors consider the following mechanism of the process. Silver interacts with the dichloroethane molecule through chlorine atoms, while palladium acts as a hydrogen accumulator and donor, with the help of which the silver chloride formed is reduced to Ag (0). The palladium content in the Pd-Ag alloy determines the amount of excess adsorbed hydrogen, which interacts with the formed adsorbed ethylene to produce ethane.
A kinetic model of the process on the Pd-Ag catalyst was presented in [139,140]. The model corresponds to the proposed by the authors mechanism including sequential separation of chlorine atoms above the silver surface followed by desorption of the formed ethylene. The role of palladium is to promote dissociative chemisorption of hydrogen followed by regeneration of silver surface as a catalyst. Hydrogen adsorbed on palladium also results in a significant decrease in the proportion of the ethylene hydrogenation reaction with the ethane formation.
However, the authors of [139,140] do not discuss the reason for ethyl chloride absence in the reaction products. In the presence of a catalyst containing only palladium, 1,2-dichloroethane hydrodechlorination proceeds through the intermediate formation of ethyl chloride, which corresponds to the mechanism of sequential removal of chlorine atoms in the 1,2-dichloroethane molecule. The selective ethylene formation in the presence of Pd-Ag alloys, in our opinion, suggests a different mechanism, which consists in the multiple separation of chlorine atoms. This makes the ethyl chloride formation impossible and involves the formation of ethylene, which desorption into the gas phase excludes the possibility of its further hydrogenation to ethane. An increase in the reaction temperature, as well as an increase in the proportion of silver in the alloy, accelerates the ethylene desorption, which, in turn, explains a decrease in the ethane content in the reaction products.
On the basis of the above it can be seen that the most feasible catalysts for the selective hydrodechlorination of the main components of vinyl chloride wastes may be the systems containing various metals of variable valency. The most promising are catalysts that retain activity for a long time. These include systems containing Ni, Ni-Cr, Cu-Cr, and Ni-Pd (palladium content at the level of 0.01 wt %).
Hydrodechlorination in the Presence of a Nickel-Chromium Catalyst
In [23], the organochlorine compounds hydrodechlorination in the presence of the Ni-Cr catalyst with a nickel content of at least 48% and chromium (Cr2O3) of at least 27% based on graphite was studied. The capabilities of this system in the selective hydrodechlorination are shown by an example of optimizing 1,2-dichloroethane and 1,1,2-trichloroethane processing conditions.
It was shown [23] that 1,2-dichloroethane hydrodechlorination process proceeds with a high rate and selective formation of ethylene at the temperatures above 300 °C and the molar ratio of H2:DCE = 1÷2:1. The products of the process are ethylene, ethane, vinyl chloride, and ethyl chloride, whereas the selectivity for the ethylene formation is 89–95% over the entire temperature range.
The formation of these products suggests a series of parallel-sequential reactions, the contribution of which to the process result can largely depend on the conditions of its implementation.
In particular, an increase in the temperature results in a slight increase in the selectivity for the ethylene formation and simultaneous decreasing of the selectivity for ethane, vinyl chloride, and ethyl chloride formation.
An increase in the H2:DCE ratio promotes an increase in the conversion of 1,2-dichloroethane and a slight increase in the selectivity of the ethylene formation. These changes occur, first, due to a decrease in the selectivity of the vinyl chloride and ethyl chloride formation, while the selectivity of the ethane formation slightly increases. It should be noted that, in the entire investigated range of variation in the ratios of the initial reagents, the selectivity of the process on ethylene remains at a high (85–90%) level.
In [5], considerations were given on a possible scheme for 1,2-dichloroethane hydrodechlorination. Given the high rate of the vinyl chloride formation on heterogeneous catalysts, it can be assumed that the first stage in the hydrodechlorination of 1,2-dichloroethane is its dehydrochlorination on the catalyst, obtaining the vinyl chloride. The next stage may be substitutional hydrogenation of vinyl chloride to ethylene. To clarify the chemistry of the hydrodechlorination process, 1,2-DCE dehydrochlorination was studied in the presence of the nickel-chromium catalyst at the temperature of 350 °C and the residence time of 10 s [5].
It was found that the conversion of 1,2-dichloroethane into vinyl chloride was only 5.7%. Under the same conditions, the conversion of 1,2-dichloroethane in the hydrodechlorination reaction exceeds 70%. Obviously, the dehydrochlorination reaction cannot be responsible for the first stage of 1,2-dichloroethane hydrodechlorination.
The obtained experimental data and, in particular, the observed high selectivity of the ethylene formation are in satisfactory agreement with the scheme, which suggests the so-called ‘dechlorination’ reaction or the multiple separation of chlorine with the formation of ethylene and ethane as the first stage of 1,2-dichloroethane hydrodechlorination:
C2H4Cl2 + H2 → C2H4 + 2HCl
C2H4Cl2 + 2H2 → C2H6 + 2HCl
Multiplet separation of chlorine is quite feasible at a high level of the catalyst surface saturation with hydrogen. Obtaining ethylene and ethane to a small extent, obviously, also proceeds through the formation of vinyl and ethyl chlorides with hydrogenation of ethylene to ethane:

The low selectivity of the ethane formation during hydrodechlorination suggests that the route of its formation through ethylene is not the basic one.
It can be assumed that the multiplet separation of chlorine atoms from the adsorbed 1,2-dichloroethane molecule results in the formation of a residual fragment (●CH2 –CH2●), a significant part of which is released from the catalyst surface in the form of ethylene, thus being removed from the chain of further transformations. The remainder of the fragment (●CH2–CH2●) is then hydrogenated with ethane formation and its removal from the catalyst surface. The observed decrease in the ethane formation selectivity with increasing temperature agrees well with published data [53,141] on a decrease in the ethylene hydrogenation rate under the temperatures exceeding 250 °C. In our opinion, the observed effect can be justified by an increase in the desorption ability of the fragment (●CH2–CH2●) in the form of ethylene as a result of an increase in the reaction temperature, which ultimately results in a decrease in the ethane formation selectivity.
Direct experiments on the ethylene hydrogenation on the considered catalyst showed that in the absence of 1,2-dichloroethane, ethylene is hydrogenated to ethane at a high rate [5].
It was shown that during 1,1,2-trichloroethane hydrodechlorination under the temperature range of 280–350 °C, the molar ratio of H2:TCE = 3:1 and the residence times of 5 and 10 s the obtained products are ethane, ethylene, vinyl chloride, and dichloroethenes, whereas the vinyl chloride formation selectivity is at the level of 52–81% depending on the temperature. The conversion of 1,1,2-trichloroethane in the temperature range of 300–350 °C is significantly higher than the conversion of 1,2-dichloroethane under the same conditions.
Taking into account that vinyl chloride is the basic component of the obtained products, it can be assumed that it is mainly formed according to the multiplet chlorine separation scheme, as in the case of 1,2-dichloroethane hydrodechlorination:

The presence of a significant amount of dichloroethenes (5–12%, depending on temperature) in the products suggests a somewhat more significant role of 1,1,2-trichloroethane dehydrochlorination in the hydrodechlorination process with dichloroethenes obtaining and their subsequent conversion.
The ethane formation can occur both during substitution hydrodechlorination of intermediate organochlorine saturated compounds, multiplet separation of chlorine, and during ethylene hydrogenation.
With such a process scheme, it is assumed that the conditions for its implementation can have a significant impact on the results. The authors of [5] indicate that for vinyl chloride and ethylene selective production using the Ni-Cr catalyst, the process should be carried out at 300–350 °C, the residence time of 10–15 sec, and the hydrogen concentration of 1 mol of hydrogen per a chlorine gram-atom contained in the processed chlorohydrocarbons. An increase in temperature or a decrease in the hydrogen content at the system inlet can result in catalyst deactivation. The authors of [5] conclude that there is a high probability of the process proceeding according to the scheme with multiplet separation of chlorine from 1,1,2-trichloroethane molecule to vinyl chloride and substitution hydrogenation of vinyl chloride to ethylene.
Kinetics studies are mainly restricted to the rate of C2 chlorohydrocarbons consumption in the hydrodechlorination process [48,49,63]. The data obtained can be used primarily for calculating the reactor unit and some refining of the process mechanism.
It was found that dichloroethane and dichloroethenes hydrodechlorination rate at the temperature of 300–350 °C does not depend on the concentration of these chlorohydrocarbons in the reaction mixture and is directly proportional to hydrogen concentration. The kinetic equation for the rate of their loss during hydrodechlorination is
The observed zero order on chlorohydrocarbons indicates the adsorption saturation of the catalytic surface by them. Table 14 shows the Arrhenius parameters for 1,2-dichloroethane and 1,2-dichloroethene hydrodechlorination processes.
Table 15 shows the Arrhenius parameters for 1,1,2-trichloroethane and trichloroethene hydrodechlorination processes.
The found values of the activation energy are quite close to and are in good agreement with the data obtained in the study of other catalytic systems [45,64,66].
A higher value of the activation energy for trichloroethene is probably due to the asymmetry of its molecule, the conversion of which requires more energy to create an activated complex on the catalyst surface.
The data on C2 chlorohydrocarbons hydrodechlorination on a catalyst containing 30% nickel oxide on calcium mono- and dialuminate are also presented in [5], where the potential for practical using this catalyst are shown as well (the ethylene formation selectivity during 1,2-dichloroethane hydrodechlorination is ~90% and virtually independent of temperature). The results obtained suggest that catalysts of this type retain their activity for at least 6000 h.
The use of a chromium-copper catalyst (4 wt % Cr and 1 wt % Ni on Al2O3) may also be promising. According to [5], the conversion of the initial chlorohydrocarbon during hydrodechlorination of both di- and trichloroethane is close to 100%. The ethylene formation selectivity during 1,2-dichloroethane processing is close to 100%; the basic product in the processing of 1,1,2-trichloroethane is vinyl chloride (selectivity is up to 78%, for ethylene—13–20% depending on temperature).
A series of articles by A. Srebowata et al. [86,142,143,144,145] is devoted to the patterns studies of 1,2-dichloroethane selective hydrodechlorination on the nickel catalysts. According to [86], the ethylene formation selectivity exceeding 97% is achieved on the Ni/Sibunit catalysts under the temperature not exceeding 230 °C and a high degree of Ni dispersion on the surface. It was shown that during the hydrodechlorination process, Ni carbonization occurs with the formation of Ni3C, and this compound catalyzes the side reaction of 1,2-dichloroethane dehydrochlorination producing vinyl chloride, the selectivity of which at 230 °C can reach 30%. The addition of even small amounts of Pd to Ni catalysts results in a decrease in the ethylene formation selectivity due to its hydrogenation to ethane [142]. The activation energy of the hydrodechlorination reaction is at a level of 58–66 kJ/mol, and the addition of Pd reduces the Eact by 3–6 kJ/mol.
A possible reaction mechanism for 1,2-dichloroethane hydrodechlorination on an aluminosilicate catalyst containing 1–2 wt % of Ni was proposed in [143]. The primary stage is the adsorption of the 1,2-dichloroethane molecule at the Bronsted acid sites Al-O(H)-Si with the simultaneous release of hydrogen chloride. The second chlorine atom in the 1,2-dichloroethane molecule interacts with a Ni-containing active site, resulting in cleavage of the second C-Cl bond. At the same time, hydrogen is adsorbed accompanied with its dissociation and regeneration of the acid sites by atomic hydrogen. The adsorbed atomic hydrogen interacts with chlorine-containing organic moieties. The resulting ethylene and hydrogen chloride are desorbed from the catalyst surface. The same ideas were confirmed and expanded in [144,145] during hydrodechlorination of trichloroethene along with 1,2-dichloroethane.
Implementation of Catalytic Hydrodechlorination Process
Thanks to studies conducted in the late 1990 s and early 2000 s, the obvious progress has been made in the implementation of the hydrodechlorination processes.
In [146], a schematic diagram of the UOP pilot plant for the organochlorine wastes processing and the results obtained at this plant during hydrodechlorination of organochlorine wastes of the vinyl chloride and epichlorohydrin productions are shown. The capacity of the plant for the recyclable wastes was more than 20 tons/day. The materials presented in [146] describe the results obtained during one month of continuous plant operation using the UOP catalyst. The analysis carried out showed that the activity of the catalyst and the yield of products were kept constant for the studied time period.
Hydrodechlorination of the wastes is carried out under a significant excess of hydrogen, while unreacted hydrogen is returned to the reactor.
The installation flow chart is shown in Figure 7. It contains several basic units.
Liquid production waste is fed to the installation. Before the waste is fed to the hydrodechlorination reactor, it is treated to remove solid inclusions (primarily metals) and osmols at a special unit by supplying a stream of recycle hydrogen to the heated waste. At the purification unit, ~15 wt % of solid inclusions sent to incineration are separated from the wastes.
A mixture of wastes and hydrogen enters the hydrodechlorination reactor, in which a high conversion of wastes is achieved (99.9–99.999%).
Hydrodechlorination products containing hydrogen chloride go through the purification unit, in which hydrogen chloride is neutralized with a caustic solution.
The neutralized organic hydrodechlorination products in a mixture with hydrogen are fed to a separation unit, where streams of ‘light’ gas containing C1–C4 hydrocarbons and liquid products (C5+ hydrocarbons) are released. The ‘light’ gas is used, as a rule, as fuel.
The average experimental yield of the hydrodechlorination products, wt % obtained during the processing of vinyl chloride production wastes (relatively to wastes fed to the reactor):
HCl—74.5
Methane—0.4
Ethane—14.8
Propane—2.5
Butane—8.8
C5+—4.3
As can be seen from the above data, HCl is the basic product of hydrodechlorination, its yield is about 70%, which corresponds to the content of organically bound chlorine in the wastes.
The authors [5] considered the flow chart of the selective hydrodechlorination of vinyl chloride production wastes. The processing of liquid organochlorine production wastes is shown, but the gaseous wastes can also be used.
In the option considered by the authors, the hydrodechlorination is carried out in a vertical apparatus with two to three shelves with fixed beds of the nickel-containing catalyst, on which the hydrodechlorination occurs in an adiabatic mode. The conversion of clarified waste components is 80–100% per a run. An important element of the scheme is the unit for release of hydrogen chloride, which is then returned to the ethylene oxychlorination stage. A part of the hydrocarbons formed (ethylene, vinyl chloride) are returned to the production cycle, another part is sent to the thermal treatment unit.
4.2.6. Catalytic Methods for Organochlorine Compounds Reduction in an Aqueous Medium
The authors of [9,147] consider different schemes for treating water contaminated with volatile organic compounds (VOC). Along with hydrodechlorination, reduction with zero-valence metals, primarily Fe, was also considered. The combination of these processes is thermodynamically favorable and results in to virtually complete ‘dechlorination’ of the initial contaminants with the formation of hydrocarbons and HCl as final products. As drawbacks, the authors of [147] noted a low rate of reduction by zero-valent iron, as well as its rapid loss of activity.
X. Wang et al. [148] point to the possibility of reduction without hydrogen addition using a binary Fe-Pd catalyst. A possible transformation scheme according to [148] is:
(I) Fe + 2H+ ➔ Fe2+ + H2
Fe + 2H2O ➔ Fe2+ + H2 + 2OH-
Fe + RCl + H+ ➔ RH + Fe2+ + Cl-
(II) Pd + H2 ➔ Pd.H2
Pd + RCl ➔ Pd…Cl…R
(III) Pd.H2 + Pd…Cl…R ➔ RH + H+ + Cl- + 2Pd
Under conditions of iron corrosion in an aqueous medium, the released hydrogen is absorbed by palladium with the formation of PdH2 hydride. This compound is very active in the hydrodechlorination reactions. The decrease in pH of the medium also favorably affects the process. A similar approach was considered by the authors of [149] for the Fe-Ni systems and of [150] for the Fe-Co and Fe-Cu systems.
It was noted in [151] that nanocomposite systems containing Pd, Fe nanoparticles supported on silica gel, organobentonite, and alumina are very efficient in removing aromatic and aliphatic chlorine compounds from water and soil.
C-C Huang et al. [152] considered the catalyzed by copper nanoparticles 1,2-dichloroethane hydrodechlorination carried out in an aqueous medium in the presence of Na borohydride. At 22 °C, the 1,2-dichloroethane conversion reaches 80%, whereas the basic product is ethane, and a small amount of ethylene is also observed.
In [27], the basic patterns of the CCl4 liquid phase hydrodechlorination on a Pd catalyst in the presence of aqueous solutions of sodium hydroxide were studied. The authors indicate that the system under consideration is a heterophasic process with participation of heterogeneous catalyst (Pd/sibunit) particles, hydrogen gas, and two immiscible liquid phases—organochlorine products and an aqueous solution (NaOH and NaCl) with low mutual solubility. In the temperature range of 80–120 °C with vigorous stirring of the reaction mass, the catalyst is in the aqueous phase, and the process rate is determined by the concentration of the dissolved in it reagents. The basic reaction products are sodium formate, hexachloroethane, and perchloroethene. Based on the obtained experimental data, it was concluded in [27] that two parallel reactions of hexachloroethane and chloroform formation occur during CCl4 hydrodechlorination, whereas both of which have a common first stage of CCl4 dissociative adsorption on the active metal surface with the trichloromethyl moety formation
CCl4 ➔ CCl3 ads. + Cl. ads
Next, adsorbed trichloromethyl particles interact with hydrogen turning into chloroform
CCl3. ads + H. ads. ➔ CHCl3 ads
Chloroform desorbed from the catalyst surface in an alkaline medium is converted to sodium formate. Part of the adsorbed trichloromethyl particles recombines on the catalyst surface into hexachloroethane, which then via hydrogenation turns into perchloroethene with hydrogen chloride release
C2Cl6 + H2 ➔ C2Cl4 + 2HCl
Thus, according to [27], the products of this process are not ‘dechlorinated’ hydrocarbons, but chlorine-substituted C2 compounds being an alternative to prohibited for use tetrachloromethane.
5. Reduction of Organochlorine Compounds with Metal Hydrides and Complex Hydrides of Elements
Chlorohydrocarbon hydrogenolysis with metal hydrides and complex hydrides of elements is an important component in an array of research papers devoted to the organochlorine compounds reduction. A number of studies are devoted to the interaction of organochlorine compounds with lithium aluminum hydride and sodium boron hydride [153,154,155]. According to [153,154], the reactions of halohydrocarbons with these compounds are heterolytic in nature and belong to the nucleophilic substitution type. The reaction rate decreases from primary to secondary halogen derivatives, and the tertiary ones are mainly dehydrochlorinated [155], which corresponds to the order of reactivity of halogen derivatives in SN2-type reactions. Haszeldine and Osborne [156] believe that the hydride ion transition proceeds via a cyclic transition complex. The nucleophilic nature of haloydrocarbons reduction with sodium boron hydride is manifested, for example, in that the reduction of benzhydryl chloride to diphenylmethane in the presence of water at 45 °C proceeds 60 times faster than under the same conditions in the absence of water [157]. The authors suggest that the reaction proceeds with the intermediate formation of carbonium cations according to the scheme
RCl <-> R+ + Cl−
R+ + BH4− ➔ RH + BH3
At the same time, good results are obtained with such chlorine derivatives that can form stable enough carbonium cations. For example, triphenylchloromethane, 2-phenyl-2-chloropropane are easily reduced. Under the reaction conditions in the presence of water, the side reactions of hydrolysis and dehydrochlorination proceed, which also confirms the proposed mechanism [1].
Lithium aluminum hydride as a reducing agent combines a number of important features. It is easily accessible, can be stored for a long time at room temperature, it is soluble in ethers, in comparison with other reducing agents with the exception of hydrogen it has an advantageous ratio of reducing capacity to mass, and in some cases the reduction can be carried out at room temperature.
The efficiency of the chlorohydrocarbons reduction with lithium aluminum hydride depends on the chloroalkane structure, temperature, nature of the solvent, etc. [158]. Fatty monochlorocarbons are reduced with lithium aluminum hydride with difficulty [1]. In contrast, the reduction of unsaturated compounds containing allylic chlorine proceeds quite easy. Moreover, the recovery of the secondary chlorine derivative proceeds mainly with an allyl rearrangement, and that of the primary chlorine derivative proceeds without an allyl rearrangement [159].
Lithium aluminum hydride exerts a catalytic effect during the halohydrocarbons reduction with lithium hydride [160]: this process apparently proceeds in two stages according to the scheme:
RCl +LiAlH4 ➔ RH + AlH3 + LiCl
3RCl + AlH3 ➔ 3RH + AlCl3
The first stage proceeds much faster than the second one, therefore the recovery of chlorohydrocarbon with an equimolecular amount of lithium aluminum hydride goes fast, while at the ratio of RCl: LiAlH4 = 4:1 the recovery proceeds slowly. The aluminum hydride formed in the first stage can be easily converted to LiAlH4 using lithium hydride. Therefore, the method of halohydrocarbons reducing with lithium hydride in the presence of lithium aluminum hydride as a catalyst has advantages. In the presence of lithium hydride alone, the reaction does not occur [1].
Unlike LiAlH4, sodium boron hydride (NaBH4) is relatively stable in water and many mixed aqueous systems suitable for solvolytic generation of carbonium cations. Secondary and tertiary chlorohydrocarbons are reduced under these conditions with a high yield of hydrocarbons [161]. The basic side processes are dehydrochlorination and pyrolysis. According to [162], the reduction of CCl4 with sodium borohydride in dimethyl sulfoxide proceeds through the formation of the CCl3 anion or dichloromethane.
The reduction of many chlorine derivatives with sodium boron hydride in aprotic solvating solvents (dimethyl sulfoxide, sulfolane, dimethylformamide) proceeds much easier than in water or in aqueous-organic media [162,163,164]. It is assumed that the reduction of alkyl chloride and aromatic alkyl occurs via the SN2 mechanism [162], while tertiary alkyl chlorides are likely to be reduced according to a more complex scheme (elimination of HCl, hydroboration of olefin, protonolysis of an organoboron compound) [163,164].
Also note the method proposed in [165] for the monochloro- and hexachlorobenzene hydrodechlorination with sodium boron hydride in an aqueous ethanol solution on the Pd catalyst with the addition of Pt, Co, Fe, or Ru. Various tetraethylammonium compounds were used as a phase-transfer catalyst. At 50–55 °C, the conversion degree of the initial chloroaromatic compounds was in the range of 26–96% depending on the catalyst composition and the experiment duration.
6. Conclusions
Hydrodechlorination of chlorinated organic compounds is one of the most studied segments in the field of organochlorine synthesis. The main reasons for this are the relative simplicity of the systems under study, the availability of catalysts, and good modelability of kinetic and adsorption data during the development of enlarged reactor systems. The environmental component plays a significant role—the processing of by-products of organochlorine synthesis is not associated with the formation of dioxins and greenhouse gases. At the same time, both the chlorine and carbon components of the organochlorine wastes can potentially be returned to the production cycle, which positively affects the technical and economic indicators of the processes. The largest number of publications is devoted to studies of di- and trichloroethane hydrodechlorination patterns, as the main components of vinyl chloride production wastes, as well as tetrachloromethane, which is formed in the production of chloroform and is prohibited for use as a compound that negatively affects the ozone layer.
The most promising for these purposes is the use of complex nickel-containing catalysts, characterized by high selectivity in the formation of target products. For the practical implementation of catalytic hydrodechlorination processes, it is necessary to have reliable data on the stability of the catalysts used, as well as on the possibility of their regeneration.
A limiting point is the need for preliminary preparation of wastes for processing by their clarifying in order to avoid the rapid deactivation of the catalysts. It can be assumed that in the future the technology for catalytic hydrodechlorination of the chlorine-containing organic compounds will compete with the most currently widely accepted method of thermal treatment.
Author Contributions
Conceptualization M.R.F. and Y.A.T.; Validation M.R.F. and Y.A.T.; Investigation L.M.K. and M.R.F.; Data curation Y.A.T. and M.R.F.; Writing—original draft preparation L.M.K.; Writing—review and editing M.R.F.; Project administration M.R.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank M.V. Babotina and V.E. Bragilevsky for the significant technical assistance in the preparation of review.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
References
- Nesmeyanov, A.N.; Kocheshkov, K.A. (Eds.) Methods of Elementoorganic Chemistry; Chloroaliphatic Compounds; Nauka: Moscow, Russia, 1973; pp. 491–548. (In Russian) [Google Scholar]
- Pinder, A.R. The Hydrogenolysis of Organic Halides. Synthesis 1980, 1980, 425–452. [Google Scholar] [CrossRef]
- Johnstone, R.A.; Wilby, A.H. Heterogeneous catalytic transfer hydrogenation and its relation to other methods for reduction of organic compounds. Chem Rev. 1985, 85, 129–170. [Google Scholar] [CrossRef]
- Zanaveskin, L.N.; A Aver’Yanov, V.; A Treger, Y. Prospects for the development of methods for the processing of organohalogen waste. Characteristic features of the catalytic hydrogenolysis of halogen-containing compounds. Russ. Chem. Rev. 1996, 65, 617–624. [Google Scholar]
- Flid, M.R.; Treger, Y.A. Vinyl Chloride: Chemistry and Technology; Kalvis: Moscow, Russia, 2008; Volume 2, pp. 163–231. (In Russian) [Google Scholar]
- Otromke, H.; Theerkorn, U.; Hedemann, G. Verfahren und vorrichtung zur gezielten zeriegung (Cracken) borganisch Stoffe in der Produktion und zur umweltfeundlichen Aufbereitung der gecrackten Stosse. EU Patent 0295454, 21 December 1988. [Google Scholar]
- Zanaveskin, L.N.; Averyanov, V.A.; Popov, S.A. Method for producing chlorobenzene. RF Patent No. 2039731, 18 May 1993. [Google Scholar]
- Rylander, P.N. Catalytic Hydrogenation in Organic Syntheses; Aldrichimica Acta: St. Louis, MI, USA, 1979; Volume 12, pp. 53–58. [Google Scholar]
- Lokteva, E.; Golubina, E.; Likholobov, V.; Lunin, V. Disposal of Chlorine-Containing Wastes. In Chemistry Beyond Chlorine; Tundo, P., Lokteva, E., Liang-Nian, H., Mota, C., Eds.; Springer International Publishing: Basel, Switherland, 2016; pp. 559–584. [Google Scholar]
- Lunin, V.V.; Lokteva, E.S. Catalytic hydrodehalogenation of organic compounds. Russ. Chem. Bull. 1996, 45, 1519–1534. [Google Scholar] [CrossRef]
- Evans, M.G.; Hush, N.S. Ionogenic reactions involving bond breaking at electrodes. J. de Chim. Phys. 1952, 49, C159–C171. [Google Scholar] [CrossRef]
- Elving, P.J.; Pullman, B. Mechanisms of Organic Electrode Reactions. Adv. Chem. Phys. 1961, 3, 1–31. [Google Scholar]
- Tomilov, A.P.; Mairanovsky, S.G.; Fioshin, M.Y.; Smirnov, V.A. Electrochemistry of Organic Compounds; Chimiya: Leningrad, Russia, 1968. (In Russian) [Google Scholar]
- Popp, F.D.; Schultz, H.P. Electrolytic Reduction of Organic Compounds. Chem. Rev. 1962, 62, 19–40. [Google Scholar] [CrossRef]
- Stackelberg, M.; Stracke, W.Z. Das polarographische Verhalten ungesättigter und halogenierter Kohlenwasserstoffe. ElectrochemI 1949, 53, 118–125. [Google Scholar]
- Jura, W.H.; Gaul, R.J. Polarographic Behavior of Unsymmetrical Polyhalogenated Compounds. Polychlorinated Propionitrile and Derivatives1. J. Am. Chem. Soc. 1958, 80, 5402–5409. [Google Scholar] [CrossRef]
- Pikaev, A.K.; Ershov, B.G. Primary products of water radiolysis and their reactivity. Russ. Chem. Rev. 1967, 36, 1427–1459. (In Russian) [Google Scholar] [CrossRef]
- Anbar, M.; Neta, P. Reactions of halogenoaliphatic acids with free radicals in aqueous solution. Part I. Reactions with hydrogen atoms. J. Chem. Soc. 1967, 5, 834–837. [Google Scholar] [CrossRef]
- Anbar, M.; Meyerstein, D.; Neta, P. Reactivity of aliphatic compounds towards hydroxyl radicals. J. Chem. Soc. B 1966, 742. [Google Scholar] [CrossRef]
- Abramson, F.P.; Buckhold, B.M.; Firestone, R.F. The Effects of Temperature and Various Solutes on the Radiolysis of CCl4. J. Am. Chem. Soc. 1962, 84, 2285–2288. [Google Scholar] [CrossRef]
- Louw, R.; Dijks, H.; Mulder, P. Thermal hydro-dechlorination of (poly)chlorinated organic compounds. Chem. Ind. 1983, 10, 759–760. [Google Scholar]
- Louw, R.; Manion, J.A.; Mulder, P. Gas-phase thermal hydrogenolysis of organic chlorine compounds: An alternative to incineration. Resour. Conserv. 1987, 14, 365–368. [Google Scholar] [CrossRef]
- Flid, M.R. Sustainable chlorine-balanced technologies for the production of vinyl chloride from ethane-ethylene raw materials. Dis. Doc. Chem. Sci. 2002, 333. (In Russian) [Google Scholar]
- Treger, J.A.; Kartashov, L.M. The aim — waste-free production. Ind. Russ. 1997, 8, 51–54. (In Russian) [Google Scholar]
- Treger, J.A.; Kartashov, L.M. The problem of organochlorine production wastes processing and methods for its solving. creation of waste-free technologies. Russ. J. Gen. Chem. 1998, 42, 58–66. (In Russian) [Google Scholar]
- Zanaveskin, L.N.; Averyanov, V.A. Thermal hydrogenolysis of organochlorine compounds. Basic patterns and development prospects. Russ. Chem. Ind. 1999, 2, 3–13. (In Russian) [Google Scholar]
- Terekhov, A.V.; Zanaveskin, L.N.; Zanaveskin, K.L.; Konorev, O.A. Catalytic hydrodechlorination of chlorohydrocarbons in sodium hydroxide solutions. Catal. Ind. 2012, 6, 39–47. (In Russian) [Google Scholar]
- Zanaveskin, L.N.; Averyanov, V.A. Carbon tetrachloride. Methods of processing into environmentally friendly products and prospects for their development (review). Russ. Chem. Ind. 2002, 9, 4–21. (In Russian) [Google Scholar]
- Converti, A.; Zilli, M.; De Faveri, D.M.; Ferraiolo, G. Hydrogenolysis of organochlorinated pollutants: Kinetics and thermodynamics. J. Hazard. Mater. 1991, 27, 127–135. [Google Scholar] [CrossRef]
- Louw, R.; Senden, M.M.G.; Mulden, P.; Tels, M. Thermal hydro-dechlorination of chlorinated waste, an alternative to incineration. In Recycling International Industrial and Hazardous Waste; Thome-Kozmiensky, K.J., Ed.; E. Freitag: Berlin, Germany, 1984; pp. 999–1004. [Google Scholar]
- Louw, R.; Manion, J.A.; Mulder, P. Presented at the 3rd International Symposium on Materials and Energy from Refuses, Antwerp, Belgium, 18–20 March 1986.
- Mackie, R.; Smith, D. Guidebook for Organic Synthesis (Russian translation); Mir: Moscow, Russia, 1985. [Google Scholar]
- Manion, J.A.; Louw, R. Gas-phase hydrogenolysis of chloroethene: rates, products, and computer modelling. J. Chem. Soc. Perkin Trans. 2 1988, 2, 1547. [Google Scholar] [CrossRef]
- Manion, J.A.; Louw, R. The gas-phase thermolyses of di-, tri- and tetrachloroethene in hydrogen between 828 and 1050 K. Rec. Trav. Chim. Pays-Bas. 1989, 108, 235–241. [Google Scholar] [CrossRef]
- Kondratiev, V.N. Rate Constants of Gas Phase Reactions; Nauka: Moscow, Russia, 1970. (In Russian) [Google Scholar]
- Weiss, A.H.; Krieger, K.A. Hydrodechlorination kinetics and reaction mechanisms. J. Catal. 1969, 6, 167–185. [Google Scholar] [CrossRef]
- Louw, R.; Mulder, P. Man and His Ecosystem. In Proceedings of the 8th World Clean Air Congress, The Hague, The Netherlands, 11–15 September 1989. [Google Scholar]
- Johnson, R.W.; Hilfman, L. Non-catalytic process for the conversion of a hydrocarbonaceous stream containing halogenated organic compounds. US Patent No. 4840722, 1 April 1990. [Google Scholar]
- Moreau, C.; Durand, R.; Geneste, P. Chemical evidence for the existence of two types of catalytic sites for hydroprocessing of substituted benzenes over NiW(Mo)/γ-A12O3. Am. Chem. Soc. Div. Pet. Chem. Preprint. Amer. 1987, 32, 298–307. [Google Scholar]
- Coq, B.; Tijani, A.; Figureueras, F. Pt/γ-Al2O3 catalytic membranes vs. Pt on γ-Al3 powders in the selective hydrogenation of p-chloronitrobenzene. Mol. Catal. 1991, 2, 331–338. [Google Scholar] [CrossRef]
- Coq, B.; Hub, S.; Figueras, F.; Tournigant, D. Conversion under hydrogen of dichlorodifluoromethane over bimetallic palladium catalysts. Appl. Catal. A Gen. 1993, 101, 41–50. [Google Scholar] [CrossRef]
- Mishchenko, A.P.; Senina, E.V. Investigation of carbon tetrachloride transformations on membrane catalysts. Russ. Chem. Bull. 1987, 7, 1664–1666. (In Russian) [Google Scholar]
- Ohnishi, R.; Wang, W.-L.; Ichikawa, M. Selective hydrodechlorination of CFC-113 on Bi- and Tl-modified palladium catalysts. Appl. Catal. A: Gen. 1994, 113, 29–41. [Google Scholar] [CrossRef]
- Heisler, M.; Gabler, W.; Pichl, E.; Strasser, R. Process for the preparation of trichloroethylene. Pat. DE No. 3804265, 11 February 1988. [Google Scholar]
- Weiss, A.; Krieger, K. Hydrodechlorination kinetics and reaction mechanisms. J. Catal. 1966, 6, 167–185. [Google Scholar] [CrossRef]
- Zanaveskin, L.N. Catalytic Hydrogenolysis of Chloroethanes and Chloroethylenes; GOSNIIHLORPROEKT: Moscow, Russia, 1987. (In Russian) [Google Scholar]
- Zaidman, O.A.; Verkhutova, E.I.; Zanaveskin, L.N.; Treger, Y.A. Substitution catalytic hydrogenation of C2 chlorine derivatives. Russ. Chem. Ind. 1991, 9, 5–10. (In Russian) [Google Scholar]
- Dasaeva, G.S.; Flid, M.R.; Dmitriev, Y.K.; Kartashov, L.M.; Treger, Y.A. Catalytic hydrodechlorination of 1,2-dichloroethane. Russ. Chem. Ind. 2000, 3, 49–54. (In Russian) [Google Scholar]
- Dasaeva, G.S.; Flid, M.R.; Dmitriev, Y.K.; Kartashov, L.M.; Treger, Y.A. Catalytic hydrodechlorination of 1,1,2-trichloroethane. Russ. Chem. Ind. 2000, 4, 44–46. (In Russian) [Google Scholar]
- Ordonez, S.; Diez, F.V.; Sastre, H. Catalytic Hydrodechlorination of Chlorinated Olefins over a Pd/Al2O3 Catalyst: Kinetics and Inhibition Phenomena. Ind. Ing. Chem. Res. 2002, 41, 505–511. [Google Scholar] [CrossRef]
- Gambhir, B.S.; Weiss, A.H. Depletion of reactants on a catalytic surface during reaction. J. Catal. 1972, 26, 82–91. [Google Scholar] [CrossRef]
- Ito, L.N.; Murchion, C.B. Processes for converting chlorinated alkenes to useful, less chlorinated alkenes. US Patent No. 5476979, 14 April 1994. [Google Scholar]
- Heinrichs, B.; Delhez, P. Palladium–silver sol–gel catalysts for selective hydrodechlorination of 1,2-dichloroethane into ethylene: 1 synthesis and characterization. J. Catal. 1997, 172, 322–335. [Google Scholar] [CrossRef]
- James, R.B.; Kalnes, T.N. Process for the simultaneous hydroconversion of a first feedstock comprising unsaturated, halogenated organic compounds and a second feedstock comprising saturated, halogenated organic compounds. US Patent No. 4895995, 2 December 1988. [Google Scholar]
- Bozzelli, J.W.; Chen, Y.M.; Chuang, S.S.C. Catalytic hydrodechlorination of 1,2-dichloroethane and trichloroethylene over Rh/SiO2 catalysts. Chem. Eng. Commun. 1992, 115, 1–11. [Google Scholar] [CrossRef]
- Scharfe, G.; Wilhelms, R.-E. Catalytic conversion of hydrocarbon chlorides to hydrogen chloride and hydrocarbons. US Patent No. 3892818, 7 December 1972. [Google Scholar]
- Shapovalov, V.V.; Ivanov, A.Y. Catalytic hydrogenation of dichloroethane. J. Appl. Chem. 1996, 69, 513–514. (In Russian) [Google Scholar]
- Mishakov, I.V.; Chesnokov, V.V.; Buyanov, R.A.; Pakhomov, N.A. Regularities of chlorine organic car bons decomposition flatiron group metals. Kinet. Katal. 2001, 42, 598–603. (In Russian) [Google Scholar] [CrossRef]
- Shapovalov, V.V.; Ivanov, A.Y. Features of the catalytic dechlorination of 1,2-dichloroethane on nickel. Russ. J. Phys. Chem. 1998, 72, 1326–1327. (In Russian) [Google Scholar]
- Timmons, R.B.; Jang, W.L.; He, Y.; Houpt, D.J. Catalytic hydrodehalogenation of polyhalogenated hydrocarbons. US Patent No. 5276240, 16 October 1992. [Google Scholar]
- Ordóñez, S.; Sastre, H.; DíezF, V. Hydrodechlorination of aliphatic organochlorinated compounds over commercial hydrogenation catalysts. Appl. Catal. B: Environ. 2000, 25, 49–58. [Google Scholar] [CrossRef]
- Martino, M.; Rosal, R.; Sastre, H.; DíezF, V. Hydrodechlorination of dichloromethane, trichloroethane, trichloroethylene and tetrachloroethylene over a sulfided Ni/Mo–γ-alumina catalyst. Appl. Catal. B Environ. 1999, 20, 301–307. [Google Scholar] [CrossRef]
- Dasaeva, G.S.; Flid, M.R.; Kartashov, L.M.; Treger, Y.A. Hydrodechlorination of trichlorosubstituted C2 hydrocarbons on nickel catalyst. Catal. Ind. 2002, 5, 24–29. (In Russian) [Google Scholar]
- Kim, D.I.; Allen, D.T. Catalytic Hydroprocessing of Chlorinated Olefins. Ind. Eng. Chem. Res. 1997, 36, 3019–3026. [Google Scholar] [CrossRef]
- Weiss, A.H.; Valinski, S.; Antoshin, G.V. Hydrodechlorination and oligomerization of carbon tetrachloride over nickel Y zeolites. J. Catal. 1982, 74, 136–143. [Google Scholar] [CrossRef]
- Meyer, R.J.; Kim, D.I.; Allen, D.T.; Jo, J. Catalytic hydrodechlorination of 1,3-dichloropropene. Chem. Eng. Sci. 1999, 54, 3627–3634. [Google Scholar] [CrossRef]
- Campbell, J.S.; Kemball, C. Catalytic fission of the carbon-halogen bond. Part 1.—Reactions of ethyl chloride and ethyl bromide with hydrogen on evaporated metal films. Trans. Faraday Soc. 1961, 57, 809. [Google Scholar] [CrossRef]
- Gomez-Sainero, L.M.; Cortés, A.; Seoane, X.L.; Arcoya, A. Hydrodechlorination of Carbon Tetrachloride to Chloroform in the Liquid Phase with Metal-Supported Catalysts. Effect of the Catalyst Components. Ind. Eng. Chem. Res. 2000, 39, 2849–2854. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Seoane, X.L.; Fierro, J.L.; Arcoya, A. Liquid-Phase Hydrodechlorination of CCl4 to CHCl3 on Pd/Carbon Catalysts: Nature and Role of Pd Active Species. J. Catal. 2002, 209, 279–288. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, F.; Liao, S.; Yu, D.; Sun, N. Active catalyst for the hydrodechlorination of perchlorobenzene. React. Funct. Polym. 2000, 44, 65–69. [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Kovalchuk, V.I.; D’Itri, J.L. Oligomerization pathways of dichlorodifluoromethane hydrodechlorination catalyzed by activated carbon supported Pt-Cu, Pt-Ag, Pt-Fe, and Pt-Co. Appl. Catal. B: Environ. 2002, 36, 299–309. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; Telegina, N.S.; Stakheev, A.Y.; Tundo, P. The role of Fe addition on the activity of Pd-containing catalysts in multiphase hydrodechorination. Appl. Catal. A. Gen. 2006, 302, 32–41. [Google Scholar] [CrossRef]
- Wang, C.-B.; Lin, H.-K.; Ho, C.-M. Effects of the addition of titania on the thermal characterization of alumina-supported palladium. J. Mol. Catal. A: Chem. 2002, 180, 285–291. [Google Scholar] [CrossRef]
- Gregori, M.; Benito, P.; Fornasari, G.; Migani, M.; Millefanti, S.; Ospitali, F.; Albonetti, S. Preparation of Pd/Cu MCM-41 catalysts for hydrodechlorination: Influence of the synthesis procedure. Microporous Mesoporous Mater. 2014, 190, 1–9. [Google Scholar] [CrossRef]
- Turakulova, A.O.; Golubina, E.V.; Lokteva, E.S.; Korotkov, A.V.; Lunin, V.V. ZrO2-Al2O3 binary oxides as promising supports for palladium catalysts of hydrodechorination. Russ. J. Phys. Chem. A. 2011, 85, 402–407. (In Russian) [Google Scholar] [CrossRef]
- Dafinger, W.; Schmidhammer, L. Process for the hydrogenation of chloromethanes. EP. Patent No. 0523553, 15 July 1991. [Google Scholar]
- Ohnishi, R.; Suzuki, H.; Wang, W.-L.; Ichikawa, M. Promoting role of metal additives in modified Pd catalysts for selective hydrodechlorination of CFC-113. Prep. Catal. V - Sci. Bases Prep. Heterog. Catal. Proc. Fifth Int. Symp. 1993, 77, 429–432. [Google Scholar]
- Lingaiah, N.; Uddin, A.; Muto, A.; Sakata, Y. Hydrodechlorination of chlorinated hydrocarbons over metal–carbon composite catalysts prepared by a modified carbothermal reduction method. Chem. Commun. 1999, 1657–1658. [Google Scholar] [CrossRef]
- Bonarowska, M.; Pielaszek, J.; Semikolenov, V.A. Pd-Au/Sibunit carbon catalysts: characterization and catalytic activity in hydrodechorination of dichlorodifluoromethane (CFC-12). J Catal. 2002, 209, 528–538. [Google Scholar] [CrossRef]
- Yuan, G.; Louis, C.; Delannoy, L.; Keane, M.A. Silica- and titania-supported Ni–Au: Application in catalytic hydrodechlorination. J. Catal. 2007, 247, 256–268. [Google Scholar] [CrossRef]
- Bonarowska, M.; Machynskyy, O.; Łomot, D.; Kemnitz, E.; Karpinski, Z. Supported palladium–copper catalysts: Preparation and catalytic behavior in hydrogen-related reactions. Catal. Today 2014, 235, 144–151. [Google Scholar] [CrossRef]
- Pirard, S.L.; Mahy, J.G.; Pirard, J.-P.; Heinrichs, B.; Raskinet, L.; Lambert, S.D. Development by the sol–gel process of highly dispersed Ni–Cu/SiO2 xerogel catalysts for selective 1,2-dichloroethane hydrodechlorination into ethylene. Microporous Mesoporous Mater. 2015, 209, 197–207. [Google Scholar] [CrossRef]
- Baran, R.; Śrębowata, A.; Casale, S.; Łomot, D.; Dzwigaj, S. Hydrodechlorination of 1,2-dichloroethane on nickel loaded Beta zeolite modified by copper: Influence of nickel and copper state on product selectivity. Catal. Today 2014, 226, 134–140. [Google Scholar] [CrossRef]
- Baddeley, C.; Bloxham, L.; Laroze, S.; Raval, R.; Noakes, T.; Bailey, P. The dynamic catalytic surface: probing bimetallic active sites with medium energy ion scattering. Surf. Sci. 1999, 433, 827–832. [Google Scholar] [CrossRef]
- Setterfild, C. The Practical Course of Heterogeneous Catalysis (Russian Translation); Mir: Moscow, Russia, 1984. [Google Scholar]
- Śrębowata, A.; Juszczyk, W.; Kaszkur, Z.; Sobczak, J.W.; Kępiński, L.; Karpiński, Z. Hydrodechlorination of 1,2-dichloroethane and dichlorodifluoromethane over Ni/C catalysts: The effect of catalyst carbiding. Appl. Catal. A Gen. 2007, 319, 181–192. [Google Scholar] [CrossRef]
- Díaz, E.; Faba, L.; Ordóñez, S. Effect of carbonaceous supports on the Pd-catalyzed aqueous-phase trichloroethylene hydrodechlorination. Appl. Catal. B Environ. 2011, 104, 415–417. [Google Scholar] [CrossRef]
- Bae, J.W.; Kim, I.G.; Lee, J.S.; Lee, K.H.; Jang, E.J. Hydrodechlorination of CCl4 over Pt/Al2O3: Effects of platinum particle size on product distribution. Appl. Catal. A Gen. 2003, 240, 129–142. [Google Scholar] [CrossRef]
- Amorim, C.; Yuan, G.; Patterson, P.; Keane, M. Catalytic hydrodechlorination over Pd supported on amorphous and structured carbon. J. Catal. 2005, 234, 268–281. [Google Scholar] [CrossRef]
- Cuenya, B.R. Synthesis and catalytic properties of metal nanoparticles: size, shape support, composition, and oxidation state effects. Thin Solid Film. 2010, 518, 3127–3150. [Google Scholar] [CrossRef]
- Telas, E.; Margitfalvi, J.L.; Nedelus, M.; Godolos, S.; Ruckenbauer, A. Presented at the 6-th International Symposium on Relations between Homogeneous and Heterogeneous Catalysis, Pisa, Italy, 25–29 September 1989; p. 6959.
- Kavalerskaya, N.E.; Lokteva, E.S.; Rostovshchikova, T.N. Hydrodechlorination of chlorobenzene in the presence of Ni/Al2O3 prepared by laser electrodispersion and from a colloidal dispersion. Kinet. Catal. 2013, 54, 597–606. (In Russian) [Google Scholar] [CrossRef]
- Kulkarni, P.P.; Deshmukh, S.S.; Kovalchuk, V.I.; D’Itri, J.L. Hydrodechlorination of dichlorodifluoromethane on carbon-supported Group VIII noble metal catalysts. Catal. Lett. 1999, 61, 161–166. [Google Scholar] [CrossRef]
- Ordonez, S.; Diez, F.V.; Sastre, H. Characterization of the deactivation of platinum and palladium supported on activated carbon used as hydrodechlorination catalysts. Appl. Catal. B Environ. 2001, 31, 113–122. [Google Scholar] [CrossRef]
- Yoneda, T.; Takido, T.; Konuma, K. Hydrodechlorination reactivity of para-substituted chlorobenzenes over Pt/C catalyst. J. Mol. Catal. A Chem. 2006, 256, 80–89. [Google Scholar]
- Keane, M.A. Hydrodehalogenation of haloarenes over Silica supported Pd and Ni: a consideration of catalytic activity/selectivity and haloarene reactivity. Appl. Catal. A Gen. 2004, 271, 109–118. [Google Scholar] [CrossRef]
- Shin, E.-J.; Keane, M.A. Catalytic Hydrogen Treatment of Aromatic Alcohols. J. Catal. 1998, 173, 450–459. [Google Scholar] [CrossRef]
- Bae, J.W.; Park, E.D.; Lee, J.S. Hydrodechorination of CCl4 over Pt/Al2O3: Effects of reaction pressure and diluetnt gases on distribution of productsand catalyst stability. Appl. Catal. A Gen. 2001, 217, 79–89. [Google Scholar] [CrossRef]
- Diaz, E.; Mohedano, A.; Casas, J.A.; Calvo, L.; Gilarranz, M.; Rodriguez, J. Deactivation of a Pd/AC catalyst in the hydrodechlorination of chlorinated herbicides. Catal. Today 2015, 241, 86–91. [Google Scholar] [CrossRef]
- Campbell, J.S.; Kemball, C. Catalytic fission of the carbon-halogen bond. Part 2. Reactions of tertiary butyl chloride with hydrogen and deuterium on evaporated platinum and palladium films. Trans. Faraday Soc. 1963, 59, 2583. [Google Scholar] [CrossRef]
- Treger, Y.A.; Rozanov, V.N. Obtaining of organochlorine compounds based on monocarbon molecules. Russ. Chem. Rev. 1989, 58, 138–160. (In Russian) [Google Scholar] [CrossRef]
- Weiss, A.H.; Gambhir, B.S.; Leon, R.B. Hydrodechlorination of carbon tetrachloride. J. Catal. 1971, 22, 245–254. [Google Scholar] [CrossRef]
- Noelke, C.J.; Rase, H.F. Improved Hydrodechlorination Catalysis: Chloroform over Platinum-Alumina with Special Treatments. Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 325–328. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; O Turakulova, A.; I Simagina, V.; Stoyanova, I.V. Modification of the supported palladium catalysts surface during hydrodechlorination of carbon tetrachloride. Appl. Catal. A Gen. 2003, 241, 123–132. [Google Scholar] [CrossRef]
- Bonarowska, M.; Lin, K.-N.; Legawiec-Jarzyna, M. Multi-Wall Carbon Nanotubes as a Support for Platinum Catalysts for the Hydrodechlorination of Carbon Tetrachloride and Dichlorodifluoromethane. Solid State Phenom. 2007, 128, 261–271. [Google Scholar] [CrossRef]
- Santo, V.D.; Dossi, C.; Recchia, S.; Colavita, P.; Vlaic, G.; Psaro, R. Carbon tetrachloride hydrodechlorination with organometallics-based platinum and palladium catalysts on MgO. J. Mol. Catal. A Chem. 2002, 182, 157–166. [Google Scholar] [CrossRef]
- Rossi, M.; Rubini, C.; Pasquale, A.; Cavalli, L. Catalyst for hydrodechlorination of carbon tetrachloride to chloroform. RF Patent No. 2268773, 30 October 2001. [Google Scholar]
- Avetisov, A.K.; Tarasova, D.V.; Gelperin, E.I. Method for vapor phase hydrodechlorination of carbon tetrachloride. RF Patent No. 2125034, 27 November 1995. [Google Scholar]
- Kartashov, L.M.; Rozanov, V.N.; Treger, Y.A.; Flid, M.R.; Kalyuzhnaya, T.L.; Tkach, D.V. Processing the wastes from the production of methyl chloride in the synthesis of olefins from natural gas. Catal. Ind. 2010, 2, 230–238. [Google Scholar] [CrossRef]
- Treger, Y.A.; Rozanov, V.N.; Kartashov, L.M.; Flid, M.R. An integrated method for catalytic processing of natural gas to produce lower olefins. RF Patent No. 2451005, 30 May 2011. [Google Scholar]
- Hagh, B.F.; Allen, D.T. Catalytic hydroprocessing of chlorobenzene and 1,2-dichlorobenzene. AIChE J. 1990, 36, 773–778. [Google Scholar] [CrossRef]
- Tijani, A.; Coq, B.; Figueras, F. Hydrogenation ofpara-chloronitrobenzene over supported ruthenium-based catalysts. Appl. Catal. 1991, 76, 255–266. [Google Scholar] [CrossRef]
- Anderson, J.; McConkey, B. Reactions of methyl chloride and of methylene chloride at metal surfaces II. Reactions over evaporated films of titanium and other metals. J. Catal. 1968, 11, 54–70. [Google Scholar] [CrossRef]
- Coq, B.; Figureueras, F. Presented at the VIII Soviet-French Seminar on Catalysis, Novosibirsk, Russia, 18–21 June 1990; p. 218.
- Gurvich, L.V.; Karachevtsev, G.V.; Kondratyev, V.N. Energies of Chemical Bond Breaking. Ionization Potentials and Electron Affinity; Nauka: Moscow, Russia, 1974. (In Russian) [Google Scholar]
- Lokteva, E.S.; Simagina, V.I.; Golubina, E.V.; Stoyanova, I.V.; Lunin, V.V. Formation of C1-C5 Hydrocarbons from CCl4 in the Presence of Carbon-Supported Palladium Catalysts. Kinet. Catal. 2000, 41, 855–860. (In Russian) [Google Scholar] [CrossRef]
- Van Barneveld, W.; Ponec, V. On the apparent controversy regarding the effect of alloying on the selectivity of the Fischer-Tropsch synthesis. J. Catal. 1984, 89, 542–544. [Google Scholar] [CrossRef]
- Hagh, B.F.; Allen, D.T. Catalytic hydroprocessing of chlorinated benzenes. Chem. Eng. Sci. 1990, 45, 2695–2701. [Google Scholar] [CrossRef]
- Mishakov, I.V. Development of catalytic methods for the disposal of chlorohydrocarbons and their mixtures. Dis. Cand. Chem. 2002, 180. (In Russian) [Google Scholar]
- Kim, S.Y.; Choi, H.C.; Yanga, O.B.; Lee, K.H.; Lee, J.S.; Kim, Y.G. Hydrodechlorination of tetrachloromethane over supported Pt catalysts. J. Chem. Soc. Chem. Commun. 1995, 21, 2169. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, A.; Van De Sandt, E.; Makkee, M.; Luteijn, C.; Van Bekkum, H.; Moulijn, J. Process for the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32). Catal. Today 1996, 27, 257–264. [Google Scholar] [CrossRef]
- Choi, H.C.; Choi, S.H.; Yang, O.B.; Lee, J.S.; Lee, K.H.; Kim, Y.G. Hydrodechlorination of Carbon Tetrachloride over Pt/MgO. J. Catal. 1996, 161, 790–797. [Google Scholar] [CrossRef]
- Bucsi, I.; Beard, B.C. Selective monochlorination of methane over solid acid and zeolite catalysts. Catal. Lett. 1992, 16, 27–38. [Google Scholar] [CrossRef]
- Dasaeva, G.S.; Velichko, S.M.; Treger, Y.A.; Moiseev, I.I. Hydrodechlorination of CCl4 in the presence of Pd(OAc)2. Kinet. Catal. 1990, 31, 858–862. (In Russian) [Google Scholar]
- Dasaeva, G.S.; Treger, Y.A.; Moiseev, I.I.; Zanaveskin, L.N. Liquid phase catalytic hydrodechlorination of carbon tetrachloride. Russ. Chem. Ind. 1996, 6, 346–350. (In Russian) [Google Scholar]
- Zhang, Z. Treatment to improve the durability of a hydrodechlorination catalyst and catalyst. World Patent WO1999017876B, 10 February 1997. [Google Scholar]
- Prati, L. Reductive catalytic dehalogenation of light chlorocarbons. Appl. Catal. B Environ. 1999, 23, 135–142. [Google Scholar] [CrossRef]
- Kellner, C.S.; Lerou, J.J.; Rao, V.N.M.; Wuttke, K.G. Regeneration or activation of noble metal catalysts using fluorohalocarbons or fluorohalohydrocarbons. US Patent No. 4980324, 25 December 1990. [Google Scholar]
- Kellner, C.S. Regeneration of noble metal catalysts used in hydrodehalogenation of halogen-substituted hydrocarbons containing fluorine and at least one other halogenus. US Patent No. 5057470, 22 May 1990. [Google Scholar]
- Holbrook, M.T.; Harley, A.D. Vapor phase hydrogenation of carbon tetrachlorideus. US Patent No. 5105032, 4 October 1990. [Google Scholar]
- Bozon, A.; Lakatos, E.; Koberstein, E.; Pletka, H.-D.; Voelker, H. Process for application of a catalytically active coating containing platinum, palladium or rhodium or their mixtures to a carrier and product made by said process. US Patent No. 4374047, 15 February 1983. [Google Scholar]
- Novak, M. and Zdrazil, M. Effects of Sulfidation and Synergism in Hydrodechlorination of o-Dichlorobenzene Over NiMo/Alumina Catalyst. Bull. Soc. Chim. Belg. 1993, 102, 271–279. [Google Scholar] [CrossRef]
- Mullin, C.R.; Wymore, C.E. Hydrogenolysis of carbon tetrachloride and chloroform. US Patent No. 3579596, 29 March 1968. [Google Scholar]
- Kincey, P.M.B. 2-Aminopurine preparation process. EP Patent No. 0355986, 18 July 1990. [Google Scholar]
- Hoke, J.B.; Gramiccioni, G.A.; Balko, E.N. Catalytic hydrodechlorination of chlorophenols. Appl. Catal. B: Environ. 1992, 1, 285–296. [Google Scholar] [CrossRef]
- Simakova, I.L.; Semikolenov, V.A. Study of the principles of liquid-phase hydrodechlorination of organochlorine compounds on a Pd/C catalyst. Kinet. Catal. 1991, 32, 989–993. (In Russian) [Google Scholar]
- Sharf, V.Z.; Gurovets, A.S.; Slinyakova, I.B. Investigation of the catalytic activity of metal complexes fixed on a solid support. Message I. Hydrogenolysis of chlorobenzenes in the presence of palladium chloride complex deposited on modified silica gel. Russ. Chem. Bull. 1980, 1, 114–117. (In Russian) [Google Scholar]
- Kartashov, L.M.; Flid, M.R.; Treger, Y.A. Stability of catalytic systems in hydrodechlorination of chlorohydrocarbons and chlorocarbons C1–C6. In Proceedings of the 10th International Symposium on Catalyst Deactivation, Berlin, Germany, 5–8 February 2006. [Google Scholar]
- Heinrichs, B.; Schoebrechts, J.-P.; Pirard, J.-P. Palladium–Silver Sol–Gel Catalysts for Selective Hydrodechlorination of 1,2-Dichloroethane into Ethylene. J. Catal. 2001, 200, 309–320. [Google Scholar] [CrossRef]
- Job, N.; Heinrichs, B.; Ferauche, F.; Noville, F.; Marien, J.; Pirard, J.-P. Hydrodechlorination of 1,2-dichloroethane on Pd–Ag catalysts supported on tailored texture carbon xerogels. Catal. Today 2005, 102, 234–241. [Google Scholar] [CrossRef]
- Kiperman, S.L. Introduction to Kinetics of Heterogeneous Catalytic Reactions; Nauka: Moscow, Ruissia, 1964. (In Russian) [Google Scholar]
- Śrębowata, A.; Juszczyk, W.; Kaszkur, Z.; Karpiński, Z. Hydrodechlorination of 1,2-dichloroethane on active carbon supported palladium–nickel catalysts. Catal. Today 2007, 124, 28–35. [Google Scholar] [CrossRef]
- Baran, R.; Kamińska, I.; Śrębowata, A.; Dzwigaj, S. Selective hydrodechlorination of 1,2-dichloroethane on NiSiBEA zeolite catalyst: Influence of the preparation procedure on a high dispersion of Ni centers. Microporous Mesoporous Mater. 2013, 169, 120–127. [Google Scholar] [CrossRef]
- Śrębowata, A.; Baran, R.; Łomot, D.; Lisovytskiy, D.; Onfroy, T.; Dzwigaj, S. Remarkable effect of postsynthesis preparation procedures on catalytic properties of Ni-loaded BEA zeolites in hydrodechlorination of 1,2-dichloroethane. Appl. Catal. B Environ. 2014, 147, 208–220. [Google Scholar] [CrossRef]
- Kaminska, I.I.; Srebowata, A. Active carbon-supported nickel-palladium catalysts for hydrodechlorination of 1,2-dichloroethane and 1,1,2-trichloroethane. Rec. Chem. Intermed. 2015, 41, 2007. [Google Scholar]
- Kalnes, T.N.; James, R.B. Hydrogenation and recycle of organic waste streams. Environ. Prog. 1988, 7, 185–191. [Google Scholar] [CrossRef]
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment — sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef]
- Wang, X.; Chen, C.; Chang, Y.; Liu, H. Dechlorination of chlorinated methanes by Pd/Fe bimetallic nanoparticles. J. Hazard. Mater. 2009, 161, 815–823. [Google Scholar] [CrossRef]
- Zhang, W.-H.; Quan, X.; Zhang, Z.-Y. Catalytic reductive dechlorination of p-chlorophenol in water using Ni/Fe nanoscale particles. J. Environ. Sci. 2007, 19, 362–366. [Google Scholar] [CrossRef]
- Zhou, T.; Li, Y.; Lim, T.-T. Catalytic hydrodechlorination of chlorophenols by Pd/Fe nanoparticles: Comparisons with other bimetallic systems, kinetics and mechanism. Sep. Purif. Technol. 2010, 76, 206–214. [Google Scholar] [CrossRef]
- Trujillo-Reyes, J.; Peralta-Videa, J.; Gardea-Torresdey, J. Supported and unsupported nanomaterials for water and soil remediation: Are they a useful solution for worldwide pollution? J. Hazard. Mater. 2014, 280, 487–503. [Google Scholar] [CrossRef]
- Huang, C.-C.; Lo, S.-L.; Tsai, S.-M.; Lien, H.-L. Catalytic hydrodechlorination of 1,2-dichloroethane using copper nanoparticles under reduction conditions of sodium borohydride. J. Environ. Monit. 2011, 13, 2406. [Google Scholar] [CrossRef]
- Roginskaya, E.V. Lithium aluminum hydride and its use in organic chemistry. Russ. Chem. Rev. 1952, 21, 3–39. (In Russian) [Google Scholar]
- Brown, H.S. Organic Reactions; IL: Moscow, Russia, 1952. [Google Scholar]
- Gaylord, N.C. Reduction with complex metal hydrides. J. Chem. Educ. 1956, 34, 367. [Google Scholar] [CrossRef]
- Haszeldine, R.N.; Osborne, J.E. 13. Addition of free radicals to unsaturated systems. Part XII. Free-radical and electrophilic attack on fluoro-olefins. J. Chem. Soc. 1956, 61. [Google Scholar] [CrossRef]
- Brown, H.C.; Bell, H.M. Alkali-Catalyzed Dealkilation of Two Diastereometric Benzyl Boranes. J. Org. Chem. 1962, 27, 1928. [Google Scholar]
- Eliel, E.L.; Herrmann, C.; Traxler, J.T. The Mechanism of Halide Reductions with Lithium Aluminum Hydride. III. Reduction of α-Chloro Acids and Esters. J. Am. Chem. Soc. 1956, 78, 1193–1198. [Google Scholar] [CrossRef]
- Jacobs, T.L.; Wilcox, R.D. Dehalogenation of Propargyl and Allenyl Halides. II. J. Am. Chem. Soc. 1964, 86, 2240–2247. [Google Scholar] [CrossRef]
- Johnson, J.E.; Blizzard, R.H.; Carhart, H.W. Hydrogenolysis of Alkyl Halides by Lithium Aluminum Hydride1. J. Am. Chem. Soc. 1948, 70, 3664–3665. [Google Scholar] [CrossRef]
- Bell, H.M.; Brown, H.C. Selective Reductions. XI. The Reaction of Sodium Borohydride with Alkyl Halides under Solvolytic Conditions. Borohydride as a Convenient Trap for Carbonium Ions1,2. J. Am. Chem. Soc. 1966, 88, 1473–1477. [Google Scholar] [CrossRef]
- Bell, H.M.; Vanderslice, C.W.; Spehar, A. Reduction of organic halogen compounds by sodium borohydride. J. Org. Chem. 1969, 34, 3923–3926. [Google Scholar] [CrossRef]
- Hutchins, R.O.; Hoke, D.; Keogh, J.; Koharski, D. Sodium borohydride in dimethyl sulfoxide or sulfolane. Convenient systems for selective reductions of primary, secondary and certain tertiary halides and tosylates. Tetrahedron Lett. 1969, 10, 3495–3498. [Google Scholar] [CrossRef]
- Jacobus, J. The mechanism of the reduction of alkyl halides with sodium borohydride in dimethyl sulphoxide. Chem. Comm. 1970, 6, 338. [Google Scholar]
- Simagina, V.I.; Stojanova, I.V.; Gentsler, A.G.; Tajban, E.S. Catalyst and method for hydrodechlorination of chloroaromatic compounds. RF Patent No. 2214864, 22 July 2002. [Google Scholar]
Figure 1.
Publications on hydrodechlorination problems.

Figure 2.
Obtaining vinyl chloride from ethylene according to a balanced scheme with the stage of waste hydrodechlorination.
Figure 2.
Obtaining vinyl chloride from ethylene according to a balanced scheme with the stage of waste hydrodechlorination.

Figure 3.
Chlorohydrocarbon reactivity in the process of thermal hydrodechlorination. Hydrogen excess, atmospheric pressure, residence time 11 s.
Figure 3.
Chlorohydrocarbon reactivity in the process of thermal hydrodechlorination. Hydrogen excess, atmospheric pressure, residence time 11 s.

Figure 4.
Temperature dependence of the process rate constants for different chlorohydrocarbons.

Figure 5.
Schematic diagram of thermal hydrodechlorination of organochlorine products. 1—reactor; 2—regenerator; 3, 4—cyclones; 5—quenching column; 6—condenser; 7—hydrogen chloride absorber; 8—hydrochloric acid reservoir.
Figure 5.
Schematic diagram of thermal hydrodechlorination of organochlorine products. 1—reactor; 2—regenerator; 3, 4—cyclones; 5—quenching column; 6—condenser; 7—hydrogen chloride absorber; 8—hydrochloric acid reservoir.

Figure 6.
Dependence of the indices of 1,2-dichloroethane hydrodechlorination on a nickel catalyst on nickel concentration of microspheric alumina at 350 °C. 1—conversion of dichloroethane; 2—selectivity of ethylene formation; 3—selectivity of ethane formation; 4—selectivity of vinyl chloride formation.
Figure 6.
Dependence of the indices of 1,2-dichloroethane hydrodechlorination on a nickel catalyst on nickel concentration of microspheric alumina at 350 °C. 1—conversion of dichloroethane; 2—selectivity of ethylene formation; 3—selectivity of ethane formation; 4—selectivity of vinyl chloride formation.

Figure 7.
Flow-chart of waste hydrodechlorination with fuel obtaining.


{kind=link}
{kind=link}
{kind=link}
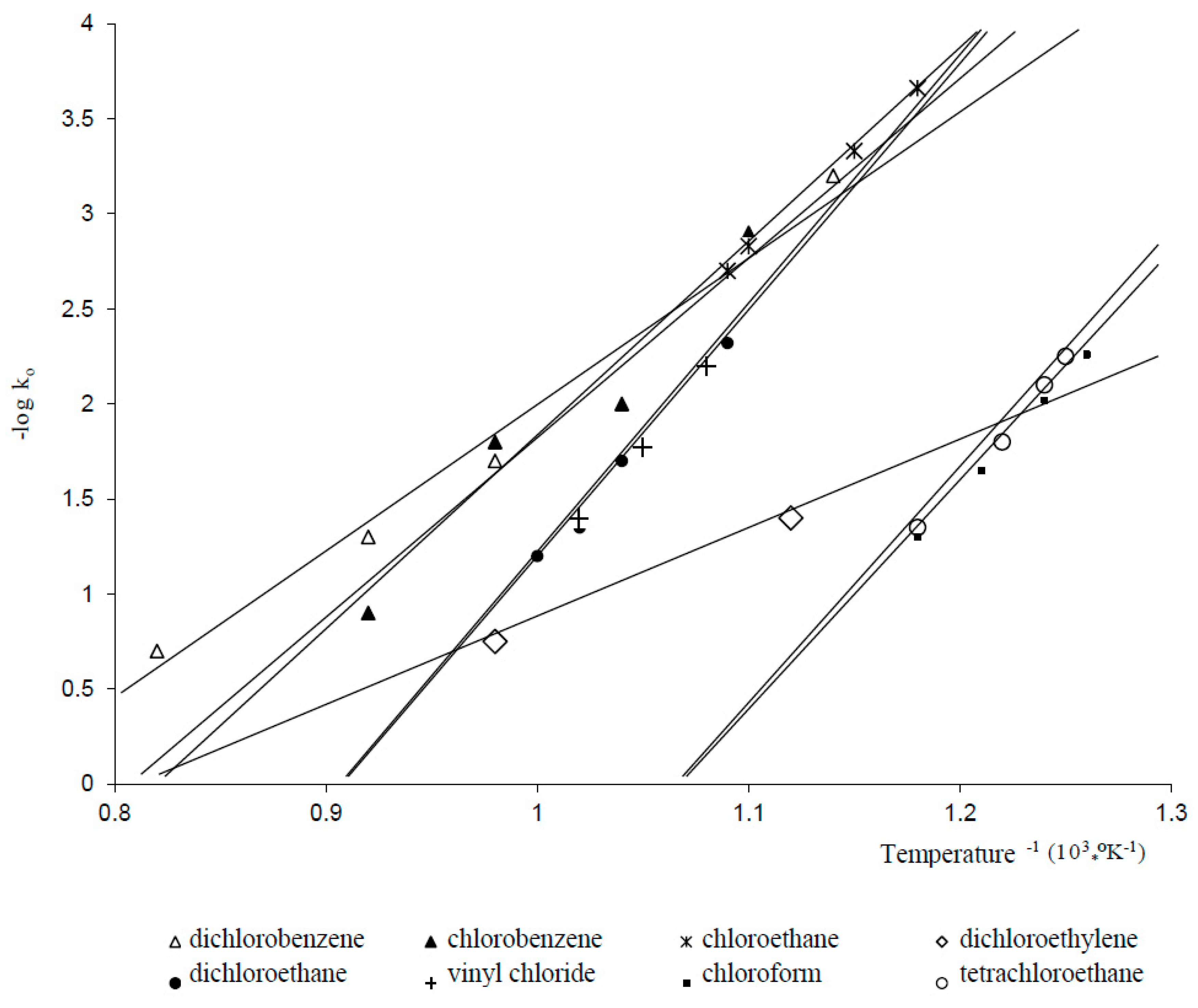{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Standard heats of formation of waste components and products of their conversion, as well as thermal effect of hydrodechlorination reactions.
Table 1.
Standard heats of formation of waste components and products of their conversion, as well as thermal effect of hydrodechlorination reactions.
| Component | Heat of Formation ∆H298, kJ/mol | Thermal Effect of the Reaction, kJ/mol | ||
|---|---|---|---|---|
| to Ethylene | to Ethane | to Methane and Benzene | ||
| Chloroform | −100.3 | - | - | +251.6 (to methane) |
| Tetrachloromethane | −105.3 | - | - | +339.0 (to methane) |
| Chlorethyl | −105.3 | −64.8 | +71.9 | - |
| Vinyl chloride | +155.5 | +77.3 | +214.0 | - |
| Vinylidene chloride | +1.3 | +133.8 | +270.4 | - |
| 1,1-dichloroethane | −130.0 | +2.9 | +139.6 | - |
| 1,2-dichloroethane | −130.0 | +2.9 | +139.6 | - |
| 1,1,2-trichloroethane | −138.4 | +86.5 | +223.2 | - |
| Perchloroethene | −15.0 | +302.2 | +438.9 | - |
| 1,1,2,2-tetrachloroethane | −152.6 | +164.7 | +301.4 | - |
| Chlorobutanes and Chlorobutenes | −191.4 | +26.8 | +27.2 | - |
| Chlorobenzene | −111.6 | - | - | +102.0 (to benzene) |
| Methane | −74.8 | - | - | - |
| Ethylene | +52.3 | - | - | - |
| Ethane | −84.4 | - | - | - |
| Butene | −125.4 | - | - | |
| Butane | −126.2 | - | - | - |
| Benzene | +82.8 | - | - | - |
| HCl | −92.4 | - | - | - |
Table 2.
Thermal hydrodechlorination of chlorohydrocarbons.
| Component | Temperature, °C | Residence Time, s | Conversion, % | Reference |
|---|---|---|---|---|
| CH3Cl | 604–840 | 11 | 19.6–98.4 | [31] |
| CH2Cl2 | 650–720 | 11 | 12.0–88.0 | [31] |
| CHCl3 | 526–550 | 11 | 44.5–78.5 | [31] |
| CCl4 | 532–573 | 11 | 16.4–74.7 | [31] |
| CH2=CH–Cl | 660–800 | 11 | 43.2–80.0 | [31] |
| Cl–CH=CH–Cl | 610–750 | 6.2–8.0 | 50.0–96.0 | [21,30] |
| CCl2=CCl2 | 750 | 6 | 96 | [32] |
| C2H4Cl2 | 580 | 10 | 8 | [32] |
| C6H5Cl | 604–840 | 7.5–11.0 | 19.6–98.4 | [21,30,31] |
| C6H4Cl2 | 600–950 | 4.6–8.8 | 0.7–93.0 | [21,30] |
Table 3.
Calculated thermodynamic characteristics for hydrodechlorination of various chlorohydrocarbons. Temperature of 700 °C.
Table 3.
Calculated thermodynamic characteristics for hydrodechlorination of various chlorohydrocarbons. Temperature of 700 °C.
| Component | Eact (kJ/mol) | ∆Sact (kJ/mol K) | ∆Gact (kJ/mol) | ko (c−1•103) | logA |
|---|---|---|---|---|---|
| CH3Cl | 184.8 | –103.9 | 285.8 | 9 | 7.88 |
| CH2Cl2 | 237.9 | –37.7 | 274.4 | 40 | 11.35 |
| CHCl3 | 243.9 | 8.4 | 235.5 | 4790 | 13.74 |
| CCl4 | 256.8 | 23 | 234.6 | 5517 | 15.52 |
| CH2=CH–Cl | 244.7 | –30.6 | 274 | 41 | 11.72 |
| Cl–CH=CH–Cl | 90.9 | –181 | 267.3 | 96 | 3.86 |
| C6H5Cl | 200.7 | –113.1 | 285.8 | 10 | 7.40 |
| C6H4Cl2 | 146.7 | –146.2 | 288.7 | 7 | 5.68 |
Table 4.
Conditions of the hydrodechlorination process of chlorohydrocarbons in the presence of various catalysts based on the noble metals.
Table 4.
Conditions of the hydrodechlorination process of chlorohydrocarbons in the presence of various catalysts based on the noble metals.
| Noble Metal Systems | ||||
| Catalyst | Raw materials | Process conditions | Products | Source |
| Pd/Al2O3, Pd/SiO2 palladium content 0.5–4 wt % | 1,2-dichloroethane | 200–350 °C | ethane, ethylene | [46,47,48] |
| 1,1,2-trichloroethane | 200–350 °C | ethane, ethylene, vinyl chloride | [46,47,49] | |
| tetrachloroethane | 200-350 | ethane, ethylene, dichloroethenes | [46,49] | |
| Pt/Al2O3, Pd/Al2O3 | dichloroethenes | 90 °C 170–350 °C, 0,5 MPa | ethane, chloroethenes ethane | [45,50,51] |
| Pd-Cu-Sn/C | Chloralkenes, (perchloroethene) | 160–250 °C 0.2–2 MPa | less chlorinated alkenes | [52] |
| Pd-Ag | 1,2-dichloroethane | 250–350 °C | ethylene | [53] |
| Pd/Al2O3 | Trichloroethene, perchloroethene | 170–350 °C 0.5 MPa | ethane | [50] |
| Pd/Al2O3 | Unsaturated halogenated hydrocarbons, saturated halogenated hydrocarbons | 80 °C, 5 MPa 320 °C, 5 MPa | saturated hydrocarbons | [54] |
| Rh/SiO2 Rh/carrier with promotor: Pd, Pt, Ru, Fe, Co, etc. | 1,2-dichloroethane Trichloroethene Hydrocarbons C1–C30 (dichloropropane) | 90–280 °C 120 °C 180–250 °C | ethane, ethyl chloride ethane (propane, propene) | [55,56] [56] |
| Pd, Pt, Rh/carrier | Dichloropropane | 200–300 °C | propene, propane | [25] |
| Nickel Based Systems | ||||
| Catalyst | Raw materials | Process conditions | Products | Source |
| 9.6% Ni on pumice | 1,2-dichloroethane | 350–500 °C | ethane | [57] |
| Nickel aluminum | 1,2-dichloroethane | 400–550 °C | methane, ethane | [58] |
| Ni on fireclay | 1,2-dichloroethane | 350–400 °C | ethylene | [59] |
| Ni-Cr on C | 1,2-dichloroethane | 280-370 °C | ethylene, ethane, monochlorinated C2 | [48] |
| Ni-Cr on C | 1,1,2-trichloroethane | 280–370 °C | ethylene, ethane, vinyl chloride, dichloroethenes | [49] |
| Ni on ZSM-5/Al2O3 | Trichloroethene Trichloroethane | 250–450 °C | non-chlorinated olefins, paraffins, aromatic compounds | [60] |
| Ni-Mo/Al2O3 | 1,1,1-trichloroethane | 300 °C | ethane, vinyl chloride, dichloroethenes | [61] |
| Ni-Mo/γ-Al2O3 sulphurized | 1,1,1-trichloroethane, per- and trichloroethene | 250–350 °C | non-chlorinated hydrocarbons | [62] |
| Ni-Mo/Al2O3 | Trichloroethene | 300 °C | ethane, dichloroethenes | [61] |
| Ni-Cr on C | Trichloroethene | 300–350 °C | ethylene, vinyl chloride | [63] |
| Ni-Mo/Al2O3 | Perchloroethene Trichloroethene | 175–275 °C | dichloroethenes, non-chlorinated aliph. compounds | [64] |
| Ni-Mo/Al2O3 | Perchloroethene | 300 °C | ethane, trichloroethene, dichloroethenes, ethyl chloride | [61] |
| Ni on Y zeolites | tetrachloromethane | 370 | chlorinated and non-chlorinated alkanes | [65] |
| Ni-Mo/γ-Al2O3 | 1,3-dichloropropene | 325 °C | propene, propane, allyl chloride | [66] |
| Ni/Al2O3/SiO2 | 1,3-dichloropropene | 325 °C | propene, chloropropenes | [66] |
| Ni on H-ZSM-5/Al2O3 | 1,3-dichloropropene | 325 °C | propene, chloropropenes | [66] |
Table 5.
Activation energy for hydrodechlorination.
| Reaction | Catalyst | E, kJ/mol | Source |
|---|---|---|---|
| CH3Cl + H2 → CH4 + HCl | Ti film | 67.3 | [113] |
| CH2Cl2 + H2 → CH3Cl + HCl | Ti film | 56.0 | [113] |
| CH2Cl2 + 2H2 → CH4 + 2HCl | Ti film | 55.6 | [113] |
| (CH3)3CCl + H2 → (CH3)3CH + HCl | Pt film | 79.4 | [100] |
| (CH3)3CCl + H2 → (CH3)3CH + HCl | Pd film | 75.2 | [100] |
| C6H5Cl + H2 → C6H6 + HCl | Pd/Al2O3 | 104.5 | [114] |
| ClHC = CHCl + H2 → H2C = CHCl + HCl | Pt/Al2O3 | 115.0 | [45] |
Table 6.
Influence of temperature and reagents ratio on composition of 1,2-dichloroethane hydrodechlorination products. Residence time: 18 s.
Table 6.
Influence of temperature and reagents ratio on composition of 1,2-dichloroethane hydrodechlorination products. Residence time: 18 s.
| Temperature, °C | Molar Ratio, H2:DCE | Composition of the Reaction Products, % | DCE Conversion Degree, % | |||
|---|---|---|---|---|---|---|
| CH2CHCl | CH3CH2Cl | C2H6 | C2H4 | |||
| 200 | 1:1 | - | - | 100 | - | 11 |
| 250 | 1:1 | 1.4 | - | 90.9 | 7.7 | 40 |
| 300 | 1:1 | 5.5 | - | 79.3 | 15.2 | 47 |
| 350 | 1:1 | 1.8 | 12.0 | 71.0 | 15.2 | 55 |
| 350 | 2:1 | 0.75 | 8.4 | 83.9 | 7.0 | 45 |
| 350 | 1:1 | 1.8 | 12.0 | 71.0 | 15.2 | 24 |
| 350 | 1:2 | 8.0 | 8.0 | 48.2 | 35.9 | 10 |
| 350 | 1:4 | 10.0 | 5.3 | 37.1 | 47.6 | 5 |
Table 7.
Influence of temperature and reagents ratio on composition of 1,1,2-trichloroethane hydrodechlorination products. Residence time: 18 s.
Table 7.
Influence of temperature and reagents ratio on composition of 1,1,2-trichloroethane hydrodechlorination products. Residence time: 18 s.
| Temperature, °C | Molar Ratio, H2:DCE | Composition of the Reaction Products, % | 1,1,2-TCE Conversion Degree, % | ||||
|---|---|---|---|---|---|---|---|
| CH2=CHCl | CH3CH2Cl | CH2CLCH2Cl | C2H6 | C2H4 | |||
| 200 | 3:1 | - | 7.7 | 1.1 | 91.0 | 0.2 | 46 |
| 250 | 3:1 | - | 15.7 | 2.4 | 81.5 | 0.4 | 58 |
| 300 | 3:1 | 2.1 | 18.0 | 0.7 | 71.8 | 9.5 | 96 |
| 350 | 3:1 | 3.2 | 20.0 | - | 53.7 | 23.1 | 99 |
| 300 | 4:1 | 1.3 | 13.3 | - | 78.5 | 6.9 | 97 |
| 300 | 3:1 | 2.1 | 18.0 | 0.7 | 71.8 | 9.5 | 96 |
| 300 | 1:2 | 9.8 | 14.3 | 0.7 | 48.8 | 26.1 | 62 |
| 300 | 1:1 | 21.3 | 5.6 | 1.0 | 32.5 | 38.7 | 21 |
Table 8.
Activity and selectivity of various catalysts in 1,2-dichloroethane hydrodechlorination residence time: 10 sec; molar ratio H2:1,2-dichloroethane = 2:1.
Table 8.
Activity and selectivity of various catalysts in 1,2-dichloroethane hydrodechlorination residence time: 10 sec; molar ratio H2:1,2-dichloroethane = 2:1.
| Catalyst | Temperature, | Conversion, | Selectivity, mol% | |||
|---|---|---|---|---|---|---|
| °C | 1,2-DCE, % of Feed | C2H4 | C2H6 | C2H3Cl | C2H5Cl | |
| 0.25% Pd on Al2O3 | 280 | 58.7 | 41.9 | 52.4 | 2.0 | 3.7 |
| 350 | 83.0 | 62.9 | 28.6 | 7.6 | 0.9 | |
| 0.5% Pd on Al2O3 | 280 | 68.7 | 28.1 | 65.8 | 1.1 | 4.9 |
| 350 | 90.3 | 55.1 | 33.5 | 9.0 | 2.3 | |
Table 9.
Activity and selectivity of various catalysts in 1,1,2-trichloroethane hydrodechlorination Residence time: 10 sec; molar ratio H2:1,1,2-trichloroethane = 3:1.
Table 9.
Activity and selectivity of various catalysts in 1,1,2-trichloroethane hydrodechlorination Residence time: 10 sec; molar ratio H2:1,1,2-trichloroethane = 3:1.
| Catalyst | Temperature, | Conversion, | Selectivity, mol% | |||
|---|---|---|---|---|---|---|
| °C | 1,1,2-TCE, % of Feed | C2H4 | C2H6 | C2H3Cl | i-C2H2Cl2 | |
| 0.25% Pd on Al2O3 | 280 | 78.7 | 7.8 | 70.2 | 19.8 | 2.2 |
| 350 | 98.9 | 23.9 | 62.1 | 11.4 | 2.6 | |
| 0.5 % Pd on Al2O3 | 280 | 89.2 | 6.2 | 75.2 | 15.8 | 2.8 |
| 350 | 99.2 | 25.6 | 67.7 | 5.6 | 1.1 | |
Table 10.
Results of 1,2-dichloroethane conversion on catalysts (Me/Al2O3) with a variable metal component.
Table 10.
Results of 1,2-dichloroethane conversion on catalysts (Me/Al2O3) with a variable metal component.
| Metal Component of Catalyst | Temperature, °C | DCE Conversion, % | Selectivity, mol% | Yield of Ethylene, % | |||
|---|---|---|---|---|---|---|---|
| Ethane | Ethylene | Propylene | Vinyl Chloride | ||||
| Ni | 250 | 3.4 | - | 63.0 | - | 100.0 | 0.0 |
| 350 | 39.0 | - | 68.7 | - | 31.2 | 26.8 | |
| Cr | 250 | 3.3 | - | 39.7 | - | 60.3 | 1.3 |
| 350 | 83.7 | - | 24.0 | 3.1 | 72.9 | 20.0 | |
| Cu | 250 | 3.9 | - | – | - | 100.0 | 0.0 |
| 350 | 21.3 | - | 35.9 | - | 64.1 | 7.6 | |
| Mn | 250 | 2.1 | - | 33.3 | - | 66.7 | 0.7 |
| 350 | 50.8 | - | 2.3 | - | 97.7 | 1.2 | |
| Cd | 250 | 2.0 | - | 39.4 | - | 60.6 | 0.8 |
| 350 | 84.3 | - | 11.3 | 1.7 | 87.0 | 9.5 | |
| Mo | 250 | 1.6 | - | 17.1 | - | 82.9 | 0.3 |
| 350 | 69.3 | - | 45.5 | 4.3 | 50.2 | 31.5 | |
| Bi | 250 | 5.4 | - | 36.5 | - | 63.5 | 2.0 |
| 350 | 82.8 | - | 5.6 | 1.3 | 93.1 | 4.6 | |
| V | 250 | 1.4 | - | 45.4 | - | 54.6 | 0.6 |
| 350 | 68.0 | - | 30.7 | 5.3 | 64.0 | 20.9 | |
Table 11.
Results of 1,2-dichloroethane hydrodechlorination on binary systems using nickel and chromium. The ratio H2:C2H4Cl2 = 2:1 (mol), residence time ~7 s. Temperature 350 °C, carrier Al2O3.
Table 11.
Results of 1,2-dichloroethane hydrodechlorination on binary systems using nickel and chromium. The ratio H2:C2H4Cl2 = 2:1 (mol), residence time ~7 s. Temperature 350 °C, carrier Al2O3.
| The Content in the Catalyst, wt% | DCE Conversion, % | Selectivity, mol% | Yield of C2H4, mol% | ||||
|---|---|---|---|---|---|---|---|
| Ni | Mo | Cr | Cu | C2H4 | VC | ||
| 4.0 | - | - | - | 39.0 | 68.7 | 31.2 | 26.8 |
| 2.0 | 2.0 | - | - | 54.7 | 63.2 | 36.0 | 34.6 |
| - | - | 4.0 | - | 83.7 | 24.0 | 72.9 | 20.0 |
| - | 2.0 | 2.0 | - | 53.9 | 60.2 | 37.8 | 32.3 |
| 2.0 | - | 2.0 | - | 58.1 | 78.1 | 20.3 | 45.3 |
| - | - | 2.0 | 2.0 | 99.0 | 99.5 | 0.5 | 98.1 |
Table 12.
Influence of different parameters on results of 1,2-dichloroethane hydrodechlorination using a catalyst containing 2% Ni and 2% Mo on microspheric alumina.
Table 12.
Influence of different parameters on results of 1,2-dichloroethane hydrodechlorination using a catalyst containing 2% Ni and 2% Mo on microspheric alumina.
| Temp., °C | Fed H2, L/h | H2: DCE, mol | Residence Time, s | DCE Conversion, % | Selectivity, mol% | Yield of Ethylene, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ethane | Ethylene | Propane | Propylene | Vinyl Chloride | ||||||
| 250 | 0.7 | 2:1 | 8.9 | 2.1 | - | 72.2 | - | - | 27.8 | 1.5 |
| 350 | 0.7 | 2:1 | 7.5 | 54.7 | - | 63.2 | - | 0.8 | 36.0 | 34.6 |
| 350 | 1.4 | 4:1 | 6.5 | 77.2 | - | 90.2 | - | 0.9 | 8.9 | 69.6 |
Table 13.
Influence of different parameters on results of 1,2-dichloroethane hydrodechlorination using a catalyst containing 2% Cr 2% Mo on microspheric alumina.
Table 13.
Influence of different parameters on results of 1,2-dichloroethane hydrodechlorination using a catalyst containing 2% Cr 2% Mo on microspheric alumina.
| Temp, °C | Fed H2, L/h | H2: DCE, mol | Residence Time, s | DCE Conversion, % | Selectivity, mol% | Yield of Ethylene, % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ethane | Ethylene | Propane | Propylene | Vinyl Chloride | ||||||
| 250 | 0.7 | 2:1 | 8.7 | 2.3 | - | 73.9 | - | 0.1 | 23.1 | 2.7 |
| 350 | 0.7 | 2:1 | 7.6 | 53.9 | - | 60.2 | - | 0.9 | 37.8 | 30.9 |
| 350 | 1.4 | 4:1 | 6.6 | 77.8 | - | 91.6 | - | 1.1 | 9.6 | 72.4 |
Table 14.
Arrhenius parameters for hydrodechlorination of 1,2-dichloroethane and 1,2-dichloroethene.
Table 14.
Arrhenius parameters for hydrodechlorination of 1,2-dichloroethane and 1,2-dichloroethene.
| A, L/Lcat sec | E/R | |
|---|---|---|
| 1,2-DCE-an | 9.4●104 | 8.51 |
| 1,2-DCE-en | 2.67●105 | 8.91 |
Table 15.
Arrhenius parameters for hydrodechlorination of 1,1,2-trichloroethane and trichloroethene.
Table 15.
Arrhenius parameters for hydrodechlorination of 1,1,2-trichloroethane and trichloroethene.
| A, L/Lcat sec | E/R | |
|---|---|---|
| 1,1,2-trichloroethane | 1.0●106 | 8.51 |
| Trichlorethylene | 4.1●106 | 10.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Flid, M.R.; Kartashov, L.M.; Treger, Y.A. Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies. Catalysts 2020, 10, 216. https://doi.org/10.3390/catal10020216
AMA Style
Flid MR, Kartashov LM, Treger YA. Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies. Catalysts. 2020; 10(2):216. https://doi.org/10.3390/catal10020216
Chicago/Turabian StyleFlid, M.R., L.M. Kartashov, and Yu.A. Treger. 2020. "Theoretical and Applied Aspects of Hydrodechlorination Processes—Catalysts and Technologies" Catalysts 10, no. 2: 216. https://doi.org/10.3390/catal10020216
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.