
HOXA1 3′UTR Methylation Is a Potential Prognostic Biomarker in Oral Squamous cell Carcinoma
 , , and
, , and
Abstract
:Simple Summary
Abstract
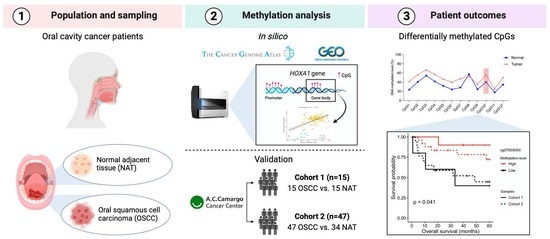
1. Introduction
2. Materials and Methods
2.1. In Silico Analysis
2.2. HOXA1 Analysis by Pyrosequencing
2.3. Statistical Analysis
3. Results
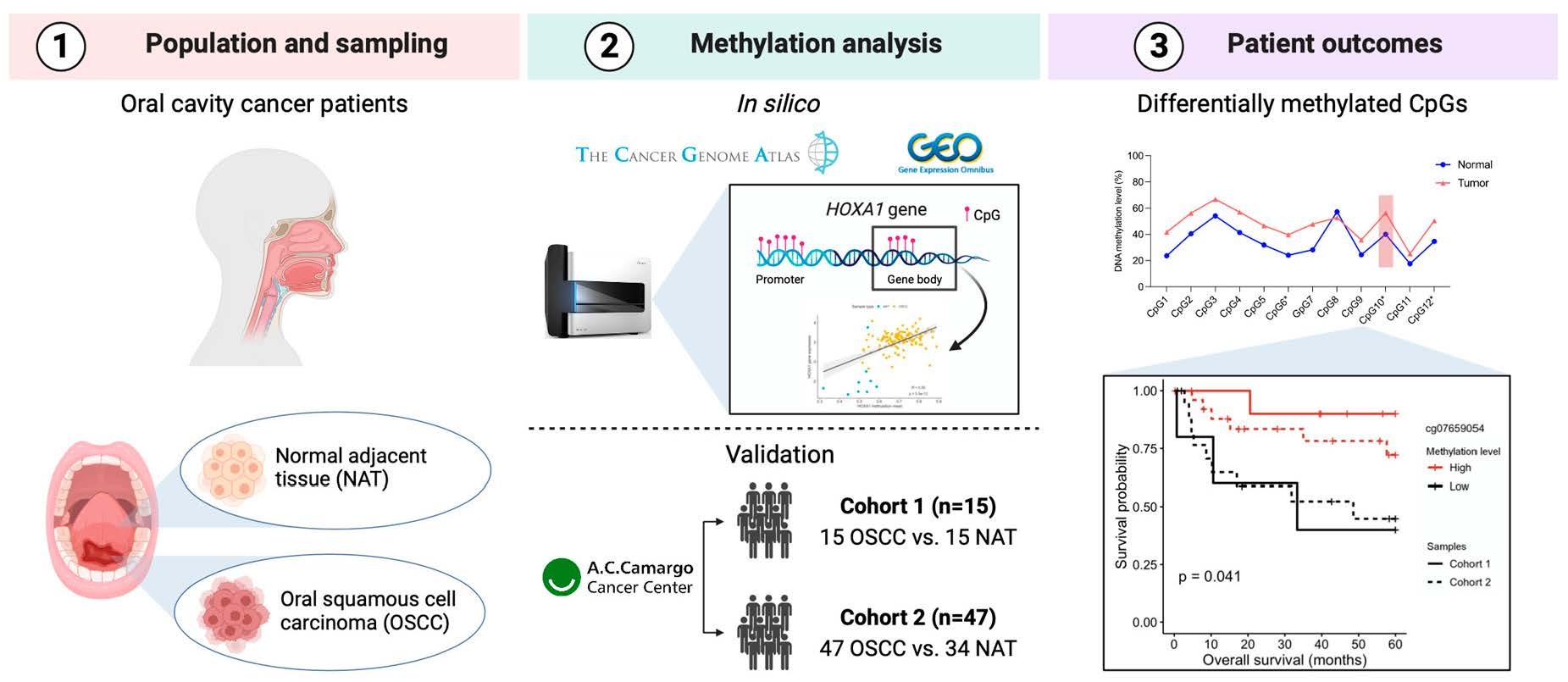
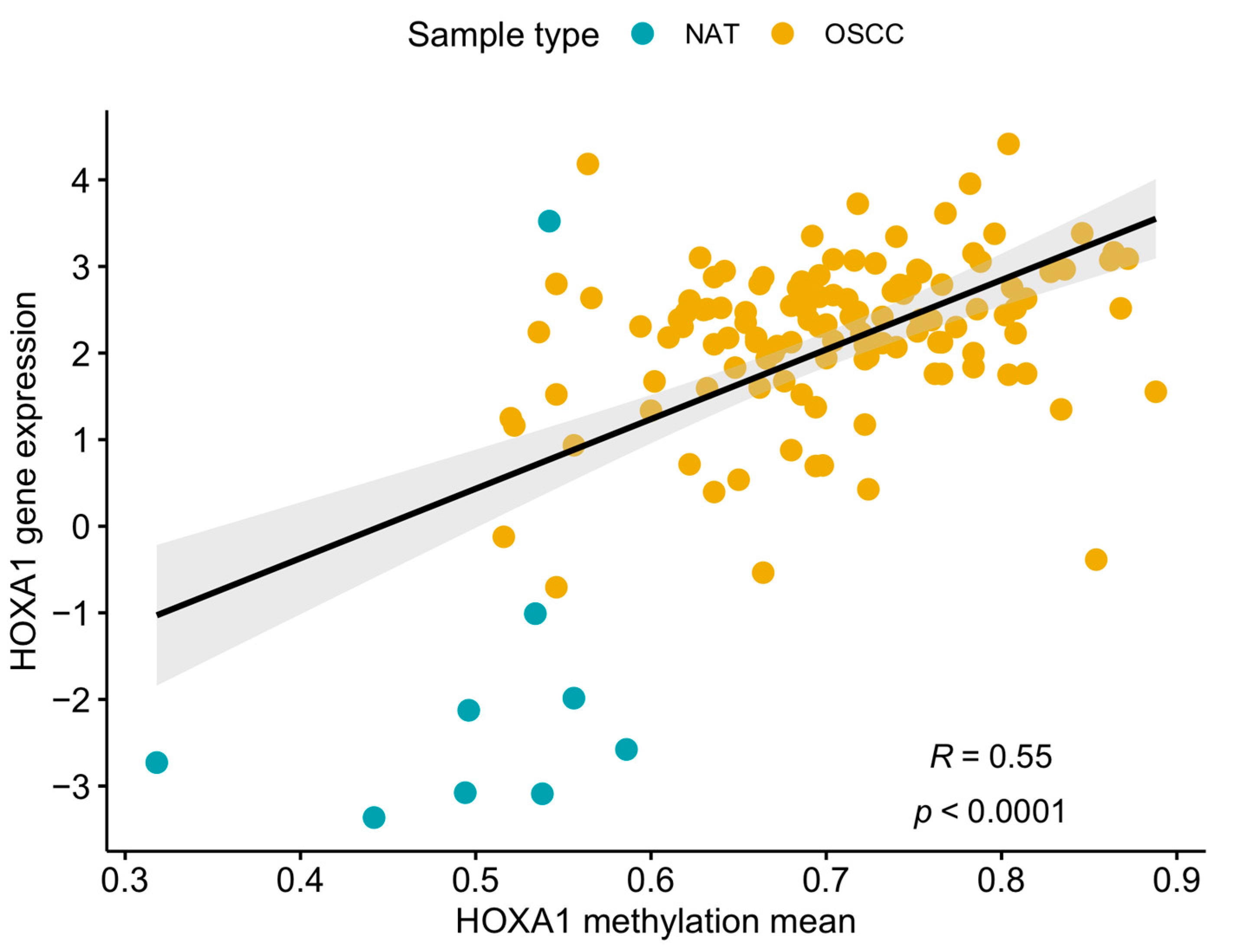
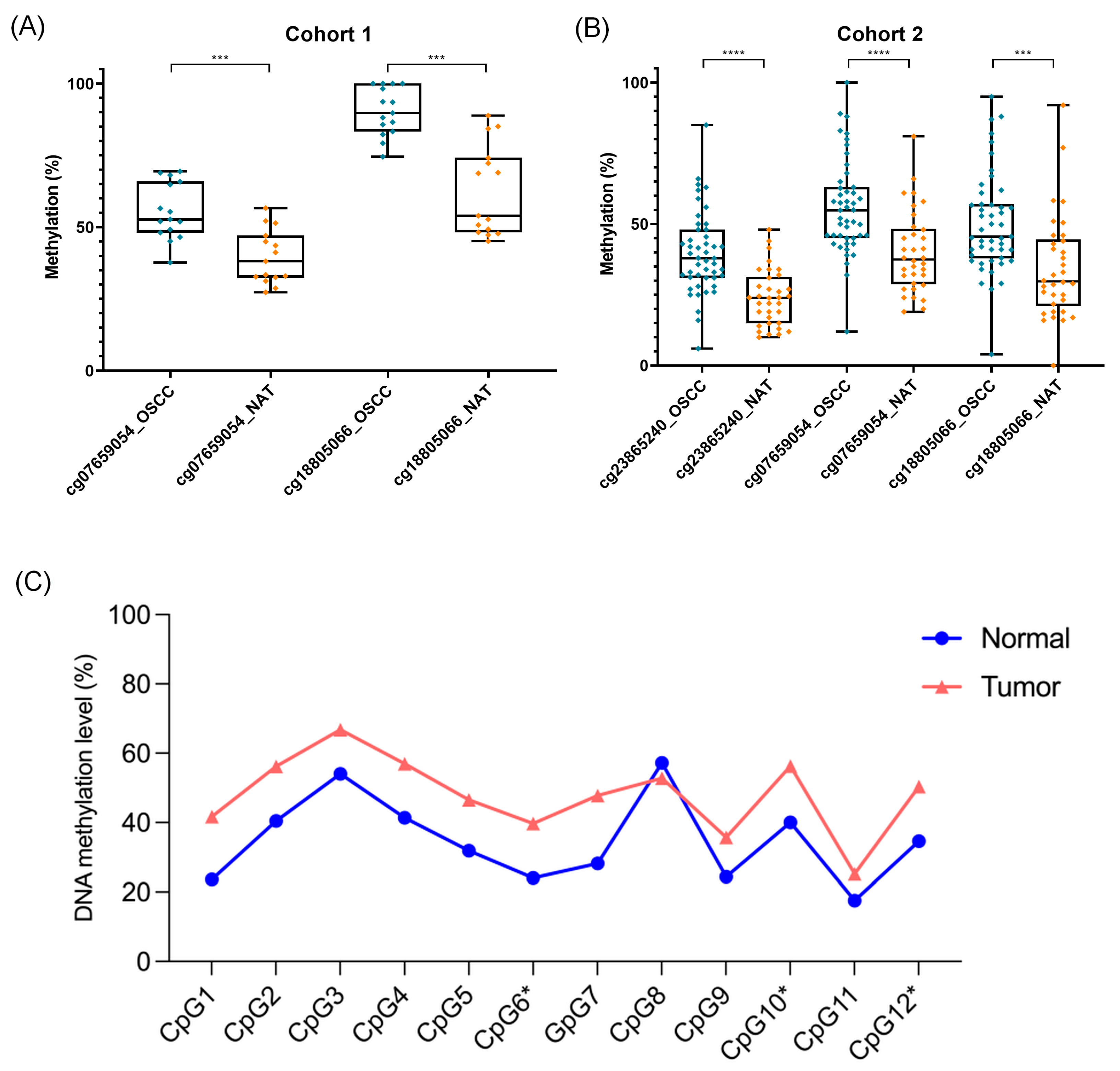
3.1. Differentially Methylated Sites between OSCC and NAT Tissues
3.2. Validation of the DMPs
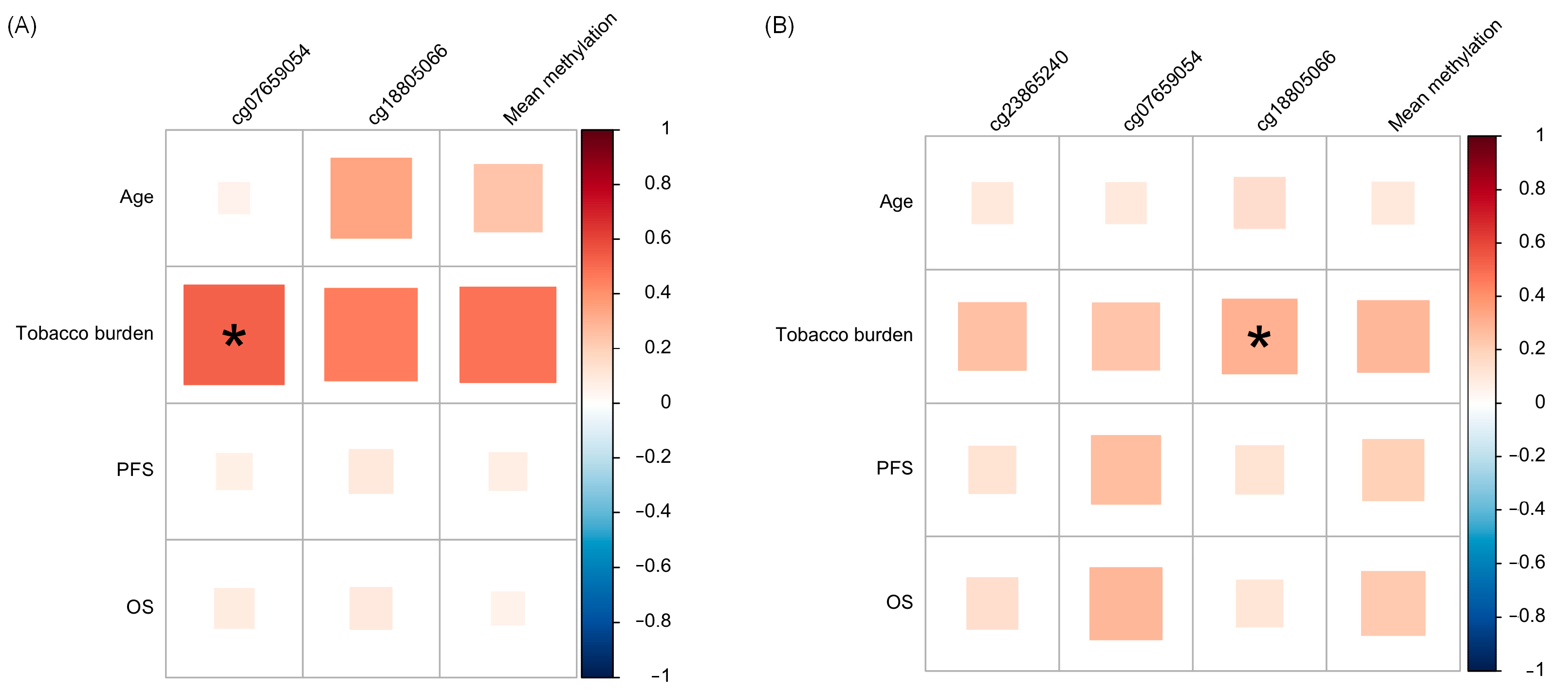
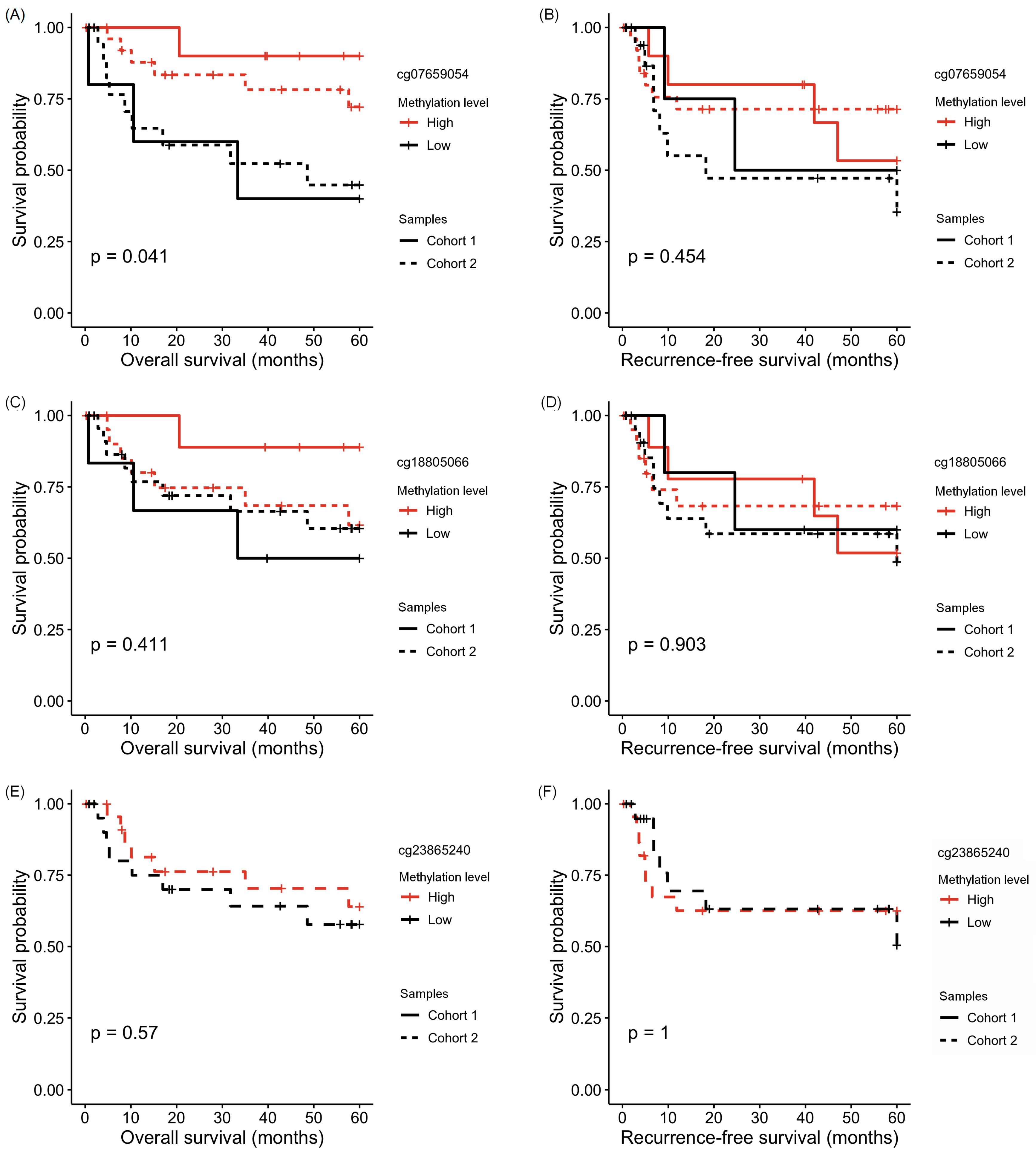
3.3. Association of HOXA1 Methylation with Clinicopathological Data
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Baj, A.; Fusco, N.; Bolzoni, A.; Carioli, D.; Mazzucato, C.; Faversani, A.; Bresciani, L.; Maggioni, M.; Capaccio, P. A novel integrated platform for the identification of surgical margins in oral squamous cell carcinoma: Results from a prospective single-institution series. BMC Cancer 2019, 19, 467. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, B.; Patil, S.; Sarode, S.C.; Sarode, G.S.; Rao, R.S. Clinico-pathological prognosticators in oral squamous cell carcinoma: An update. Transl. Res. Oral Oncol. 2017, 2, 2057178X17738912. [Google Scholar] [CrossRef]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Hall, G.L.; Woolgar, J.A.; Lowe, D.; Rogers, S.N.; Field, J.K.; Liloglou, T.; Risk, J.M. Quantitative methylation analysis of resection margins and lymph nodes in oral squamous cell carcinoma. Br. J. Oral. Maxillofac. Surg. 2007, 45, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Munoz-Alegre, M.; Henderson, S.; Tang, T.; Sun, P.; Johnson, N.; Fletcher, O.; Dos Santos Silva, I.; Peto, J.; Boshoff, C.; et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum. Mol. Genet. 2009, 18, 1332–1342. [Google Scholar] [CrossRef]
- Supic, G.; Kozomara, R.; Brankovic-Magic, M.; Jovic, N.; Magic, Z. Gene hypermethylation in tumor tissue of advanced oral squamous cell carcinoma patients. Oral. Oncol. 2009, 45, 1051–1057. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, L.; Mashrah, M.; Zhu, Y.; He, Z.; Hu, Y.; Xiang, T.; Yao, Z.; Guo, F.; Zhang, C. Expression and promoter methylation of Wnt inhibitory factor-1 in the development of oral submucous fibrosis. Oncol. Rep. 2015, 34, 2636–2642. [Google Scholar] [CrossRef]
- Papanicolau-Sengos, A.; Aldape, K. DNA Methylation Profiling: An Emerging Paradigm for Cancer Diagnosis. Annu. Rev. Pathol. 2022, 17, 295–321. [Google Scholar] [CrossRef]
- Stirzaker, C.; Taberlay, P.C.; Statham, A.L.; Clark, S.J. Mining cancer methylomes: Prospects and challenges. Trends Genet. 2014, 30, 75–84. [Google Scholar] [CrossRef]
- Basu, B.; Chakraborty, J.; Chandra, A.; Katarkar, A.; Baldevbhai, J.R.K.; Dhar Chowdhury, D.; Ray, J.G.; Chaudhuri, K.; Chatterjee, R. Genome-wide DNA methylation profile identified a unique set of differentially methylated immune genes in oral squamous cell carcinoma patients in India. Clin. Epigenet. 2017, 9, 13. [Google Scholar] [CrossRef]
- Teissandier, A.; Bourc’his, D. Gene body DNA methylation conspires with H3K36me3 to preclude aberrant transcription. EMBO J. 2017, 36, 1471–1473. [Google Scholar] [CrossRef]
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene body methylation in cancer: Molecular mechanisms and clinical applications. Clin. Epigenet. 2022, 14, 154. [Google Scholar] [CrossRef]
- Rideout, W.M., 3rd; Coetzee, G.A.; Olumi, A.F.; Jones, P.A. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science 1990, 249, 1288–1290. [Google Scholar] [CrossRef]
- Jeziorska, D.M.; Murray, R.J.S.; De Gobbi, M.; Gaentzsch, R.; Garrick, D.; Ayyub, H.; Chen, T.; Li, E.; Telenius, J.; Lynch, M.; et al. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E7526–E7535. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes. Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes. Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Ibrahim, J.; Peeters, M.; Van Camp, G.; Op de Beeck, K. Methylation biomarkers for early cancer detection and diagnosis: Current and future perspectives. Eur. J. Cancer 2023, 178, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Mamani, M.; Terrero-Perez, A.; Tucunduva, R.M.A.; Rubira, C.M.F.; Santos, P.; Honorio, H.M.; Rubira-Bullen, I.R.F. Occurrence of field cancerization in clinically normal oral mucosa: A systematic review and meta-analysis. Arch. Oral. Biol. 2022, 143, 105544. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Guerrero-Preston, R.; Okamura, J.; Michailidi, C.; Kahn, Z.; Li, X.; Ahn, J.; Goldsmith, M.; Koch, W. Innovative rapid gene methylation analysis of surgical margin tissues in head and neck cancer. Ann. Surg. Oncol. 2014, 21, 3124–3131. [Google Scholar] [CrossRef] [PubMed]
- Laytragoon-Lewin, N.; Rutqvist, L.E.; Lewin, F. DNA methylation in tumour and normal mucosal tissue of head and neck squamous cell carcinoma (HNSCC) patients: New diagnostic approaches and treatment. Med. Oncol. 2013, 30, 654. [Google Scholar] [CrossRef] [PubMed]
- Thomas Robbins, K.; Triantafyllou, A.; Suarez, C.; Lopez, F.; Hunt, J.L.; Strojan, P.; Williams, M.D.; Braakhuis, B.J.M.; de Bree, R.; Hinni, M.L.; et al. Surgical margins in head and neck cancer: Intra- and postoperative considerations. Auris Nasus Larynx 2019, 46, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Rao, K.N.; Arora, R.D.; Nagarkar, N.M.; Singh, A.; Shetty, O.S. Molecular Insights into Oral Malignancy. Indian. J. Surg. Oncol. 2022, 13, 267–280. [Google Scholar] [CrossRef]
- Lin, H.Y.; Hung, S.K.; Lee, M.S.; Chiou, W.Y.; Huang, T.T.; Tseng, C.E.; Shih, L.Y.; Lin, R.I.; Lin, J.M.; Lai, Y.H.; et al. DNA methylome analysis identifies epigenetic silencing of FHIT as a determining factor for radiosensitivity in oral cancer: An outcome-predicting and treatment-implicating study. Oncotarget 2015, 6, 915–934. [Google Scholar] [CrossRef]
- Strzelczyk, J.K.; Krakowczyk, L.; Owczarek, A.J. Methylation status of SFRP1, SFRP2, RASSF1A, RARbeta and DAPK1 genes in patients with oral squamous cell carcinoma. Arch. Oral. Biol. 2019, 98, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Bahadur, S.; Thakar, A.; Matta, A.; Macha, M.; Ralhan, R.; Gupta, S.D. Significance of promoter hypermethylation of p16 gene for margin assessment in carcinoma tongue. Head. Neck 2009, 31, 1423–1430. [Google Scholar] [CrossRef]
- Sorroche, B.P.; Talukdar, F.R.; Lima, S.C.S.; Melendez, M.E.; de Carvalho, A.C.; de Almeida, G.C.; De Marchi, P.; Lopes, M.; Ribeiro Pinto, L.F.; Carvalho, A.L.; et al. DNA Methylation Markers from Negative Surgical Margins Can Predict Recurrence of Oral Squamous Cell Carcinoma. Cancers 2021, 13, 2915. [Google Scholar] [CrossRef]
- Eljabo, N.; Nikolic, N.; Carkic, J.; Jelovac, D.; Lazarevic, M.; Tanic, N.; Milasin, J. Genetic and epigenetic alterations in the tumour, tumour margins, and normal buccal mucosa of patients with oral cancer. Int. J. Oral. Maxillofac. Surg. 2018, 47, 976–982. [Google Scholar] [CrossRef]
- Supic, G.; Kozomara, R.; Jovic, N.; Zeljic, K.; Magic, Z. Prognostic significance of tumor-related genes hypermethylation detected in cancer-free surgical margins of oral squamous cell carcinomas. Oral. Oncol. 2011, 47, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Strzelczyk, J.K.; Krakowczyk, L.; Golabek, K.; Owczarek, A.J. Expression profiles of selected genes in tumors and matched surgical margins in oral cavity cancer: Do we have to pay attention to the molecular analysis of the surgical margins? Adv. Clin. Exp. Med. 2018, 27, 833–840. [Google Scholar] [CrossRef]
- Gao, Y.; Teschendorff, A.E. Epigenetic and genetic deregulation in cancer target distinct signaling pathway domains. Nucleic Acids Res. 2017, 45, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Towle, R.; Truong, D.; Hogg, K.; Robinson, W.P.; Poh, C.F.; Garnis, C. Global analysis of DNA methylation changes during progression of oral cancer. Oral. Oncol. 2013, 49, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Lim, I.; Tan, J.; Alam, A.; Idrees, M.; Brenan, P.A.; Coletta, R.D.; Kujan, O. Epigenetics in the diagnosis and prognosis of head and neck cancer: A systematic review. J. Oral. Pathol. Med. 2024, 1–17. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Bosley, T.M.; Salih, M.A.; Alorainy, I.A.; Sener, E.C.; Nester, M.J.; Oystreck, D.T.; Chan, W.M.; Andrews, C.; Erickson, R.P.; et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat. Genet. 2005, 37, 1035–1037. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Zhang, M.; Wang, M.; Zhang, J.; Ma, S. Identification of HOXA1 as a Novel Biomarker in Prognosis of Head and Neck Squamous Cell Carcinoma. Front. Mol. Biosci. 2020, 7, 602068. [Google Scholar] [CrossRef]
- Bitu, C.C.; Destro, M.F.; Carrera, M.; da Silva, S.D.; Graner, E.; Kowalski, L.P.; Soares, F.A.; Coletta, R.D. HOXA1 is overexpressed in oral squamous cell carcinomas and its expression is correlated with poor prognosis. BMC Cancer 2012, 12, 146. [Google Scholar] [CrossRef] [PubMed]
- Khongsti, S.; Lamare, F.A.; Shunyu, N.B.; Ghosh, S.; Maitra, A.; Ghosh, S. Whole genome DNA methylation profiling of oral cancer in ethnic population of Meghalaya, North East India reveals novel genes. Genomics 2018, 110, 112–123. [Google Scholar] [CrossRef]
- Krishnan, N.M.; Dhas, K.; Nair, J.; Palve, V.; Bagwan, J.; Siddappa, G.; Suresh, A.; Kekatpure, V.D.; Kuriakose, M.A.; Panda, B. A Minimal DNA Methylation Signature in Oral Tongue Squamous Cell Carcinoma Links Altered Methylation with Tumor Attributes. Mol. Cancer Res. 2016, 14, 805–819. [Google Scholar] [CrossRef]
- Pidsley, R.; Wong, C.C.Y.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McGuire, M.H.; Herbrich, S.M.; Dasari, S.K.; Wu, S.Y.; Wang, Y.; Rupaimoole, R.; Lopez-Berestein, G.; Baggerly, K.A.; Sood, A.K. Pan-cancer genomic analysis links 3’UTR DNA methylation with increased gene expression in T cells. EBioMedicine 2019, 43, 127–137. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Mohanta, S.; Sekhar Khora, S.; Suresh, A. Cancer Stem Cell based molecular predictors of tumor recurrence in Oral squamous cell carcinoma. Arch. Oral. Biol. 2019, 99, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Gonzalez-Arriagada, W.A.; Loyola-Brambilla, M.; de Almeida, O.P.; Coletta, R.D.; Venegas, B. Clinicopathological and immunohistochemical evaluation of oral and oropharyngeal squamous cell carcinoma in Chilean population. Int. J. Clin. Exp. Pathol. 2014, 7, 5968–5977. [Google Scholar] [PubMed]
- Xiao, F.; Bai, Y.; Chen, Z.; Li, Y.; Luo, L.; Huang, J.; Yang, J.; Liao, H.; Guo, L. Downregulation of HOXA1 gene affects small cell lung cancer cell survival and chemoresistance under the regulation of miR-100. Eur. J. Cancer 2014, 50, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ma, J.; Wang, T.; Tian, J.; Zhang, Y.; Mao, L.; Xu, H.; Wang, S. Long Non-Coding RNA HOXA Transcript Antisense RNA Myeloid-Specific 1-HOXA1 Axis Downregulates the Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in Lung Cancer. Front. Immunol. 2018, 9, 473. [Google Scholar] [CrossRef]
- Prachayakul, V.; Kanchanapermpoon, J.; Thuwajit, C.; Boonyaarunnate, T.; Pongpaibul, A.; Chobson, P.; Thuwajit, P. DNA Methylation Markers Improve the Sensitivity of Endoscopic Retrograde Cholangiopancreatography-Based Brushing Cytology in Extrahepatic Cholangiocarcinoma. Technol. Cancer Res. Treat. 2017, 16, 1252–1258. [Google Scholar] [CrossRef]
- Chang, H.; Shin, B.K.; Kim, A.; Kim, H.K.; Kim, B.H. DNA methylation analysis for the diagnosis of thyroid nodules—A pilot study with reference to BRAF(V) (600E) mutation and cytopathology results. Cytopathology 2016, 27, 122–130. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Lee, S.H.; Jones, A.; Fiegl, H.; Kalwa, M.; Wagner, W.; Chindera, K.; Evans, I.; Dubeau, L.; Orjalo, A.; et al. HOTAIR and its surrogate DNA methylation signature indicate carboplatin resistance in ovarian cancer. Genome Med. 2015, 7, 108. [Google Scholar] [CrossRef]
- Callahan, C.L.; Wang, Y.; Marian, C.; Weng, D.Y.; Eng, K.H.; Tao, M.H.; Ambrosone, C.B.; Nie, J.; Trevisan, M.; Smiraglia, D.; et al. DNA methylation and breast tumor clinicopathological features: The Western New York Exposures and Breast Cancer (WEB) study. Epigenetics 2016, 11, 643–652. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Jagannathan, N. Oral field cancerization: An update on current concepts. Oncol. Rev. 2014, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | No. of Samples | ||
|---|---|---|---|
| OSCC | NAT | Total | |
| TCGA | 127 | 9 | 136 |
| GSE75537 | 54 | 29 | 83 |
| GSE97784 | 12 | 12 | 24 |
| GSE87053 | 11 | 10 | 21 |
| Forward (5′-3′) | Reverse (5′-3′) | Sequencing (5′-3′) | |
|---|---|---|---|
| Cohort 1 | TAGTGGGAGGTAGTTAGAGTG | [Btn]CCAACTTCACTACCAAACAAC | TTTTGTTTTATT |
| Cohort 2 | AGGTAGTTAGAGTGTTTGAGGTAGAA | [Btn]AAAAAATTCCACTTCAACAAATACCT | AGTGTTTGAGGTAGAAG |
| CpG ID | Location | TCGA | GSE75537 | GSE97784 | GSE87053 | ||||
|---|---|---|---|---|---|---|---|---|---|
| ∆β | adj. p-val | ∆β | adj. p-val | ∆β | adj. p-val | ∆β | adj. p-val | ||
| cg18805066 | 3′UTR; Body | 0.32 | 1.04 × 10−38 | 0.14 | 6.73 × 10−7 * | 0.20 | 2.28 × 10−3 * | 0.13 | 1.15 × 10−2 |
| cg07659054 | 3′UTR; Body | 0.24 | 3.46 × 10−31 | 0.11 | 5.36 × 10−6 * | 0.17 | 3.91 × 10−3 * | 0.13 | 2.44 × 10−3 * |
| cg23865240 | 3′UTR; Body | 0.24 | 3.11 × 10−33 | 0.12 | 1.13 × 10−7 * | 0.19 | 2.00 × 10−3 * | 0.14 | 2.89 × 10−3 * |
| cg22154659 | 3′UTR; Body | 0.23 | 5.13 × 10−36 | 0.08 | 1.08 × 10−4 | 0.16 | 4.96 × 10−3 * | 0.08 | 6.35 × 10−2 |
| cg08941355 | 3′UTR | 0.11 | 1.06 × 10−11 | 0.06 | 1.88 × 10−3 | 0.15 | 4.55 × 10−3 * | 0.11 | 4.78 × 10−3 * |
| cg10636403 | TSS1500 | 0.02 | 2.27 × 10−1 | 0.01 | 5.92 × 10−1 | 0.03 | 3.14 × 10−2 | 0.01 | 3.11 × 10−1 |
| cg06715976 | TSS200 | 0.02 | 2.99 × 10−1 | 0.02 | 2.49 × 10−1 | −0.01 | 7.52 × 10−1 | −0.01 | 3.34 × 10−1 |
| cg02978421 | TSS200 | 0.01 | 2.92 × 10−1 | 0.02 | 2.29 × 10−1 | 0.00 | 7.07 × 10−1 | 0.00 | 9.27 × 10−1 |
| cg04673590 | TSS1500 | 0.01 | 4.14 × 10−1 | 0.01 | 4.41 × 10−1 | 0.00 | 8.16 × 10−1 | 0.00 | 9.05 × 10−1 |
| cg15218775 | TSS200 | 0.01 | 5.81 × 10−1 | 0.02 | 2.01 × 10−1 | −0.01 | 5.21 × 10−1 | 0.00 | 8.38 × 10−1 |
| cg12686016 | 1stExon | 0.00 | 9.92 × 10−1 | −0.01 | 5.21 × 10−1 | −0.02 | 8.56 × 10−2 | −0.02 | 2.52 × 10−1 |
| cg04167903 | TSS200 | 0.00 | 9.40 × 10−1 | 0.00 | 8.30 × 10−1 | −0.01 | 4.05 × 10−1 | 0.00 | 7.72 × 10−1 |
| cg07450037 | Body; 1stExon | 0.00 | 9.43 × 10−1 | −0.08 | 1.33 × 10−3 | −0.06 | 1.61 × 10−1 | −0.07 | 2.67 × 10−1 |
| cg09420439 | TSS1500 | 0.00 | 8.22 × 10−1 | 0.00 | 9.22 × 10−1 | −0.01 | 1.79 × 10−1 | −0.02 | 9.49 × 10−2 |
| cg00288327 | 5′UTR; 1stExon | 0.00 | 7.38 × 10−1 | −0.01 | 7.00 × 10−1 | −0.01 | 3.66 × 10−2 | −0.01 | 3.46 × 10−1 |
| cg03700462 | TSS200 | −0.01 | 5.97 × 10−1 | 0.01 | 5.32 × 10−1 | 0.00 | 7.19 × 10−1 | −0.01 | 1.18 × 10−1 |
| cg26744375 | TSS200 | −0.01 | 4.67 × 10−1 | 0.01 | 6.87 × 10−1 | −0.01 | 2.31 × 10−1 | −0.01 | 1.67 × 10−1 |
| cg03116258 | 1stExon | −0.07 | 2.21 × 10−4 | −0.10 | 1.19 × 10−8 | −0.07 | 7.91 × 10−3 | −0.09 | 1.33 × 10−3 |
| CpG ID | Sample | Cohort 1 | Cohort 2 | ||||
|---|---|---|---|---|---|---|---|
| Mean ± SD | Δβ (%) | p * | Mean ± SD | Δβ (%) | p * | ||
| cg23865240 | OSCC | NA | NA | NA | 39.6 ± 14.5 | 15.6 | <0.001 |
| NAT | NA | 24.0 ± 9.9 | |||||
| cg07659054 | OSCC | 55.5 ± 9.9 | 15.9 | <0.001 | 56.1 ± 16.7 | 16.0 | <0.001 |
| NAT | 39.6 ± 9.3 | 40.1 ± 14.4 | |||||
| cg18805066 | OSCC | 90.3 ± 8.4 | 27.8 | <0.001 | 50.3 ± 17.8 | 15.8 | <0.001 |
| NAT | 62.5 ± 15.7 | 34.5 ± 18.4 | |||||
| Characteristic | Category | Cohort 1 n = 15 (%) | Cohort 2 n = 47 (%) |
|---|---|---|---|
| Sex | Male | 11 (73.3) | 35 (76.1) |
| Female | 4 (26.7) | 11 (23.9) | |
| Tobacco consumption | No | 7 (46.7) | 13 (28.3) |
| Yes | 6 (40.0) | 24 (52.2) | |
| Former (>2 years) | 2 (13.3) | 9 (19.6) | |
| Alcohol consumption | No | 6 (40.0) | 8 (17.4) |
| Yes/social | 7 (46.7) | 33 (71.8) | |
| Former (>2 years) | 2 (13.3) | 5 (10.9) | |
| Anatomic site | Tongue | 12 (80.0) | 24 (52.2) |
| Floor of the mouth | 3 (20.0) | 6 (13.0) | |
| Gingiva | 0 (0.0) | 9 (19.6) | |
| Other | 0 (0.0) | 7 (15.2) | |
| Clinical staging | I/II | 6 (40.0) | 13 (28.9) |
| III/IV | 9 (60.0) | 32 (71.1) | |
| Recurrence | No | 9 (60.0) | 29 (65.9) |
| Yes | 6 (40.0) | 15 (34.1) | |
| Patient status | Alive | 11 (73.3) | 29 (63.0) |
| Dead | 4 (26.7) | 17 (36.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorroche, B.P.; Miranda, K.C.; Beltrami, C.M.; Arantes, L.M.R.B.; Kowalski, L.P.; Marchi, F.A.; Rogatto, S.R.; Almeida, J.D. HOXA1 3′UTR Methylation Is a Potential Prognostic Biomarker in Oral Squamous cell Carcinoma. Cancers 2024, 16, 874. https://doi.org/10.3390/cancers16050874
Sorroche BP, Miranda KC, Beltrami CM, Arantes LMRB, Kowalski LP, Marchi FA, Rogatto SR, Almeida JD. HOXA1 3′UTR Methylation Is a Potential Prognostic Biomarker in Oral Squamous cell Carcinoma. Cancers. 2024; 16(5):874. https://doi.org/10.3390/cancers16050874
Chicago/Turabian StyleSorroche, Bruna Pereira, Keila Cristina Miranda, Caroline Moraes Beltrami, Lidia Maria Rebolho Batista Arantes, Luiz Paulo Kowalski, Fabio Albuquerque Marchi, Silvia Regina Rogatto, and Janete Dias Almeida. 2024. "HOXA1 3′UTR Methylation Is a Potential Prognostic Biomarker in Oral Squamous cell Carcinoma" Cancers 16, no. 5: 874. https://doi.org/10.3390/cancers16050874