
Alternative mRNA Splicing Controls the Functions of the Histone H3K27 Demethylase UTX/KDM6A
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples, Cell Lines and Cloning
2.2. Immunofluorescence and Confocal Microscopy
2.3. Immunoblotting Analyses
2.4. GFP Affinity Purification and MS Sample Preparation
2.5. Quantitative Mass Spectrometry Analysis
2.6. RT-PCR, Gel Electrophoresis and Nanopore Analysis of the UTX Isoforms
2.7. Public Dataset Analysis
2.8. GreenCUT&RUN
2.9. Bioinformatic Analyses of GreenCUT&RUN
3. Results
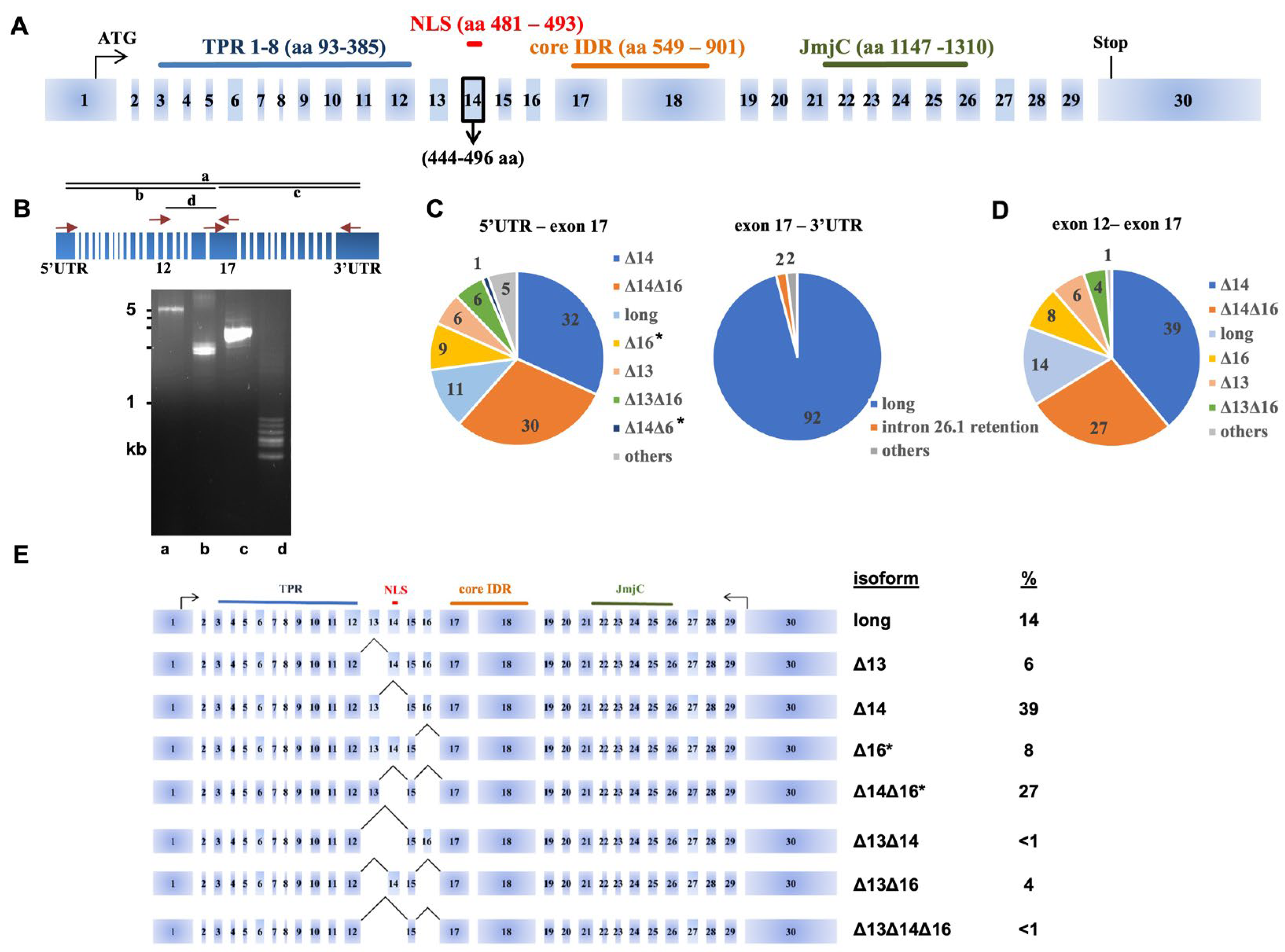
3.1. Characterization of the UTX Alternative Splicing Events
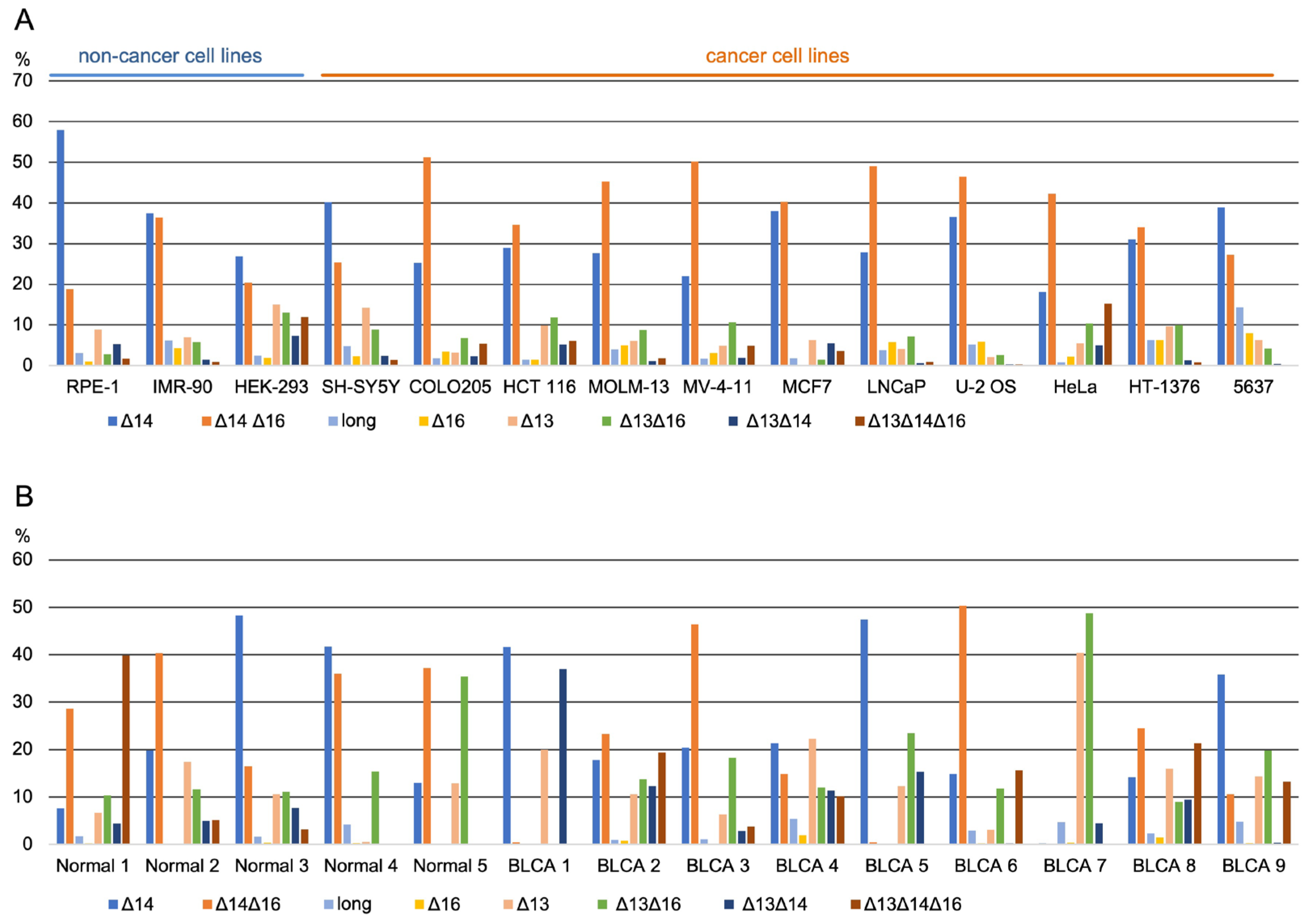
3.2. Long-Read Analysis of UTX mRNAs across A Panel of Cell Lines and Bladder Samples
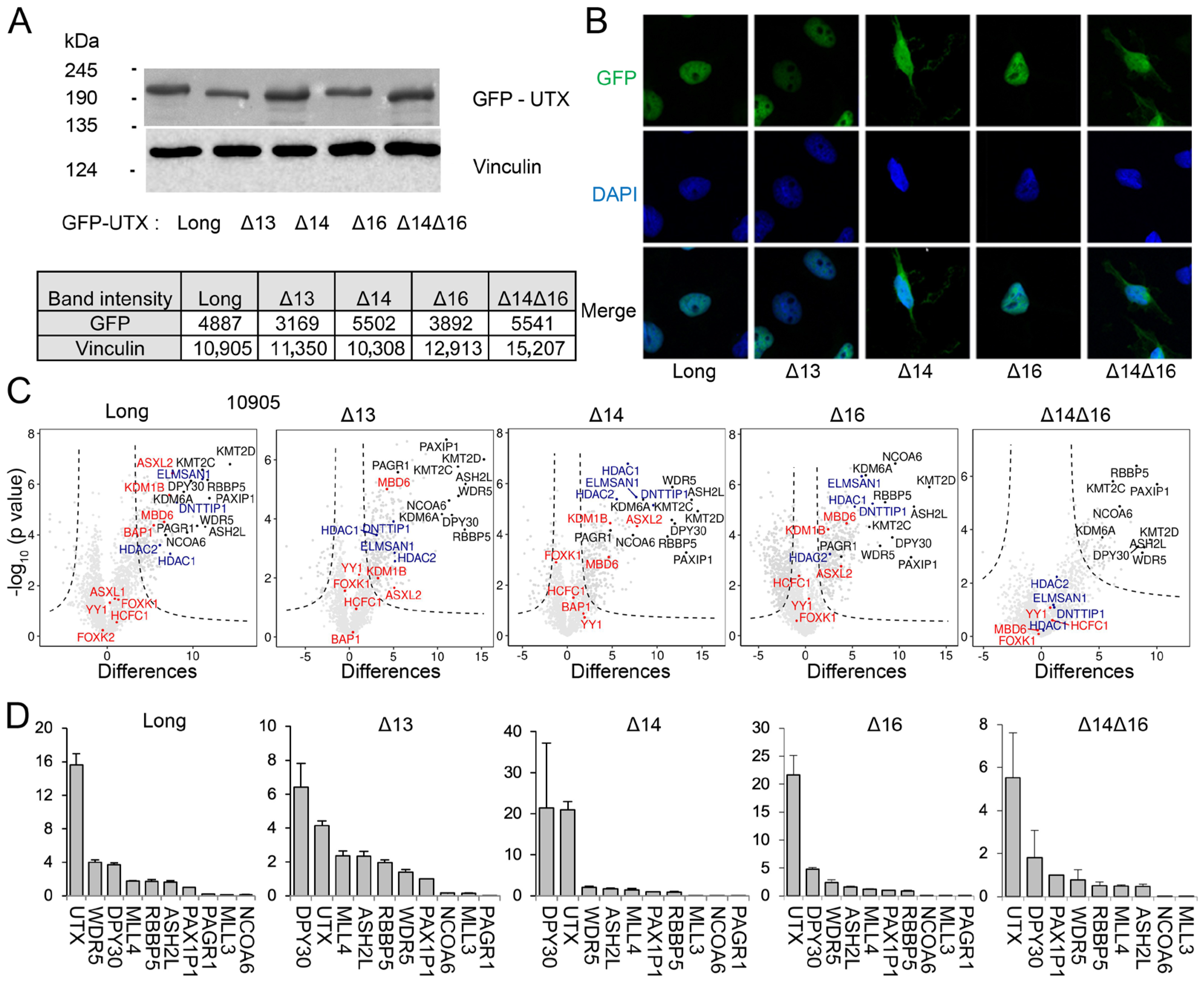
3.3. UTX Isoforms Display Different Protein–Protein Interactions
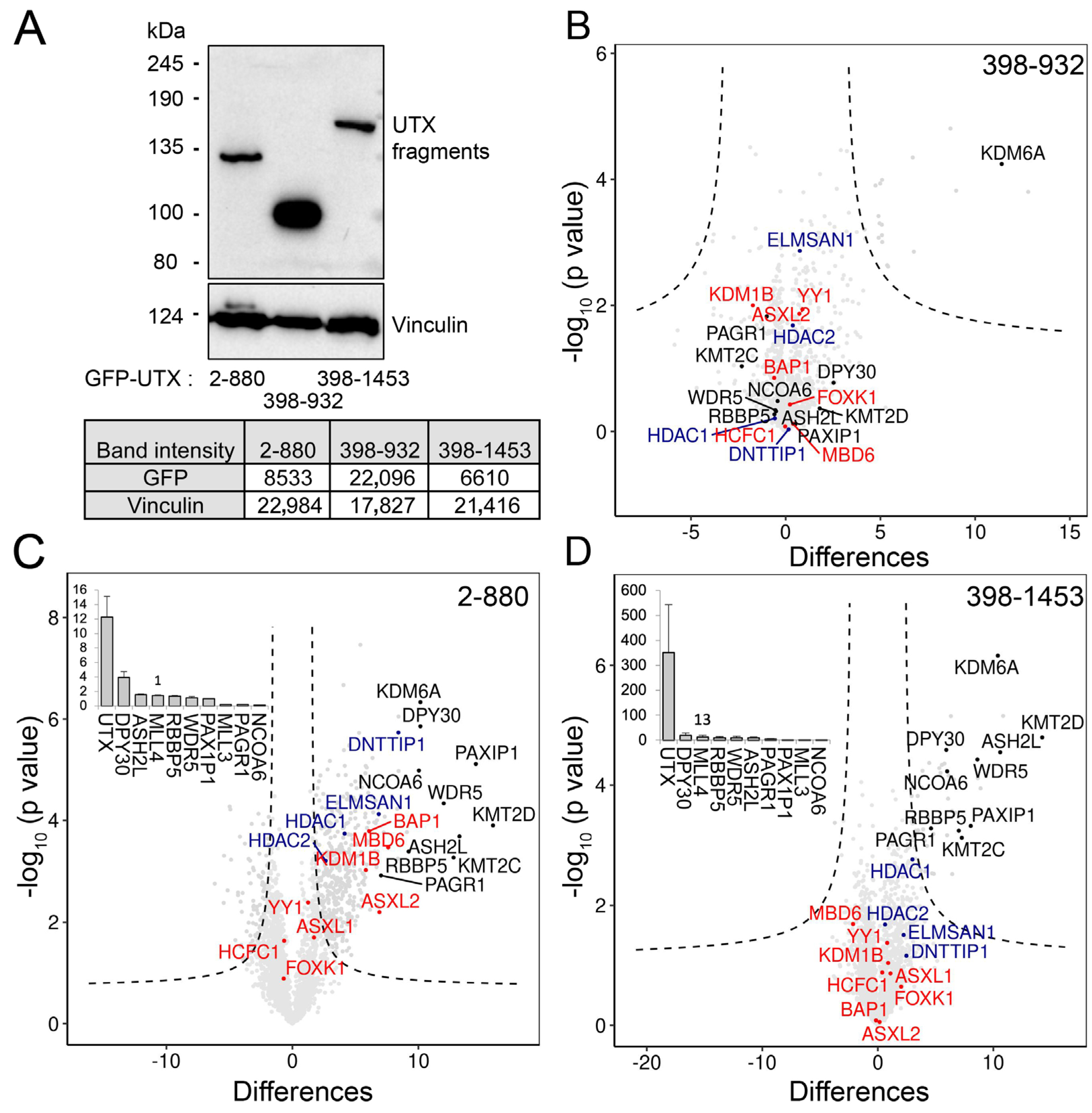
3.4. The Combination of Middle Part with TPR Is Required for Proper Protein–Protein Interaction of UTX
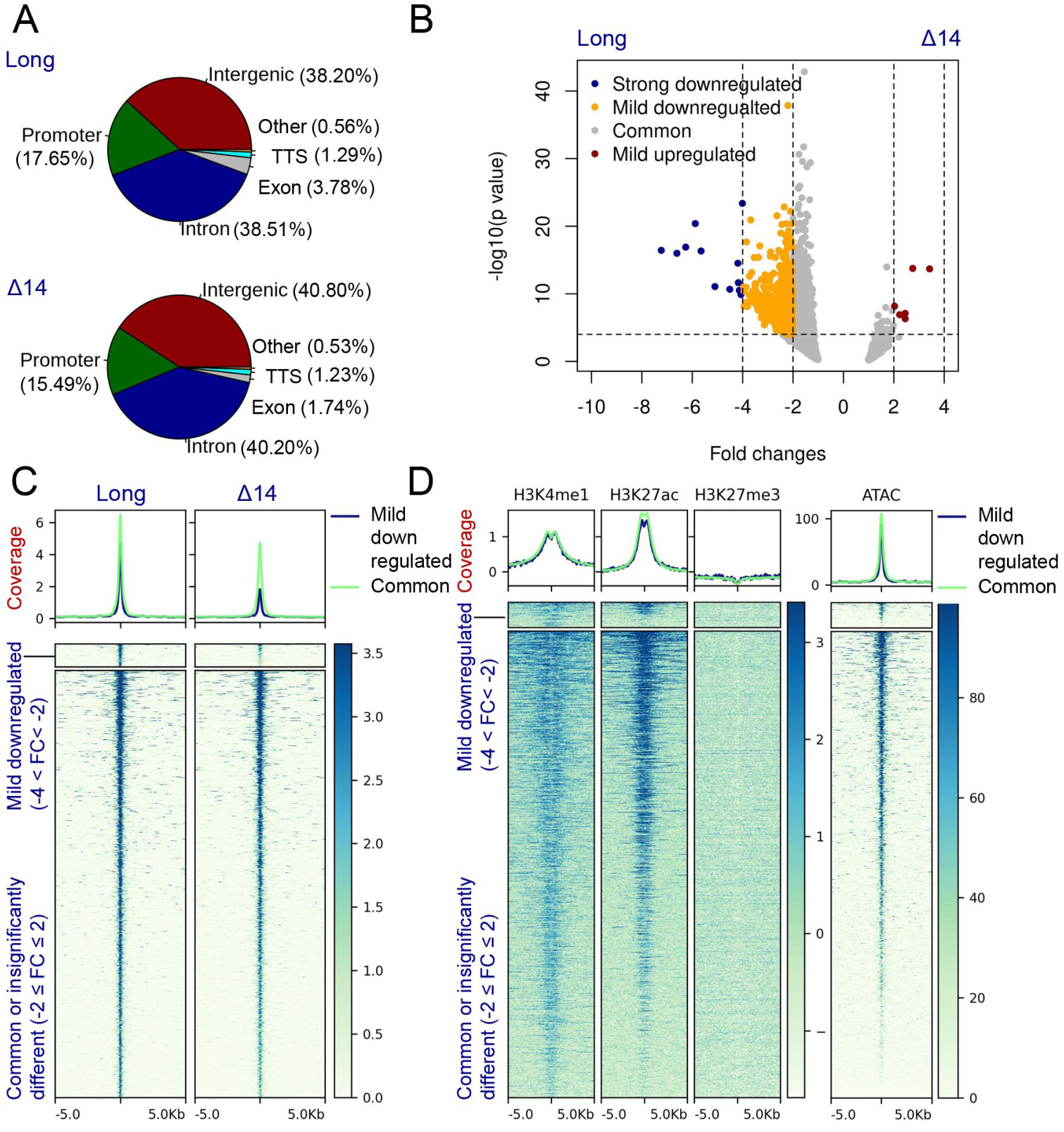
3.5. The UTX Long Isoform Displays A Stronger Chromatin Association When Compared to Canonical Isoform Δ14
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [Green Version]
- Glaser, A.P.; Fantini, D.; Shilatifard, A.; Schaeffer, E.M.; Meeks, J.J. The Evolving Genomic Landscape of Urothelial Carcinoma. Nat. Rev. Urol. 2017, 14, 215–229. [Google Scholar] [CrossRef]
- Lawson, A.R.J.; Abascal, F.; Coorens, T.H.H.; Hooks, Y.; O’Neill, L.; Latimer, C.; Raine, K.; Sanders, M.A.; Warren, A.Y.; Mahbubani, K.T.A.; et al. Extensive Heterogeneity in Somatic Mutation and Selection in the Human Bladder. Science 2020, 370, 75–82. [Google Scholar] [CrossRef]
- Li, R.; Du, Y.; Chen, Z.; Xu, D.; Lin, T.; Jin, S.; Wang, G.; Liu, Z.; Lu, M.; Chen, X.; et al. Macroscopic Somatic Clonal Expansion in Morphologically Normal Human Urothelium. Science 2020, 370, 82–89. [Google Scholar] [CrossRef]
- Kato, H.; Asamitsu, K.; Sun, W.; Kitajima, S.; Yoshizawa-Sugata, N.; Okamoto, T.; Masai, H.; Poellinger, L. Cancer-Derived UTX TPR Mutations G137V and D336G Impair Interaction with MLL3/4 Complexes and Affect UTX Subcellular Localization. Oncogene 2020, 39, 3322–3335. [Google Scholar] [CrossRef]
- Shi, B.; Li, W.; Song, Y.; Wang, Z.; Ju, R.; Ulman, A.; Hu, J.; Palomba, F.; Zhao, Y.; Le, J.P.; et al. UTX Condensation Underlies Its Tumour-Suppressive Activity. Nature 2021, 597, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 Are Histone H3K27 Demethylases Involved in HOX Gene Regulation and Development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Issaeva, I.; Zonis, Y.; Rozovskaia, T.; Orlovsky, K.; Croce, C.M.; Nakamura, T.; Mazo, A.; Eisenbach, L.; Canaani, E. Knockdown of ALR (MLL2) Reveals ALR Target Genes and Leads to Alterations in Cell Adhesion and Growth. Mol. Cell. Biol. 2007, 27, 1889–1903. [Google Scholar] [CrossRef] [Green Version]
- Van Nuland, R.; Smits, A.H.; Pallaki, P.; Jansen, P.W.; Vermeulen, M.; Timmers, H.T. Quantitative Dissection and Stoichiometry Determination of the Human SET1/MLL Histone Methyltransferase Complexes. Mol. Cell. Biol. 2013, 33, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, Z.; Ozark, P.A.; Fantini, D.; Marshall, S.A.; Rendleman, E.J.; Cozzolino, K.A.; Louis, N.; He, X.; Morgan, M.A.; et al. Resetting the Epigenetic Balance of Polycomb and COMPASS Function at Enhancers for Cancer Therapy. Nat. Med. 2018, 24, 758–769. [Google Scholar] [CrossRef]
- Wang, X.; Rosikiewicz, W.; Sedkov, Y.; Mondal, B.; Martinez, T.; Kallappagoudar, S.; Tvardovskiy, A.; Bajpai, R.; Xu, B.; Pruett-Miller, S.M.; et al. The MLL3/4 Complexes and MiDAC Co-Regulate H4K20ac to Control a Specific Gene Expression Program. Life Sci. Alliance 2022, 5, e202201572. [Google Scholar] [CrossRef] [PubMed]
- Mondal, B.; Jin, H.; Kallappagoudar, S.; Sedkov, Y.; Martinez, T.; Sentmanat, M.F.; Poet, G.J.; Li, C.; Fan, Y.; Pruett-Miller, S.M.; et al. The Histone Deacetylase Complex MiDAC Regulates a Neurodevelopmental Gene Expression Program to Control Neurite Outgrowth. eLife 2020, 9, e57519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, A.; Carrel, L.; Pennisi, D.; Philippe, C.; Quaderi, N.; Siggers, P.; Steiner, K.; Tam, P.P.; Monaco, A.P.; Willard, H.F.; et al. The UTX Gene Escapes X Inactivation in Mice and Humans. Hum. Mol. Genet. 1998, 7, 737–742. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, R.; Nicolas, E.; Hall, M.J.; Golemis, E.A. X- and Y-Linked Chromatin-Modifying Genes as Regulators of Sex-Specific Cancer Incidence and Prognosis. Clin. Cancer Res. 2020, 26, 5567–5578. [Google Scholar] [CrossRef]
- Walport, L.J.; Hopkinson, R.J.; Vollmar, M.; Madden, S.K.; Gileadi, C.; Oppermann, U.; Schofield, C.J.; Johansson, C. Human UTY(KDM6C) Is a Male-Specific Nϵ-Methyl Lysyl Demethylase. J. Biol. Chem. 2014, 289, 18302–18313. [Google Scholar] [CrossRef] [Green Version]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY Demonstrate Histone Demethylase-Independent Function in Mouse Embryonic Development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lee, J.E.; Cho, Y.W.; Xiao, Y.; Jin, Q.; Liu, C.; Ge, K. UTX Regulates Mesoderm Differentiation of Embryonic Stem Cells Independent of H3K27 Demethylase Activity. Proc. Natl. Acad. Sci. USA 2012, 109, 15324–15329. [Google Scholar] [CrossRef] [Green Version]
- Barrows, D.; Feng, L.; Carroll, T.S.; Allis, C.D. Loss of UTX/KDM6A and the Activation of FGFR3 Converge to Regulate Differentiation Gene-Expression Programs in Bladder Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 25732–25741. [Google Scholar] [CrossRef]
- Yang, L.; Song, L.; Liu, X.; Bai, L.; Li, G. KDM6A and KDM6B Play Contrasting Roles in Nuclear Transfer Embryos Revealed by MERVL Reporter System. EMBO Rep. 2018, 19, e46240. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Song, L.S.; Liu, X.F.; Xia, Q.; Bai, L.G.; Gao, L.; Gao, G.Q.; Wang, Y.; Wei, Z.Y.; Bai, C.L.; et al. The Maternal Effect Genes UTX and JMJD3 Play Contrasting Roles in Mus Musculus Preimplantation Embryo Development. Sci. Rep. 2016, 6, 26711. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting Roles of Histone 3 Lysine 27 Demethylases in Acute Lymphoblastic Leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.; Ochoa, A.; McConkey, D.J.; Aine, M.; Höglund, M.; Kim, W.Y.; Real, F.X.; Kiltie, A.E.; Milsom, I.; Dyrskjøt, L.; et al. Genetic Alterations in the Molecular Subtypes of Bladder Cancer: Illustration in the Cancer Genome Atlas Dataset. Eur. Urol. 2017, 72, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonova, S.V.; Haffke, M.; Corradini, E.; Mikuciunas, M.; Low, T.Y.; Signor, L.; van Es, R.M.; Gupta, K.; Scheer, E.; Vos, H.R.; et al. Chaperonin CCT Checkpoint Function in Basal Transcription Factor TFIID Assembly. Nat. Struct. Mol. Biol. 2018, 25, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Nizamuddin, S.; Koidl, S.; Bhuiyan, T.; Werner, T.V.; Biniossek, M.L.; Bonvin, A.M.J.J.; Lassmann, S.; Timmers, H. Integrating Quantitative Proteomics with Accurate Genome Profiling of Transcription Factors by GreenCUT&RUN. Nucleic Acids Res. 2021, 49, e49. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Systematic Identification of Cell Cycle-Dependent Yeast Nucleocytoplasmic Shuttling Proteins by Prediction of Composite Motifs. Proc. Natl. Acad. Sci. USA 2009, 106, 10171–10176. [Google Scholar] [CrossRef] [Green Version]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Cancer Genome Atlas Research, N.; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e4. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Koidl, S.; Timmers, H.T.M. GreenCUT&RUN: Efficient Genomic Profiling of GFP-Tagged Transcription Factors and Chromatin Regulators. Curr. Protoc. 2021, 1, e266. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 Regulates Polycomb Recruitment and H2A Ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC Domain-Containing UTX and JMJD3 as Histone H3 Lysine 27 Demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, M.L.; Witte, N.; Im, K.M.; Turan, S.; Owens, C.; Misner, K.; Tsang, S.X.; Cai, Z.; Wu, S.; Dean, M.; et al. Molecular Analysis of Urothelial Cancer Cell Lines for Modeling Tumor Biology and Drug Response. Oncogene 2017, 36, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative Splicing as a Regulator of Development and Tissue Identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Muller, J. Histone H2A Deubiquitinase Activity of the Polycomb Repressive Complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics Profiling of HDAC Inhibitors Reveals Selective Targeting of HDAC Complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Rickels, R.; Wang, L.; Iwanaszko, M.; Ozark, P.A.; Morgan, M.A.; Piunti, A.; Khalatyan, N.; Soliman, S.H.A.; Rendleman, E.J.; Savas, J.N.; et al. A Small UTX Stabilization Domain of Trr Is Conserved within Mammalian MLL3-4/COMPASS and Is Sufficient to Rescue Loss of Viability in Null Animals. Genes Dev. 2020, 34, 1493–1502. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.Y.; Lee, C.F.; Lai, H.T.; Yu, C.T.; Lee, J.E.; Zuo, H.; Tsai, S.Y.; Tsai, M.J.; Ge, K.; Wan, Y.; et al. Opposing Functions of BRD4 Isoforms in Breast Cancer. Mol. Cell 2020, 78, 1114–1132.e10. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, K.; Hu, G.; Babarinde, I.A.; Li, Y.; Dong, X.; Chen, Y.-S.; Shang, L.; Guo, W.; Wang, J.; et al. An Alternative CTCF Isoform Antagonizes Canonical CTCF Occupancy and Changes Chromatin Architecture to Promote Apoptosis. Nat. Commun. 2019, 10, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Light, S.; Elofsson, A. The Impact of Splicing on Protein Domain Architecture. Curr. Opin. Struct. Biol. 2013, 23, 451–458. [Google Scholar] [CrossRef]
- Wiedemuth, R.; Thieme, S.; Navratiel, K.; Dorschner, B.; Brenner, S. UTX-Moonlighting in the Cytoplasm? Int. J. Biochem. Cell Biol. 2018, 97, 78–82. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.; Wong, W.C.; Brown, R.; Akbani, R.; Su, X.; Broom, B.; Melott, J.; Weinstein, J. TCGASpliceSeq a Compendium of Alternative MRNA Splicing in Cancer. Nucleic Acids Res. 2016, 44, D1018–D1022. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform % | % of Exon Skipping | Exon Psi | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Δ14 | Δ14Δ16 | Long | Δ16 | Δ13 | Δ13 Δ16 | Δ13 Δ14 | Δ13Δ14Δ16 | No Ex 13 | No Ex 14 | No Ex 16 | Exon 13 | Exon 14 | Exon 16 | |
| Normal 1 | 8 | 29 | 2 | 0 | 7 | 10 | 4 | 40 | 61 | 81 | 79 | 38 | 19 | 20 |
| Normal 2 | 20 | 40 | 0 | 0 | 17 | 12 | 5 | 5 | 39 | 70 | 57 | 60 | 29 | 42 |
| Normal 3 | 48 | 16 | 2 | 0 | 11 | 11 | 8 | 3 | 33 | 76 | 31 | 67 | 24 | 68 |
| Normal 4 | 42 | 36 | 4 | 0 | 1 | 15 | 0 | 0 | 16 | 78 | 52 | 82 | 21 | 47 |
| Normal 5 | 13 | 37 | 0 | 0 | 13 | 35 | 0 | 0 | 49 | 50 | 73 | 51 | 49 | 26 |
| mean | 26 | 32 | 2 | 0 | 10 | 17 | 3 | 10 | 40 | 71 | 58 | 60 | 28 | 41 |
| BLCA 1 | 42 | 0 | 0 | 0 | 20 | 0 | 37 | 0 | 57 | 79 | 1 | 42 | 20 | 99 |
| BLCA 2 | 18 | 23 | 1 | 1 | 11 | 14 | 12 | 19 | 56 | 73 | 57 | 43 | 26 | 42 |
| BLCA 3 | 20 | 46 | 1 | 0 | 6 | 18 | 3 | 4 | 31 | 74 | 69 | 68 | 26 | 31 |
| BLCA 4 | 21 | 15 | 5 | 2 | 22 | 12 | 11 | 10 | 56 | 58 | 39 | 44 | 42 | 60 |
| BLCA 5 | 47 | 1 | 0 | 0 | 12 | 23 | 15 | 0 | 51 | 63 | 24 | 48 | 36 | 75 |
| BLCA 6 | 15 | 50 | 3 | 0 | 3 | 12 | 0 | 16 | 31 | 81 | 78 | 68 | 18 | 21 |
| BLCA 7 | 0 | 0 | 5 | 0 | 40 | 49 | 5 | 0 | 94 | 5 | 50 | 6 | 94 | 50 |
| BLCA 8 | 14 | 24 | 2 | 2 | 16 | 9 | 9 | 21 | 56 | 69 | 56 | 43 | 29 | 42 |
| BLCA 9 | 36 | 11 | 5 | 0 | 14 | 20 | 0 | 13 | 48 | 60 | 44 | 52 | 39 | 55 |
| mean | 24 | 19 | 3 | 1 | 16 | 17 | 10 | 9 | 53 | 63 | 46 | 46 | 37 | 53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fotouhi, O.; Nizamuddin, S.; Falk, S.; Schilling, O.; Knüchel-Clarke, R.; Biniossek, M.L.; Timmers, H.T.M. Alternative mRNA Splicing Controls the Functions of the Histone H3K27 Demethylase UTX/KDM6A. Cancers 2023, 15, 3117. https://doi.org/10.3390/cancers15123117
Fotouhi O, Nizamuddin S, Falk S, Schilling O, Knüchel-Clarke R, Biniossek ML, Timmers HTM. Alternative mRNA Splicing Controls the Functions of the Histone H3K27 Demethylase UTX/KDM6A. Cancers. 2023; 15(12):3117. https://doi.org/10.3390/cancers15123117
Chicago/Turabian StyleFotouhi, Omid, Sheikh Nizamuddin, Stephanie Falk, Oliver Schilling, Ruth Knüchel-Clarke, Martin L. Biniossek, and H. T. Marc Timmers. 2023. "Alternative mRNA Splicing Controls the Functions of the Histone H3K27 Demethylase UTX/KDM6A" Cancers 15, no. 12: 3117. https://doi.org/10.3390/cancers15123117