

Role of Mitochondrial Transporters on Metabolic Rewiring of Pancreatic Adenocarcinoma: A Comprehensive Review

 , , , ,
, , , ,  , , and
, , and 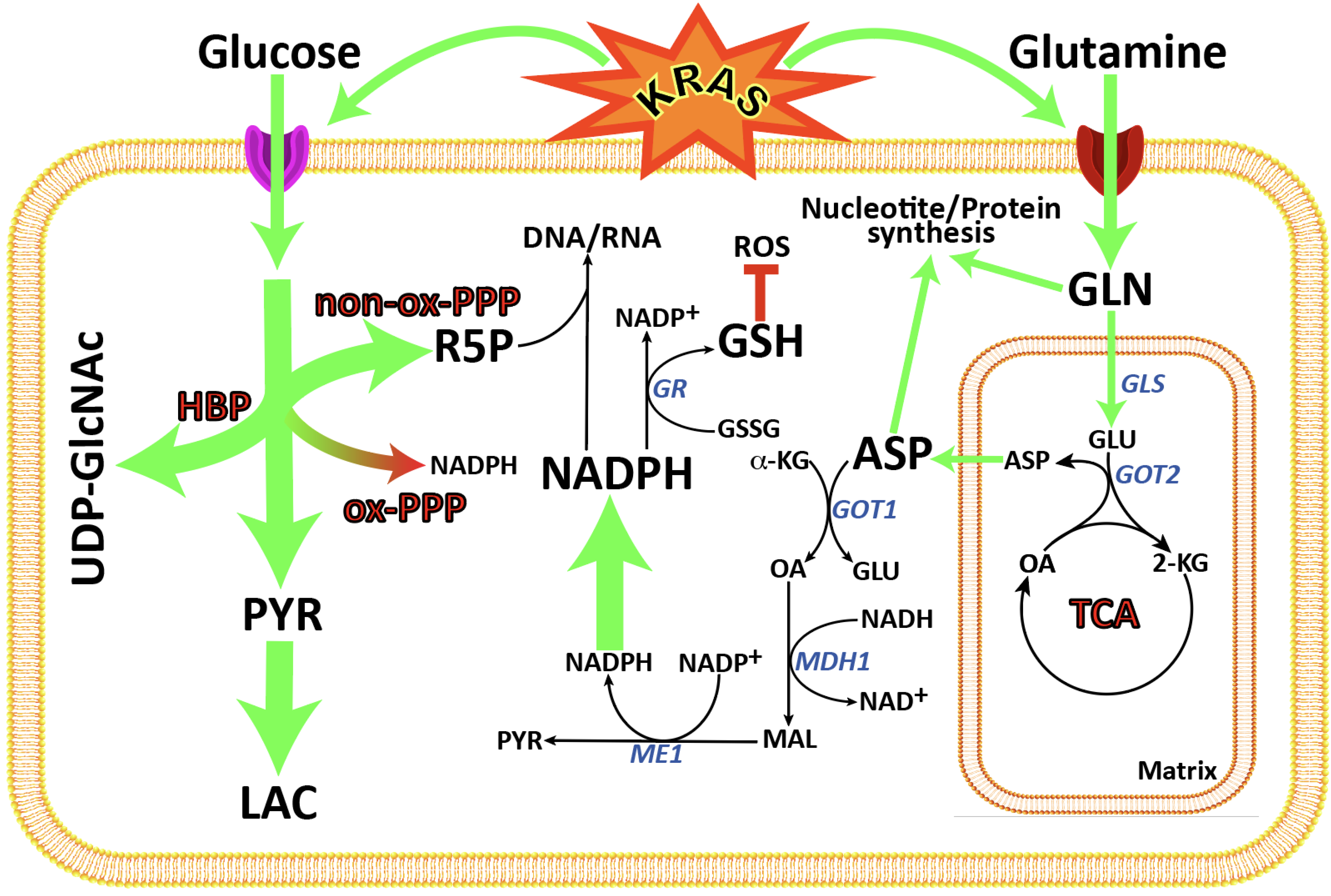{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
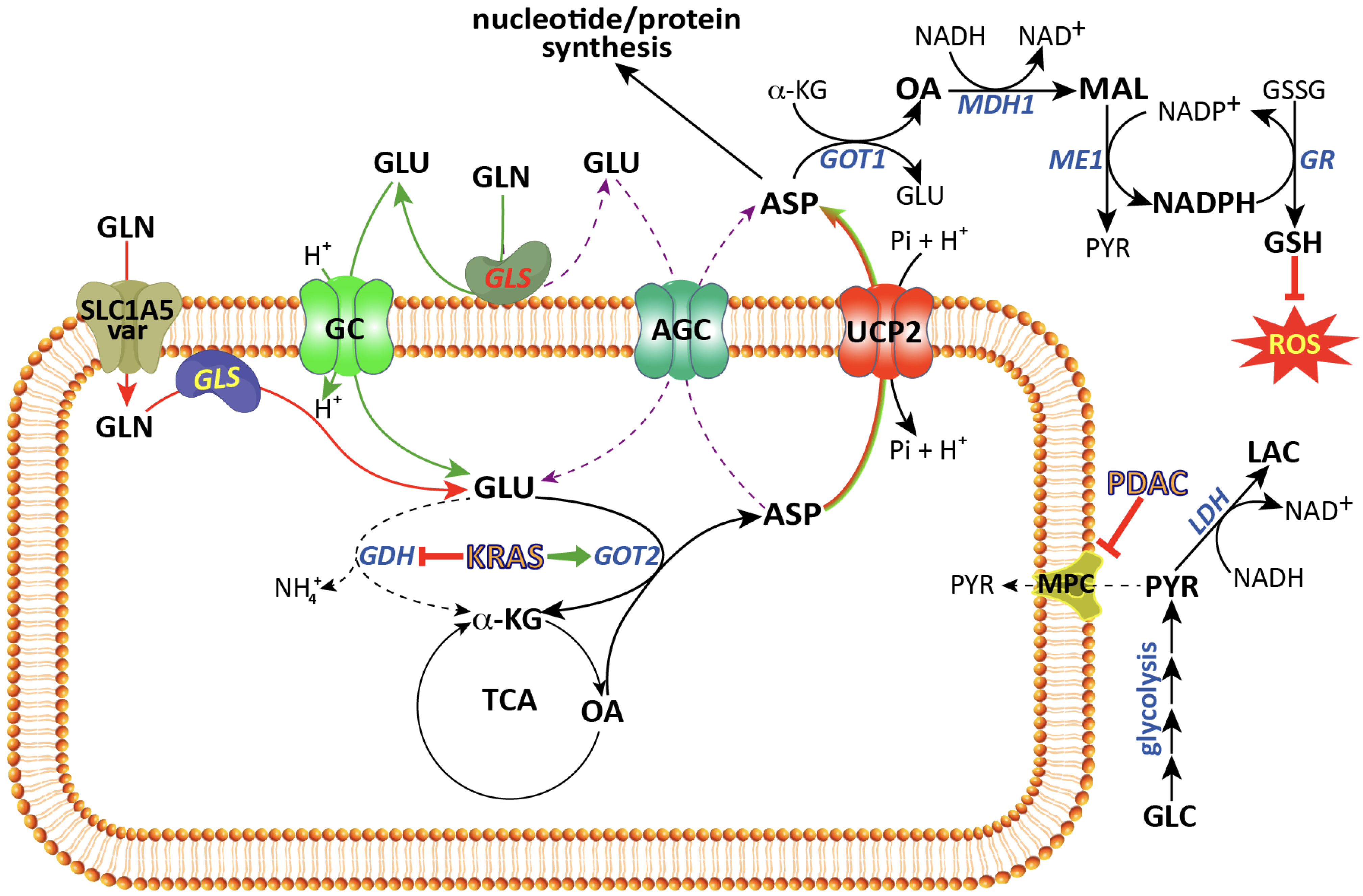
2. The Glutamine Transporter SLC1A5_Var Is at the Centre of Glutaminolysis
3. Mitochondrial Glutamate Carriers: SLC25A22 and SLC25A18
4. The Aspartate–Glutamate Carriers SLC25A12 and SLC25A13
5. UCP2: A Mitochondrial Aspartate Transporter
6. The Mitochondrial Pyruvate Carrier
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Quinonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The challenge of drug resistance in pancreatic ductal adenocarcinoma: A current overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Metzger, P.; Gerum, S.; Mayerle, J.; Schneider, G.; Belka, C.; Schnurr, M.; Lauber, K. Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat. Oncol. 2019, 14, 141. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.Q.; Wang, Z.F.; Wu, X.L.; Zhou, C.H.; Yan, J.Y.; Hu, B.Y.; et al. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal Transduct. Target. Ther. 2021, 6, 249. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Hu, H.F.; Ye, Z.; Qin, Y.; Xu, X.W.; Yu, X.J.; Zhuo, Q.F.; Ji, S.R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharm. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Armitage, E.G.; Southam, A.D. Monitoring cancer prognosis, diagnosis and treatment efficacy using metabolomics and lipidomics. Metabolomics 2016, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Olivares, O.; Vasseur, S. Metabolic rewiring of pancreatic ductal adenocarcinoma: New routes to follow within the maze. Int J. Cancer 2016, 138, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Reske, S.N.; Grillenberger, K.G.; Glatting, G.; Port, M.; Hildebrandt, M.; Gansauge, F.; Beger, H.G. Overexpression of glucose transporter 1 and increased FDG uptake in pancreatic carcinoma. J. Nucl Med. 1997, 38, 1344–1348. [Google Scholar]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ren, B.; Yang, G.; Wang, H.; Chen, G.; You, L.; Zhang, T.; Zhao, Y. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell. Mol. Life Sci. 2020, 77, 305–321. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Iwadate, D.; Kato, H.; Nakai, Y.; Tateishi, K.; Fujishiro, M. Targeting the Metabolic Rewiring in Pancreatic Cancer and Its Tumor Microenvironment. Cancers 2022, 14, 4351. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Kong, S.C.; Nohr-Nielsen, A.; Zeeberg, K.; Reshkin, S.J.; Hoffmann, E.K.; Novak, I.; Pedersen, S.F. Monocarboxylate Transporters MCT1 and MCT4 Regulate Migration and Invasion of Pancreatic Ductal Adenocarcinoma Cells. Pancreas 2016, 45, 1036–1047. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Zuzcak, M.; Trnka, J. Cellular metabolism in pancreatic cancer as a tool for prognosis and treatment (Review). Int. J. Oncol. 2022, 61, 93. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, Y.R.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef]
- Xu, R.; Yang, J.; Ren, B.; Wang, H.; Yang, G.; Chen, Y.; You, L.; Zhao, Y. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Front. Oncol. 2020, 10, 572722. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Konno, M.; Koseki, J.; Nishida, N.; Kawamoto, K.; Yamada, D.; Asaoka, T.; Noda, T.; Wada, H.; Gotoh, K.; et al. The mitochondrial one-carbon metabolic pathway is associated with patient survival in pancreatic cancer. Oncol. Lett. 2018, 16, 1827–1834. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial Metabolism in PDAC: From Better Knowledge to New Targeting Strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Hewton, K.G.; Johal, A.S.; Parker, S.J. Transporters at the Interface between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites 2021, 11, 112. [Google Scholar] [CrossRef]
- Wu, Z.; Xu, J.; Liang, C.; Meng, Q.; Hua, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Emerging roles of the solute carrier family in pancreatic cancer. Clin. Transl. Med. 2021, 11, e356. [Google Scholar] [CrossRef] [PubMed]
- Lemstrova, R.; Soucek, P.; Melichar, B.; Mohelnikova-Duchonova, B. Role of solute carrier transporters in pancreatic cancer: A review. Pharmacogenomics 2014, 15, 1133–1145. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rufian, M.; Tosina, M.; Campos-Sandoval, J.A.; Manzanares, E.; Lobo, C.; Segura, J.A.; Alonso, F.J.; Mates, J.M.; Marquez, J. Mammalian glutaminase Gls2 gene encodes two functional alternative transcripts by a surrogate promoter usage mechanism. PLoS ONE 2012, 7, e38380. [Google Scholar] [CrossRef] [Green Version]
- Szeliga, M.; Bogacinska-Karas, M.; Kuzmicz, K.; Rola, R.; Albrecht, J. Downregulation of GLS2 in glioblastoma cells is related to DNA hypermethylation but not to the p53 status. Mol. Carcinog. 2016, 55, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Majewska, E.; Marquez, J.; Albrecht, J.; Szeliga, M. Transfection with GLS2 Glutaminase (GAB) Sensitizes Human Glioblastoma Cell Lines to Oxidative Stress by a Common Mechanism Involving Suppression of the PI3K/AKT Pathway. Cancers 2019, 11, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, J.; Cho, Y.-R.; Lee, S.-Y.; Sung, G.-J.; Shin, D.-M.; Choi, K.-C.; Son, J. TFEB Supports Pancreatic Cancer Growth through the Transcriptional Regulation of Glutaminase. Cancers 2021, 13, 483. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Guo, D.; Lin, S.H.; Liang, J.; Yang, D.; Ma, C.; Shao, F.; Li, M.; Yu, Q.; Jiang, Y.; et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol. Cell 2021, 81, 2303–2316.e2308. [Google Scholar] [CrossRef] [PubMed]
- Cassago, A.; Ferreira, A.P.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberg, B.; Torgner, I.A.; Kvamme, E. The orientation of phosphate activated glutaminase in the inner mitochondrial membrane of synaptic and non-synaptic rat brain mitochondria. Neurochem. Int. 1995, 27, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Gorgoglione, R.; Impedovo, V.; Riley, C.L.; Fratantonio, D.; Tiziani, S.; Palmieri, L.; Dolce, V.; Fiermonte, G. Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives. Cancers 2022, 14, 245. [Google Scholar] [CrossRef]
- Simpson, D.P.; Adam, W. Glutamine transport and metabolism by mitochondria from dog renal cortex. General properties and response to acidosis and alkalosis. J. Biol. Chem. 1975, 250, 8148–8158. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell. Metab. 2020, 31, 267–283. [Google Scholar] [CrossRef]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.L.; Zamboni, N.; Westermann, B.; Kunji, E.R.; Martinou, J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Monne, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Kunji, E.R.S.; King, M.S.; Ruprecht, J.J.; Thangaratnarajah, C. The SLC25 Carrier Family: Important Transport Proteins in Mitochondrial Physiology and Pathology. Physiology 2020, 35, 302–327. [Google Scholar] [CrossRef]
- Palmieri, F.; Monne, M.; Fiermonte, G.; Palmieri, L. Mitochondrial transport and metabolism of the vitamin B-derived cofactors thiamine pyrophosphate, coenzyme A, FAD and NAD(+), and related diseases: A review. IUBMB Life 2022, 74, 592–617. [Google Scholar] [CrossRef] [PubMed]
- Curcio, R.; Lunetti, P.; Zara, V.; Ferramosca, A.; Marra, F.; Fiermonte, G.; Cappello, A.R.; De Leonardis, F.; Capobianco, L.; Dolce, V. Drosophila melanogaster Mitochondrial Carriers: Similarities and Differences with the Human Carriers. Int. J. Mol. Sci. 2020, 21, 6052. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef]
- Helenius, I.T.; Madala, H.R.; Yeh, J.J. An Asp to Strike Out Cancer? Therapeutic Possibilities Arising from Aspartate’s Emerging Roles in Cell Proliferation and Survival. Biomolecules 2021, 11, 1666. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Sancerni, T.; Renoult, O.; Luby, A.; Caradeuc, C.; Lenoir, V.; Croyal, M.; Ransy, C.; Aguilar, E.; Postic, C.; Bertho, G.; et al. UCP2 silencing restrains leukemia cell proliferation through glutamine metabolic remodeling. Front. Immunol. 2022, 13, 960226. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, G.; Hediger, M.A. Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules 2020, 10, 1611. [Google Scholar] [CrossRef]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef] [Green Version]
- Lunetti, P.; Cappello, A.R.; Marsano, R.M.; Pierri, C.L.; Carrisi, C.; Martello, E.; Caggese, C.; Dolce, V.; Capobianco, L. Mitochondrial glutamate carriers from Drosophila melanogaster: Biochemical, evolutionary and modeling studies. Biochim. Biophys. Acta 2013, 1827, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Goubert, E.; Mircheva, Y.; Lasorsa, F.M.; Melon, C.; Profilo, E.; Sutera, J.; Becq, H.; Palmieri, F.; Palmieri, L.; Aniksztejn, L.; et al. Inhibition of the Mitochondrial Glutamate Carrier SLC25A22 in Astrocytes Leads to Intracellular Glutamate Accumulation. Front. Cell. Neurosci. 2017, 11, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimir, M.; Lasorsa, F.M.; Rubi, B.; Caille, D.; Palmieri, F.; Meda, P.; Maechler, P. Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2009, 284, 25004–25014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Qian, Y.; Li, X.; Xu, J.; Kang, W.; Tong, J.H.; To, K.F.; Jin, Y.; Li, W.; Chen, H.; et al. SLC25A22 Promotes Proliferation and Survival of Colorectal Cancer Cells With KRAS Mutations and Xenograft Tumor Progression in Mice via Intracellular Synthesis of Aspartate. Gastroenterology 2016, 151, 945–960.e946. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.W.; Wu, X.J. SLC25A22 Promotes Proliferation and Metastasis of Osteosarcoma Cells via the PTEN Signaling Pathway. Technol. Cancer Res. Treat. 2018, 17, 1533033818811143. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Liang, H.; Fu, X.; Wu, P.; Wang, C.; Chen, H.; Zheng, B.; Zhang, J.; Hu, S.; Zeng, R.; et al. SLC25A22 promotes proliferation and metastasis by activating MAPK/ERK pathway in gallbladder cancer. Cancer Cell Int. 2019, 19, 33. [Google Scholar] [CrossRef]
- Liang, L.; Chen, Y.; Yu, Y.; Pan, W.; Cui, Y.; Xu, X.; Peng, K.; Liu, M.; Rashid, K.; Hou, Y.; et al. SLC25A18 has prognostic value in colorectal cancer and represses Warburg effect and cell proliferation via Wnt signaling. Am. J. Cancer Res. 2020, 10, 1548–1567. [Google Scholar]
- Monne, M.; Vozza, A.; Lasorsa, F.M.; Porcelli, V.; Palmieri, F. Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review. Int. J. Mol. Sci. 2019, 20, 4456. [Google Scholar] [CrossRef] [Green Version]
- Lunetti, P.; Marsano, R.M.; Curcio, R.; Dolce, V.; Fiermonte, G.; Cappello, A.R.; Marra, F.; Moschetti, R.; Li, Y.; Aiello, D.; et al. The mitochondrial aspartate/glutamate carrier (AGC or Aralar1) isoforms in D. melanogaster: Biochemical characterization, gene structure, and evolutionary analysis. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129854. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrustegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Contreras, L.; Gomez-Puertas, P.; Iijima, M.; Kobayashi, K.; Saheki, T.; Satrustegui, J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: Role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 2007, 282, 7098–7106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Arco, A.; Morcillo, J.; Martinez-Morales, J.R.; Galian, C.; Martos, V.; Bovolenta, P.; Satrustegui, J. Expression of the aspartate/glutamate mitochondrial carriers aralar1 and citrin during development and in adult rat tissues. Eur. J. Biochem. 2002, 269, 3313–3320. [Google Scholar] [CrossRef]
- Borst, P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life 2020, 72, 2241–2259. [Google Scholar] [CrossRef]
- Curcio, R.; Muto, L.; Pierri, C.L.; Montalto, A.; Lauria, G.; Onofrio, A.; Fiorillo, M.; Fiermonte, G.; Lunetti, P.; Vozza, A.; et al. New insights about the structural rearrangements required for substrate translocation in the bovine mitochondrial oxoglutarate carrier. Biochim. Biophys. Acta 2016, 1864, 1473–1480. [Google Scholar] [CrossRef]
- Broeks, M.H.; van Karnebeek, C.D.M.; Wanders, R.J.A.; Jans, J.J.M.; Verhoeven-Duif, N.M. Inborn disorders of the malate aspartate shuttle. J. Inherit. Metab. Dis. 2021, 44, 792–808. [Google Scholar] [CrossRef]
- Pardo, B.; Herrada-Soler, E.; Satrustegui, J.; Contreras, L.; Del Arco, A. AGC1 Deficiency: Pathology and Molecular and Cellular Mechanisms of the Disease. Int. J. Mol. Sci. 2022, 23, 528. [Google Scholar] [CrossRef]
- Tavoulari, S.; Lacabanne, D.; Thangaratnarajah, C.; Kunji, E.R.S. Pathogenic variants of the mitochondrial aspartate/glutamate carrier causing citrin deficiency. Trends. Endocrinol. Metab. 2022, 33, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Profilo, E.; Pena-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-regulation of the mitochondrial aspartate-glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys Acta Mol. Basis. Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Yuan, C.H.; Han, L.Y.; Huang, G.R.; Ju, L.C.; Chen, L.H.; Han, H.Y.; Zhang, C.; Zeng, L.H. The Overexpression of SLC25A13 Predicts Poor Prognosis and Is Correlated with Immune Cell Infiltration in Patients with Skin Cutaneous Melanoma. Dis. Mrk. 2022, 2022, 4091978. [Google Scholar] [CrossRef]
- Rabinovich, S.; Silberman, A.; Adler, L.; Agron, S.; Levin-Zaidman, S.; Bahat, A.; Porat, Z.; Ben-Zeev, E.; Geva, I.; Itkin, M.; et al. The mitochondrial carrier Citrin plays a role in regulating cellular energy during carcinogenesis. Oncogene 2020, 39, 164–175. [Google Scholar] [CrossRef]
- Miyo, M.; Konno, M.; Nishida, N.; Sueda, T.; Noguchi, K.; Matsui, H.; Colvin, H.; Kawamoto, K.; Koseki, J.; Haraguchi, N.; et al. Metabolic Adaptation to Nutritional Stress in Human Colorectal Cancer. Sci. Rep. 2016, 6, 38415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkan, H.F.; Bogner-Strauss, J.G. Maintaining cytosolic aspartate levels is a major function of the TCA cycle in proliferating cells. Mol. Cell Oncol. 2019, 6, e1536843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Esteves, P.; Pecqueur, C.; Alves-Guerra, M.C. UCP2 induces metabolic reprogramming to inhibit proliferation of cancer cells. Mol. Cell Oncol. 2015, 2, e975024. [Google Scholar] [CrossRef] [Green Version]
- Sreedhar, A.; Petruska, P.; Miriyala, S.; Panchatcharam, M.; Zhao, Y. UCP2 overexpression enhanced glycolysis via activation of PFKFB2 during skin cell transformation. Oncotarget 2017, 8, 95504–95515. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.; Oliver, J.; Roca, P. Role of uncoupling proteins in cancer. Cancers 2010, 2, 567–591. [Google Scholar] [CrossRef] [Green Version]
- Donadelli, M.; Dando, I.; Dalla Pozza, E.; Palmieri, M. Mitochondrial uncoupling protein 2 and pancreatic cancer: A new potential target therapy. World J. Gastroenterol. 2015, 21, 3232–3238. [Google Scholar] [CrossRef]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Lunetti, P.; Gorgoglione, R.; Curcio, R.; Marra, F.; Pignataro, A.; Vozza, A.; Riley, C.L.; Capobianco, L.; Palmieri, L.; Dolce, V.; et al. Drosophila melanogaster Uncoupling Protein-4A (UCP4A) Catalyzes a Unidirectional Transport of Aspartate. Int. J. Mol. Sci. 2022, 23, 1020. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Urbankova, E. Specific sequence of motifs of mitochondrial uncoupling proteins. IUBMB Life 2000, 49, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Holendova, B.; Garlid, K.D.; Jaburek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling. Antioxid Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef] [Green Version]
- Ricquier, D. UCP1, the mitochondrial uncoupling protein of brown adipocyte: A personal contribution and a historical perspective. Biochimie 2017, 134, 3–8. [Google Scholar] [CrossRef]
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta 2016, 1863, 2443–2456. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Souza, B.M.; Assmann, T.S.; Kliemann, L.M.; Gross, J.L.; Canani, L.H.; Crispim, D. The role of uncoupling protein 2 (UCP2) on the development of type 2 diabetes mellitus and its chronic complications. Arq. Bras. Endocrinol. Metab. 2011, 55, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Pecqueur, C.; Alves-Guerra, M.C.; Gelly, C.; Levi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J. Biol. Chem. 2001, 276, 8705–8712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves-Guerra, M.C.; Rousset, S.; Pecqueur, C.; Mallat, Z.; Blanc, J.; Tedgui, A.; Bouillaud, F.; Cassard-Doulcier, A.M.; Ricquier, D.; Miroux, B. Bone marrow transplantation reveals the in vivo expression of the mitochondrial uncoupling protein 2 in immune and nonimmune cells during inflammation. J. Biol. Chem. 2003, 278, 42307–42312. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Olejar, T.; Smolkova, K.; Jezek, J.; Dlaskova, A.; Plecita-Hlavata, L.; Zelenka, J.; Spacek, T.; Engstova, H.; Pajuelo Reguera, D.; et al. Antioxidant and regulatory role of mitochondrial uncoupling protein UCP2 in pancreatic beta-cells. Physiol. Res. 2014, 63, S73–S91. [Google Scholar] [CrossRef] [PubMed]
- Donadelli, M.; Dando, I.; Fiorini, C.; Palmieri, M. UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol. Life Sci. 2014, 71, 1171–1190. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front. Physiol. 2018, 9, 1060. [Google Scholar] [CrossRef] [Green Version]
- Sayeed, A.; Meng, Z.; Luciani, G.; Chen, L.C.; Bennington, J.L.; Dairkee, S.H. Negative regulation of UCP2 by TGFbeta signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis. 2010, 1, e53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-beta/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, A.; Moldzio, R.; Modl, B.; Pohl, E.E. Glutamine regulates mitochondrial uncoupling protein 2 to promote glutaminolysis in neuroblastoma cells. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 391–401. [Google Scholar] [CrossRef]
- Hurtaud, C.; Gelly, C.; Chen, Z.; Levi-Meyrueis, C.; Bouillaud, F. Glutamine stimulates translation of uncoupling protein 2mRNA. Cell Mol. Life Sci. 2007, 64, 1853–1860. [Google Scholar] [CrossRef]
- Azzu, V.; Brand, M.D. Degradation of an intramitochondrial protein by the cytosolic proteasome. J. Cell Sci. 2010, 123, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Chen, D.; Zheng, R.H.; Zhang, H.; Chen, Y.P.; Xiang, Z. miRNA-133a-UCP2 pathway regulates inflammatory bowel disease progress by influencing inflammation, oxidative stress and energy metabolism. World J. Gastroenterol. 2017, 23, 76–86. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K.; Chen, J.; Guo, J.; Yin, Y.; Cai, X.; Guo, X.; Wang, G.; Yang, R.; Zhu, L.; et al. In vitro evidence suggests that miR-133a-mediated regulation of uncoupling protein 2 (UCP2) is an indispensable step in myogenic differentiation. J. Biol. Chem. 2009, 284, 5362–5369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.L.; Jiang, B.G.; Li, W.T.; Zou, J.J.; Shi, Y.Q.; Liu, Z.M. MicroRNA-15a positively regulates insulin synthesis by inhibiting uncoupling protein-2 expression. Diabetes Res. Clin. Pr. 2011, 91, 94–100. [Google Scholar] [CrossRef]
- Guo, S.; Fesler, A.; Huang, W.; Wang, Y.; Yang, J.; Wang, X.; Zheng, Y.; Hwang, G.R.; Wang, H.; Ju, J. Functional Significance and Therapeutic Potential of miR-15a Mimic in Pancreatic Ductal Adenocarcinoma. Mol. Nucleic Acids 2020, 19, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.T.; Xu, W.X.; Li, H.; Xia, M. Emerging roles of MiR-133a in human cancers. J. Cancer 2021, 12, 198–206. [Google Scholar] [CrossRef]
- Yang, S.; Fei, X.; Lu, Y.; Xu, B.; Ma, Y.; Wan, H. miRNA-214 suppresses oxidative stress in diabetic nephropathy via the ROS/Akt/mTOR signaling pathway and uncoupling protein 2. Exp. Med. 2019, 17, 3530–3538. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Han, L.H.; Zhao, H.J.; Li, H.; Li, J.B. Abnormal alterations of miR-1 and miR-214 are associated with clinicopathological features and prognosis of patients with PDAC. Oncol. Lett. 2017, 14, 4605–4612. [Google Scholar] [CrossRef] [Green Version]
- Kaul, D.; Sharma, S.; Garg, A. Mitochondrial uncoupling protein (UCP2) gene expression is regulated by miR-2909. Blood Cells Mol. Dis. 2015, 55, 89–93. [Google Scholar] [CrossRef]
- Zammarchi, F.; Morelli, M.; Menicagli, M.; Di Cristofano, C.; Zavaglia, K.; Paolucci, A.; Campani, D.; Aretini, P.; Boggi, U.; Mosca, F.; et al. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. Am. J. Pathol. 2011, 178, 361–372. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Seifert, E.L.; Bouillaud, F.; Aguer, C.; Collins, S.; Harper, M.E. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J. Biol. Chem. 2011, 286, 21865–21875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfefferle, A.; Mailloux, R.J.; Adjeitey, C.N.; Harper, M.E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim. Biophys. Acta 2013, 1, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Fu, A.; Robson-Doucette, C.; Allister, E.M.; Wheeler, M.B.; Screaton, R.; Harper, M.E. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2012, 287, 39673–39685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Shi, L.; Lin, W.; Lu, B.; Zhao, Y. UCP2 promotes proliferation and chemoresistance through regulating the NF-κB/β-catenin axis and mitochondrial ROS in gallbladder cancer. Biochem. Pharm. 2020, 172, 5. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Fiorini, C.; Dando, I.; Menegazzi, M.; Sgarbossa, A.; Costanzo, C.; Palmieri, M.; Donadelli, M. Role of mitochondrial uncoupling protein 2 in cancer cell resistance to gemcitabine. Biochim. Biophys. Acta 2012, 1823, 1856–1863. [Google Scholar] [CrossRef] [Green Version]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E.; Fiermonte, G.; Steinkrauss, L.J.; Topor, L.S.; Quintos, J.B.; Ganguly, A.; De Leon, D.D.; Palmieri, F.; et al. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillaud, F. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta 2009, 1787, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef]
- Vernucci, E.; Abrego, J.; Gunda, V.; Shukla, S.K.; Dasgupta, A.; Rai, V.; Chaika, N.; Buettner, K.; Illies, A.; Yu, F.; et al. Metabolic Alterations in Pancreatic Cancer Progression. Cancers 2019, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Kerk, S.A.; Lin, L.; Myers, A.L.; Sutton, D.J.; Andren, A.; Sajjakulnukit, P.; Zhang, L.; Zhang, Y.; Jimenez, J.A.; Nelson, B.S.; et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife 2022, 11, 73245. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hwang, S.; Kim, M.; Seo, S.B.; Lee, J.H.; Jeong, S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Esteves, P.; Sancerni, T.; Lenoir, V.; Aparicio, T.; Bouillaud, F.; Dentin, R.; Prip-Buus, C.; Ricquier, D.; Pecqueur, C.; et al. UCP2 Deficiency Increases Colon Tumorigenesis by Promoting Lipid Synthesis and Depleting NADPH for Antioxidant Defenses. Cell Rep. 2019, 28, 2306–2316.e2305. [Google Scholar] [CrossRef] [Green Version]
- Rigaud, V.O.; Zarka, C.; Kurian, J.; Harlamova, D.; Elia, A.; Kasatkin, N.; Johnson, J.; Behanan, M.; Kraus, L.; Pepper, H.; et al. UCP2 modulates cardiomyocyte cell cycle activity, acetyl-CoA, and histone acetylation in response to moderate hypoxia. JCI Insight 2022, 7, 155475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Yang, Y.; Han, Y.; Li, X.; Wang, X.; Li, X.; Zhang, Z.; Wang, Y. UCP2 inhibits ROS-mediated apoptosis in A549 under hypoxic conditions. PLoS ONE 2012, 7, e30714. [Google Scholar] [CrossRef] [Green Version]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1α/UCP2 axis increasing mitochondrial O(2-) production in cancer cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Rauckhorst, A.J.; Taylor, E.B. Mitochondrial pyruvate carrier function and cancer metabolism. Curr. Opin. Genet. Dev. 2016, 38, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Proft, M.; Pascual-Ahuir, A. Differential regulation of mitochondrial pyruvate carrier genes modulates respiratory capacity and stress tolerance in yeast. PLoS ONE 2013, 8, e79405. [Google Scholar] [CrossRef] [Green Version]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagampalli, R.S.K.; Quesnay, J.E.N.; Adamoski, D.; Islam, Z.; Birch, J.; Sebinelli, H.G.; Girard, R.; Ascencao, C.F.R.; Fala, A.M.; Pauletti, B.A.; et al. Human mitochondrial pyruvate carrier 2 as an autonomous membrane transporter. Sci. Rep. 2018, 8, 3510. [Google Scholar] [CrossRef]
- Suzuki, T.; Otsuka, M.; Seimiya, T.; Iwata, T.; Kishikawa, T.; Koike, K. The biological role of metabolic reprogramming in pancreatic cancer. MedComm 2020, 1, 302–310. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, S.; Yu, D. Protein kinase function of pyruvate kinase M2 and cancer. Cancer Cell Int. 2020, 20, 523. [Google Scholar] [CrossRef]
- Ruiz-Iglesias, A.; Manes, S. The Importance of Mitochondrial Pyruvate Carrier in Cancer Cell Metabolism and Tumorigenesis. Cancers 2021, 13, 1488. [Google Scholar] [CrossRef] [PubMed]
- Zangari, J.; Petrelli, F.; Maillot, B.; Martinou, J.C. The Multifaceted Pyruvate Metabolism: Role of the Mitochondrial Pyruvate Carrier. Biomolecules 2020, 10, 1068. [Google Scholar] [CrossRef]
- Sutendra, G.; Michelakis, E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol. Cancer 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [Green Version]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: Emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Cui, Y.; Zhou, H.; Li, Q. Function of mitochondrial pyruvate carriers in hepatocellular carcinoma patients. Oncol. Lett. 2018, 15, 9110–9116. [Google Scholar] [CrossRef] [Green Version]
- Schell, J.C.; Olson, K.A.; Jiang, L.; Hawkins, A.J.; Van Vranken, J.G.; Xie, J.; Egnatchik, R.A.; Earl, E.G.; DeBerardinis, R.J.; Rutter, J. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 2014, 56, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Kuerbanjiang, M.; Gu, L.; Xu, C.; Xu, W.T.; Wen, S.; Xue, H.; Xu, Q. Decreased Expression of MPC2 Contributes to Aerobic Glycolysis and Colorectal Cancer Proliferation by Activating mTOR Pathway. J. Immunol. Res. 2021, 2021, 6618837. [Google Scholar] [CrossRef] [PubMed]
- Bensard, C.L.; Wisidagama, D.R.; Olson, K.A.; Berg, J.A.; Krah, N.M.; Schell, J.C.; Nowinski, S.M.; Fogarty, S.; Bott, A.J.; Wei, P.; et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell Metab 2020, 31, 284–300.e287. [Google Scholar] [CrossRef]
- Cui, J.; Quan, M.; Xie, D.; Gao, Y.; Guha, S.; Fallon, M.B.; Chen, J.; Xie, K. A novel KDM5A/MPC-1 signaling pathway promotes pancreatic cancer progression via redirecting mitochondrial pyruvate metabolism. Oncogene 2020, 39, 1140–1151. [Google Scholar] [CrossRef]
- Yang, G.J.; Zhu, M.H.; Lu, X.J.; Liu, Y.J.; Lu, J.F.; Leung, C.H.; Ma, D.L.; Chen, J. The emerging role of KDM5A in human cancer. J. Hematol. Oncol. 2021, 14, 30. [Google Scholar] [CrossRef]
- Takaoka, Y.; Konno, M.; Koseki, J.; Colvin, H.; Asai, A.; Tamari, K.; Satoh, T.; Mori, M.; Doki, Y.; Ogawa, K.; et al. Mitochondrial pyruvate carrier 1 expression controls cancer epithelial-mesenchymal transition and radioresistance. Cancer Sci. 2019, 110, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Wang, Q.; Tajima, K.; Matsushita, M.; Maki, H.; Igarashi, K.; Dai, Z.; White, P.J.; McGarrah, R.W.; Ilkayeva, O.R.; et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 2019, 572, 614–619. [Google Scholar] [CrossRef]
- Dolce, V.; Cappello, A.R.; Capobianco, L. Mitochondrial tricarboxylate and dicarboxylate-tricarboxylate carriers: From animals to plants. IUBMB Life 2014, 66, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Bonofiglio, D.; Santoro, A.; Martello, E.; Vizza, D.; Rovito, D.; Cappello, A.R.; Barone, I.; Giordano, C.; Panza, S.; Catalano, S.; et al. Mechanisms of divergent effects of activated peroxisome proliferator-activated receptor-gamma on mitochondrial citrate carrier expression in 3T3-L1 fibroblasts and mature adipocytes. Biochim. Biophys. Acta 2013, 1831, 1027–1036. [Google Scholar] [CrossRef]
- Carrisi, C.; Madeo, M.; Morciano, P.; Dolce, V.; Cenci, G.; Cappello, A.R.; Mazzeo, G.; Iacopetta, D.; Capobianco, L. Identification of the Drosophila melanogaster mitochondrial citrate carrier: Bacterial expression, reconstitution, functional characterization and developmental distribution. J. Biochem. 2008, 144, 389–392. [Google Scholar] [CrossRef]
- Kim, A.; Ha, J.; Kim, J.; Cho, Y.; Ahn, J.; Cheon, C.; Kim, S.H.; Ko, S.G.; Kim, B. Natural Products for Pancreatic Cancer Treatment: From Traditional Medicine to Modern Drug Discovery. Nutrients 2021, 13, 3801. [Google Scholar] [CrossRef]
- Dando, I.; Fiorini, C.; Pozza, E.D.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta 2013, 1833, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Brandi, J.; Cecconi, D.; Cordani, M.; Torrens-Mas, M.; Pacchiana, R.; Dalla Pozza, E.; Butera, G.; Manfredi, M.; Marengo, E.; Oliver, J.; et al. The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic. Biol. Med. 2016, 101, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal. Transduct Target. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Ye, H.; Zeng, C.W.; He, B.; Zhang, H.; Chen, Y.Q. Dysregulation of miR-15a and miR-214 in human pancreatic cancer. J. Hematol. Oncol. 2010, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Dang, X.; Li, W.; Ma, Q. miR-133a functions as a tumor suppressor and directly targets FSCN1 in pancreatic cancer. Oncol. Res. 2013, 21, 353–363. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauria, G.; Curcio, R.; Lunetti, P.; Tiziani, S.; Coppola, V.; Dolce, V.; Fiermonte, G.; Ahmed, A. Role of Mitochondrial Transporters on Metabolic Rewiring of Pancreatic Adenocarcinoma: A Comprehensive Review. Cancers 2023, 15, 411. https://doi.org/10.3390/cancers15020411
Lauria G, Curcio R, Lunetti P, Tiziani S, Coppola V, Dolce V, Fiermonte G, Ahmed A. Role of Mitochondrial Transporters on Metabolic Rewiring of Pancreatic Adenocarcinoma: A Comprehensive Review. Cancers. 2023; 15(2):411. https://doi.org/10.3390/cancers15020411
Chicago/Turabian StyleLauria, Graziantonio, Rosita Curcio, Paola Lunetti, Stefano Tiziani, Vincenzo Coppola, Vincenza Dolce, Giuseppe Fiermonte, and Amer Ahmed. 2023. "Role of Mitochondrial Transporters on Metabolic Rewiring of Pancreatic Adenocarcinoma: A Comprehensive Review" Cancers 15, no. 2: 411. https://doi.org/10.3390/cancers15020411