
Analysis of Cancer Genomic Amplifications Identifies Druggable Collateral Dependencies within the Amplicon
 , , , ,
, , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Copy Number Amplifications in Tumors and Cell Lines
2.2. Overall Survival and Disease-Specific Survival in Tumor Samples
2.3. Comparison of Similarity in Genomic Amplification between Tumors and Cell Lines
2.4. Screening of Gene Dependencies Associated with Gene Amplifications
2.5. Prioritization of Candidate Targets for Each Chromosome Amplification
2.6. Correlation between Gene Dependencies and Gene Expression
3. Results
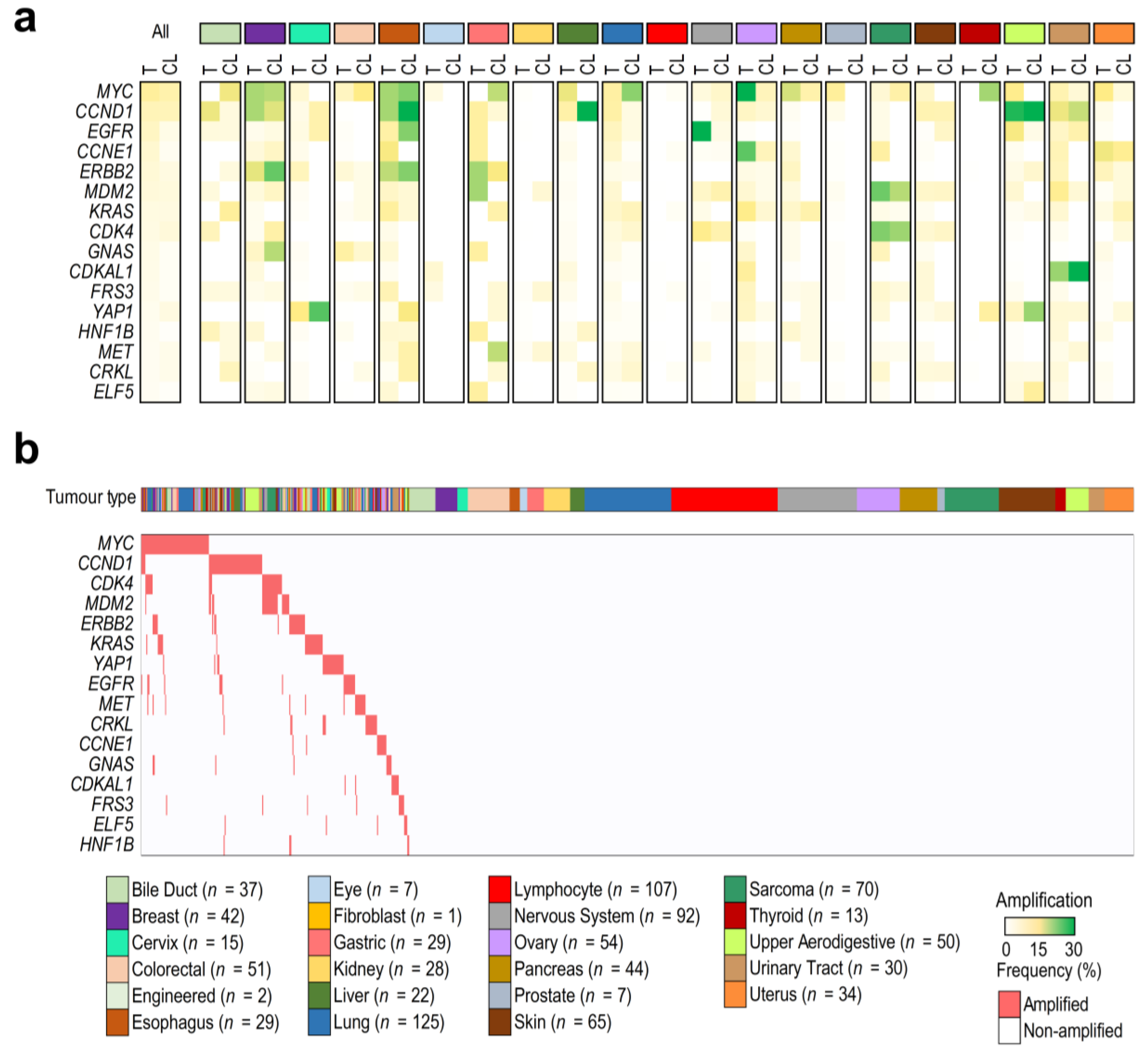
3.1. Cancer Cell Lines as a Model to Study Tumor Gene Amplifications
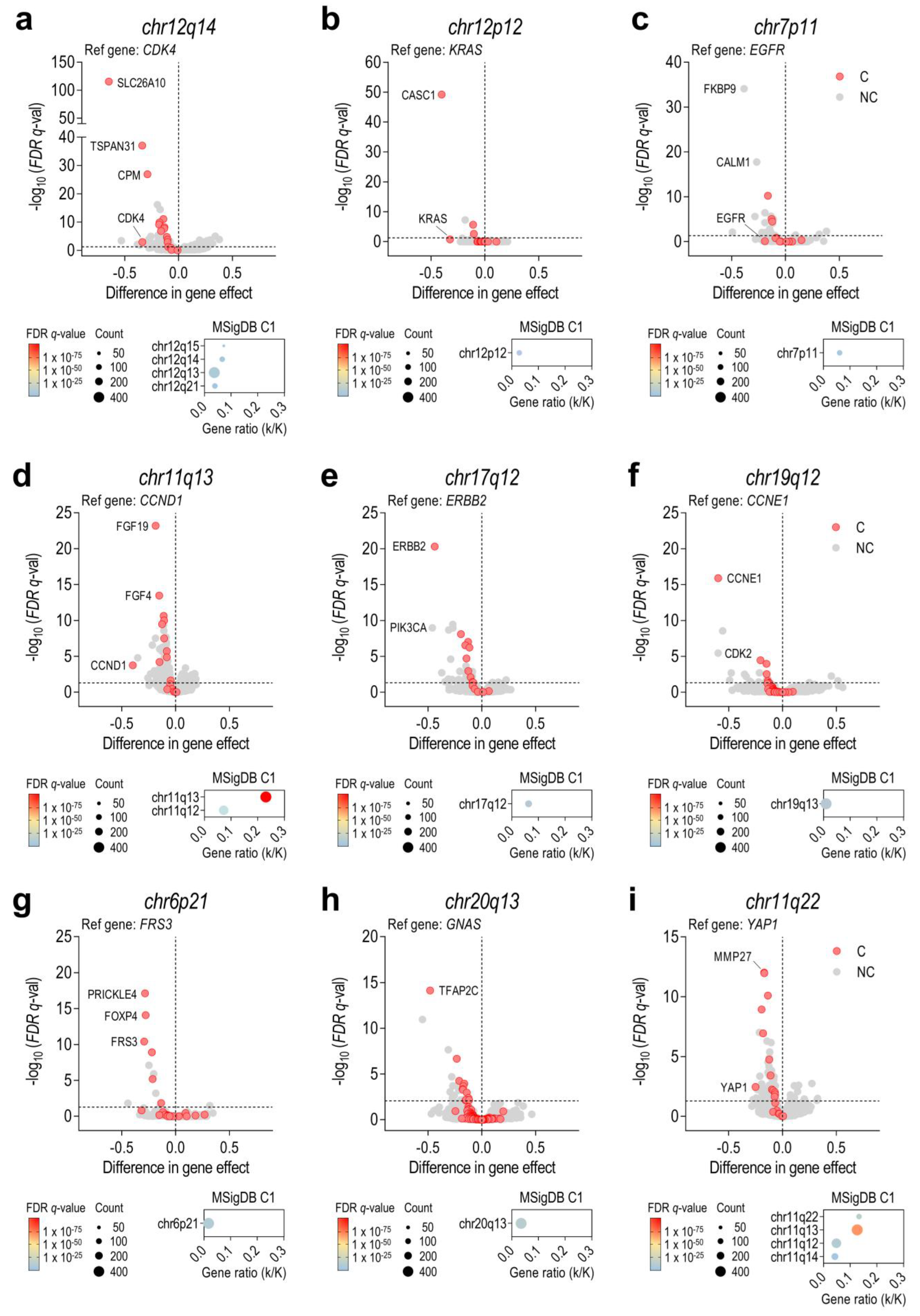
3.2. Chromosome Amplifications Generate Collateral Dependencies within the Amplicon
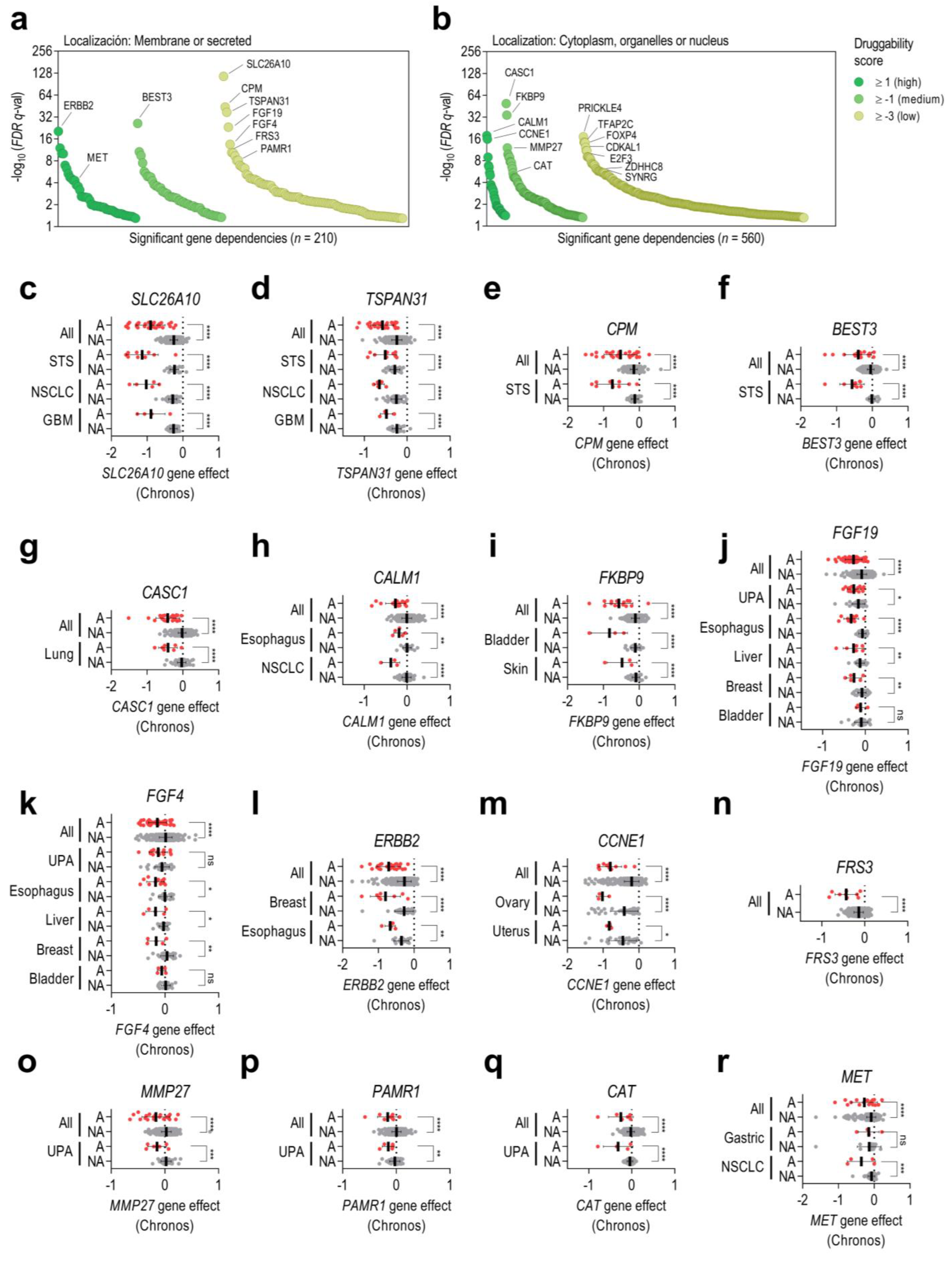
3.3. Some Collateral Dependencies Generated by Amplification Are Druggable
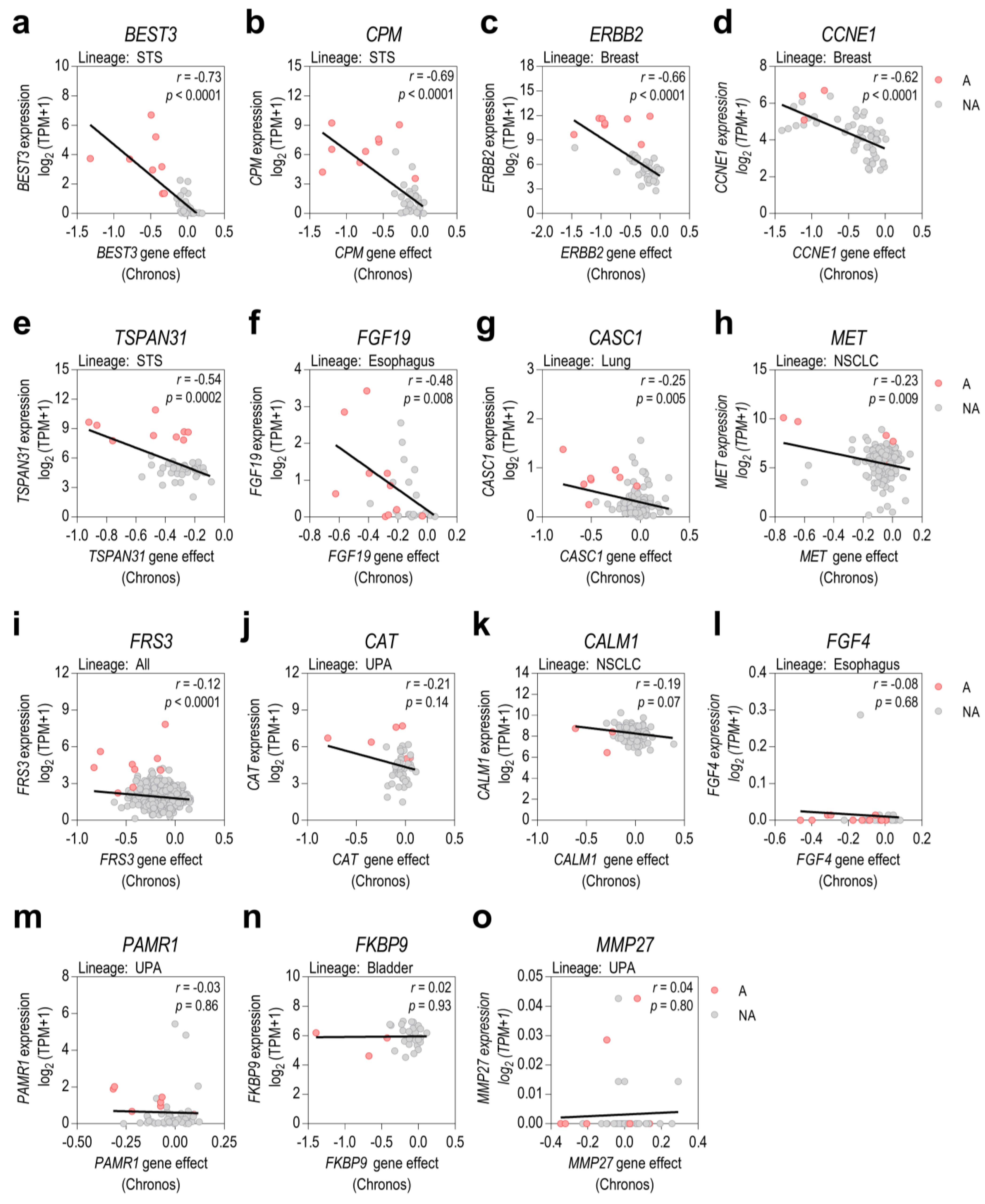
3.4. mRNA Gene Expression Levels Only Correlate with Gene Dependency in Some Prioritized Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, L.; Jiang, Y.; Lei, X.; Yang, X. Amazing Roles of Extrachromosomal DNA in Cancer Progression. Biochim. Biophys. Acta-Rev. Cancer 2023, 1878, 188843. [Google Scholar] [CrossRef] [PubMed]
- van Leen, E.; Brückner, L.; Henssen, A.G. The Genomic and Spatial Mobility of Extrachromosomal DNA and Its Implications for Cancer Therapy. Nat. Genet. 2022, 54, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, R.; Kohno, T.; Kato, M.; Shiraishi, K.; Tsuta, K.; Noguchi, M.; Ogawa, S.; Yokota, J. MYC Amplification as a Prognostic Marker of Early-Stage Lung Adenocarcinoma Identified by Whole Genome Copy Number Analysis. Clin. Cancer Res. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- Saâda-Bouzid, E.; Burel-Vandenbos, F.; Ranchère-Vince, D.; Birtwisle-Peyrottes, I.; Chetaille, B.; Bouvier, C.; Château, M.-C.; Peoc’h, M.; Battistella, M.; Bazin, A.; et al. Prognostic Value of HMGA2, CDK4, and JUN Amplification in Well-Differentiated and Dedifferentiated Liposarcomas. Mod. Pathol. 2015, 28, 1404–1414. [Google Scholar] [CrossRef] [Green Version]
- Borg, Å.; Tandon, A.K.; Sigurdsson, H.; Clark, G.M.; Fernö, M.; Fuqua, S.A.W.; Killander, D.; McGuire, W.L. HER-2 Amplification Predicts Poor Survival in Node-Positive Breast Cancer. Cancer Res. 1990, 50, 4332–4337. [Google Scholar] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after Adjuvant Chemotherapy in HER2-Positive Breast Cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR–Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A First-Generation Pediatric Cancer Dependency Map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef]
- Gillani, R.; Seong, B.K.A.; Crowdis, J.; Conway, J.R.; Dharia, N.V.; Alimohamed, S.; Haas, B.J.; Han, K.; Park, J.; Dietlein, F.; et al. Gene Fusions Create Partner and Collateral Dependencies Essential to Cancer Cell Survival. Cancer Res. 2021, 81, 3971–3984. [Google Scholar] [CrossRef]
- Munoz, D.M.; Cassiani, P.J.; Li, L.; Billy, E.; Korn, J.M.; Jones, M.D.; Golji, J.; Ruddy, D.A.; Yu, K.; McAllister, G.; et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016, 6, 900–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.-Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A Cell Population Dynamics Model of CRISPR Experiments That Improves Inference of Gene Fitness Effects. Genome Biol. 2021, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High-Content CRISPR Screening. Nat. Rev. Methods Prim. 2022, 2, 8. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Jianjiong, G.; Arman, A.B.; Ugur, D.; Gideon, D.; Benjamin, G.; Onur, S.S.; Yichao, S.; Anders, J.; Rileen, S.; Erik, L.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Rosen, N.; Chalifa-Caspi, V.; Shmueli, O.; Adato, A.; Lapidot, M.; Stampnitzky, J.; Safran, M.; Lancet, D. GeneLoc: Exon-Based Integration of Human Genome Maps. Bioinformatics 2003, 19, i222–i224. [Google Scholar] [CrossRef] [Green Version]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Stein, T.I.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite. In Practical Guide to Life Science Databases; Abugessaisa, I., Kasukawa, T., Eds.; Springer: Singapore, 2021; pp. 27–56. ISBN 978-981-16-5812-9. [Google Scholar]
- Pacini, C.; Dempster, J.M.; Boyle, I.; Gonçalves, E.; Najgebauer, H.; Karakoc, E.; van der Meer, D.; Barthorpe, A.; Lightfoot, H.; Jaaks, P.; et al. Integrated Cross-Study Datasets of Genetic Dependencies in Cancer. Nat. Commun. 2021, 12, 1661. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational Correction of Copy Number Effect Improves Specificity of CRISPR–Cas9 Essentiality Screens in Cancer Cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsopoulos, C.; Di Micco, P.; Fernandez, E.V.; Dolciami, D.; Holt, E.; Mica, I.L.; Coker, E.A.; Tym, J.E.; Campbell, J.; Che, K.H.; et al. CanSAR: Update to the Cancer Translational Research and Drug Discovery Knowledgebase. Nucleic Acids Res. 2021, 49, D1074–D1082. [Google Scholar] [CrossRef]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.M.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR Knockout Screening Outperforms ShRNA and CRISPRi in Identifying Essential Genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef]
- Tsurutani, J.; Iwata, H.; Krop, I.; Jänne, P.A.; Doi, T.; Takahashi, S.; Park, H.; Redfern, C.; Tamura, K.; Wise-Draper, T.M.; et al. Targeting HER2 with Trastuzumab Deruxtecan: A Dose-Expansion, Phase I Study in Multiple Advanced Solid Tumors. Cancer Discov. 2020, 10, 688–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Luo, J.; Chang, J.; Rekhtman, N.; Arcila, M.; Drilon, A. MET-Dependent Solid Tumours—Molecular Diagnosis and Targeted Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a Therapeutic Strategy Targeting Amplified FGF19 in Liver Cancer by Oncogenomic Screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.L.; Schuler, M.; Kang, Y.-K.; Yen, C.-J.; Edeline, J.; Choo, S.P.; Lin, C.-C.; Okusaka, T.; Weiss, K.-H.; Macarulla, T.; et al. A First-in-Human Phase 1/2 Study of FGF401 and Combination of FGF401 with Spartalizumab in Patients with Hepatocellular Carcinoma or Biomarker-Selected Solid Tumors. J. Exp. Clin. Cancer Res. 2022, 41, 189. [Google Scholar] [CrossRef]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Gallo, D.; Young, J.T.F.; Fourtounis, J.; Martino, G.; Álvarez-Quilón, A.; Bernier, C.; Duffy, N.M.; Papp, R.; Roulston, A.; Stocco, R.; et al. CCNE1 Amplification Is Synthetic Lethal with PKMYT1 Kinase Inhibition. Nature 2022, 604, 749–756. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pons, G.; Gallo-Oller, G.; Navarro, N.; Zarzosa, P.; Sansa-Girona, J.; García-Gilabert, L.; Magdaleno, A.; Segura, M.F.; Sánchez de Toledo, J.; Gallego, S.; et al. Analysis of Cancer Genomic Amplifications Identifies Druggable Collateral Dependencies within the Amplicon. Cancers 2023, 15, 1636. https://doi.org/10.3390/cancers15061636
Pons G, Gallo-Oller G, Navarro N, Zarzosa P, Sansa-Girona J, García-Gilabert L, Magdaleno A, Segura MF, Sánchez de Toledo J, Gallego S, et al. Analysis of Cancer Genomic Amplifications Identifies Druggable Collateral Dependencies within the Amplicon. Cancers. 2023; 15(6):1636. https://doi.org/10.3390/cancers15061636
Chicago/Turabian StylePons, Guillem, Gabriel Gallo-Oller, Natalia Navarro, Patricia Zarzosa, Júlia Sansa-Girona, Lia García-Gilabert, Ainara Magdaleno, Miguel F. Segura, Josep Sánchez de Toledo, Soledad Gallego, and et al. 2023. "Analysis of Cancer Genomic Amplifications Identifies Druggable Collateral Dependencies within the Amplicon" Cancers 15, no. 6: 1636. https://doi.org/10.3390/cancers15061636