
Integrative Methylome and Transcriptome Characterization Identifies SERINC2 as a Tumor-Driven Gene for Papillary Thyroid Carcinoma
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort and Design
2.2. DNA Differential Methylation Analysis and RNA Array
2.3. Cell Lines and Culture
2.4. Reagents and Transfection
2.5. Cell Proliferation Assay
2.6. Cell Apoptosis Analysis
2.7. RNA Extraction and Real-Time Quantitative RT-PCR (qRT-PCR) Assay
2.8. Western Blotting
2.9. The Cancer Dependency Map Database
2.10. Statistical Analysis
3. Results
3.1. Hypomethylated and Up-Regulated Genes Predominate in Papillary Thyroid Carcinoma
3.2. SERINC2 Is Up-Regulated in Thyroid Cancer
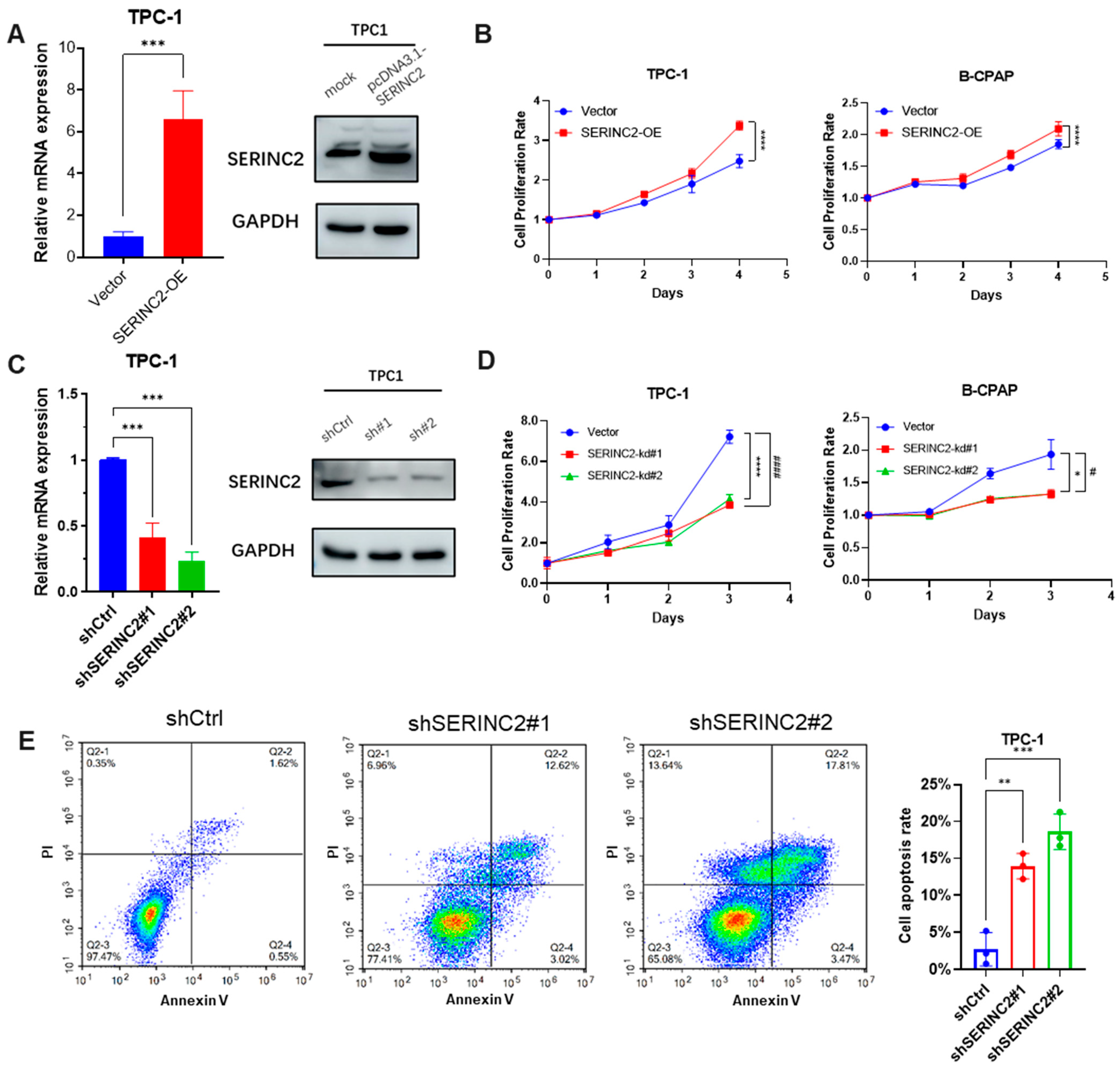
3.3. Regulation of SERINC2 Influences Papillary Thyroid Cancer Progression
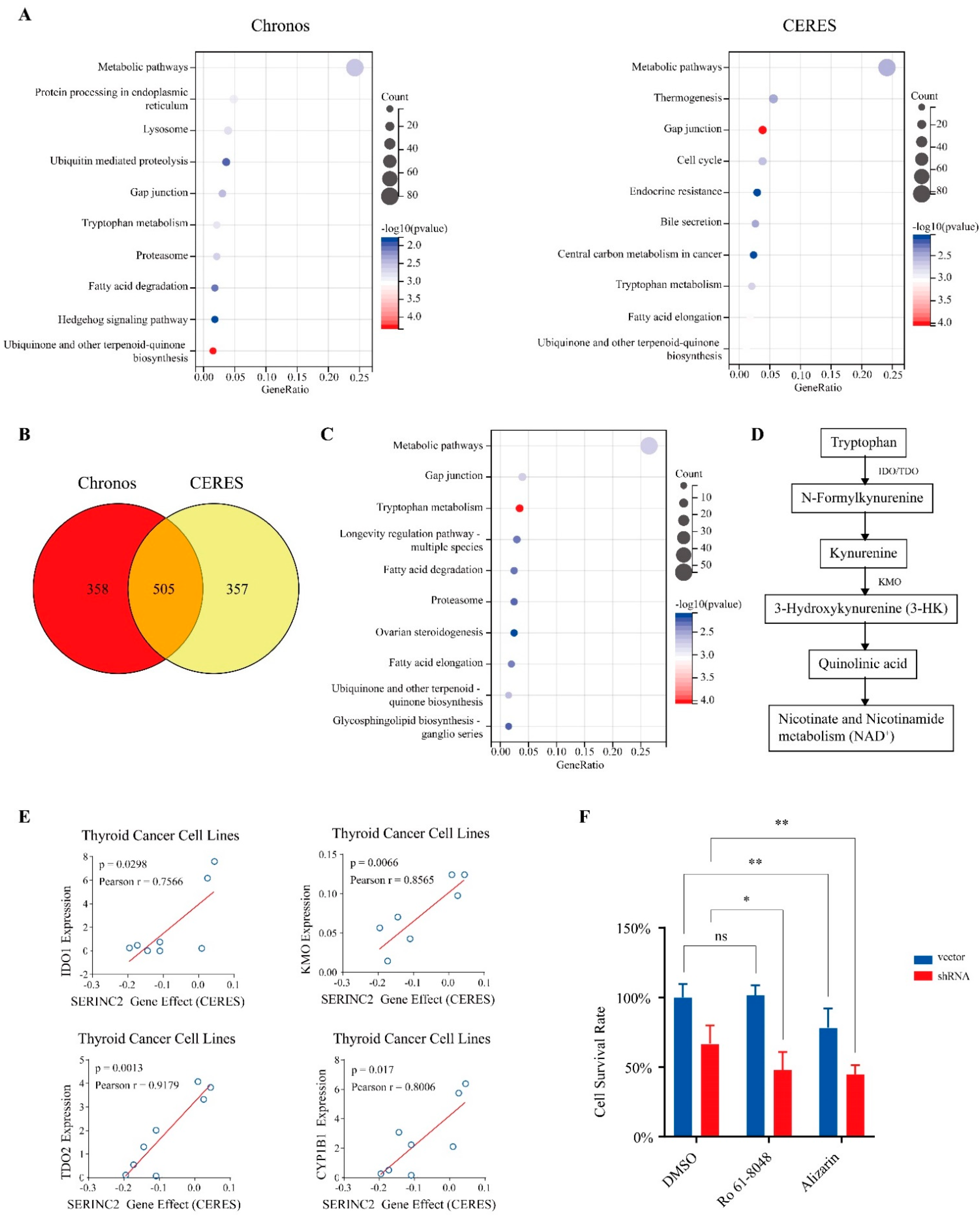
3.4. Activation of Tryptophan Metabolic Pathways Reduces the Dependence on SERINC2 in Thyroid Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Tuttle, R.M.; Davies, L. Changing Trends in the Incidence of Thyroid Cancer in the United States. JAMA Otolaryngol. Head Neck Surg. 2016, 142, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on Fundamental Mechanisms of Thyroid Cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Strickaert, A.; Corbet, C.; Spinette, S.-A.; Craciun, L.; Dom, G.; Andry, G.; Larsimont, D.; Wattiez, R.; Dumont, J.E.; Feron, O.; et al. Reprogramming of Energy Metabolism: Increased Expression and Roles of Pyruvate Carboxylase in Papillary Thyroid Cancer. Thyroid 2019, 29, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Xu, T.; Lu, X.; Huang, P.; Pan, Z.; Ge, M. Metabolic Reprogramming of Thyroid Cancer Cells and Crosstalk in Their Microenvironment. Front. Oncol. 2021, 11, 773028. [Google Scholar] [CrossRef]
- Wang, T.S.; Sosa, J.A. Thyroid surgery for differentiated thyroid cancer—Recent advances and future directions. Nat. Rev. Endocrinol. 2018, 14, 670–683. [Google Scholar] [CrossRef]
- Nabhan, F.; Ringel, M.D. Thyroid nodules and cancer management guidelines: Comparisons and controversies. Endocr. Relat. Cancer 2017, 24, R13–R26. [Google Scholar] [CrossRef]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafon, C.; Gil, J.; Perez-Gonzalez, B.; Jorda, M. DNA methylation in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef] [Green Version]
- Barros-Filho, M.C.; dos Reis, M.B.; Beltrami, C.M.; de Mello, J.B.H.; Marchi, F.A.; Kuasne, H.; Drigo, S.A.; de Andrade, V.P.; Saieg, M.A.; Pinto, C.A.L.; et al. DNA Methylation-Based Method to Differentiate Malignant from Benign Thyroid Lesions. Thyroid 2019, 29, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, P.; Ying, T.; Wang, Y.; Wang, X.; Shu, L.; Mo, Z.; Chen, Z.; Wang, X.; Wang, W.; et al. Integrative Analysis of DNA Methylation and Gene Expression Identified Follicular Thyroid Cancer-Specific Diagnostic Biomarkers. Front. Endocrinol. 2021, 12, 736068. [Google Scholar] [CrossRef]
- Inuzuka, M.; Hayakawa, M.; Ingi, T. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem. 2005, 280, 35776–35783. [Google Scholar] [CrossRef]
- Xu, S.; Zheng, Z.; Pathak, J.L.; Cheng, H.; Zhou, Z.; Chen, Y.; Wu, Q.; Wang, L.; Zeng, M.; Wu, L. The Emerging Role of the Serine Incorporator Protein Family in Regulating Viral Infection. Front. Cell Dev. Biol. 2022, 10, 856468. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Q.; Fan, B.; Gong, J.; Sun, L.; Hu, B.; Wang, D. Machine learning-based screening of the diagnostic genes and their relationship with immune-cell infiltration in patients with lung adenocarcinoma. J. Thorac. Dis. 2022, 14, 699–711. [Google Scholar] [CrossRef]
- Lim, J.; Han, Y.B.; Park, S.Y.; Ahn, S.; Kim, H.; Kwon, H.J.; Lee, C.-T.; Cho, S.; Chung, J.-H. Gene Expression Profiles of Multiple Synchronous Lesions in Lung Adenocarcinoma. Cells 2021, 10, 3484. [Google Scholar] [CrossRef]
- Nomiri, S.; Karami, H.; Baradaran, B.; Javadrashid, D.; Derakhshani, A.; Nourbakhsh, N.S.; Shadbad, M.A.; Solimando, A.G.; Tabrizi, N.J.; Brunetti, O.; et al. Exploiting systems biology to investigate the gene modules and drugs in ovarian cancer: A hypothesis based on the weighted gene co-expression network analysis. Biomed. Pharmacother. 2022, 146, 112537. [Google Scholar] [CrossRef]
- Qi, C.; Lei, L.; Hu, J.; Wang, G.; Liu, J.; Ou, S. Serine Incorporator 2 (SERINC2) Expression Predicts an Unfavorable Prognosis of Low-Grade Glioma (LGG): Evidence from Bioinformatics Analysis. J. Mol. Neurosci. 2020, 70, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Xiao, D.; He, H.; He, J.; Pan, H.; Yang, W.; Chen, Y.; He, J. SERINC2-knockdown inhibits proliferation, migration and invasion in lung adenocarcinoma. Oncol. Lett. 2018, 16, 5916–5922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, K.; Huang, W.; Sun, Y.M.; Chen, T.Q.; Zeng, Z.C.; Yang, Q.Q.; Pan, Q.; Han, C.; Sun, L.Y.; Luo, X.Q.; et al. Cis-acting lnc-eRNA SEELA directly binds histone H4 to promote histone recognition and leukemia progression. Genome. Biol. 2020, 21, 269. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: A cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome. Biol. 2021, 22, 1–23. [Google Scholar] [CrossRef]
- Meng, H.; Cao, Y.; Qin, J.; Song, X.; Zhang, Q.; Shi, Y.; Cao, L. DNA methylation, its mediators and genome integrity. Int. J. Biol. Sci. 2015, 11, 604–617. [Google Scholar] [CrossRef] [Green Version]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA methylation landscape in cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.A.; Li, M.; Lin, J.; Zhang, G.; Chen, M.; Moazzam, N.F.; Qian, W. Preliminary Study on the Sequencing of Whole Genomic Methylation and Transcriptome-Related Genes in Thyroid Carcinoma. Cancers 2022, 14, 1163. [Google Scholar] [CrossRef]
- Mancikova, V.; Buj, R.; Castelblanco, E.; Inglada, L.; Diez, A.; De Cubas, A.A.; Curras-Freixes, M.; Maravall, F.X.; Mauricio, D.; Matias-Guiu, X.; et al. DNA methylation profiling of well-differentiated thyroid cancer uncovers markers of recurrence free survival. Int. J. Cancer 2014, 135, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Gandhi, M.; Edreira, M.M.; Hochbaum, D.; Nimgaonkar, V.L.; Zhang, P.; DiPaola, J.; Evdokimova, V.; Altschuler, D.L.; Nikiforov, Y.E. Downregulation of Rap1GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. Cancer Res. 2010, 70, 1389–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Gao, M.; Qin, D.; Wang, H.; Lu, Q. Serous BMP8A has Clinical Significance in the Ultrasonic Diagnosis of Thyroid Cancer and Promotes Thyroid Cancer Cell Progression. Endocr. Metab. Immune Disord. Drug. Targets 2020, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Lv, J.; Chen, M.; Guo, N.; Fang, Y.; Tong, J.; He, X.; Wu, G.; Wang, Z. Serinc2 deficiency causes susceptibility to sepsis-associated acute lung injury. J. Inflamm. 2022, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.J.; Rendina, L.M. Targeting key dioxygenases in tryptophan-kynurenine metabolism for immunomodulation and cancer chemotherapy. Drug Discov. Today 2015, 20, 609–617. [Google Scholar] [CrossRef]
- Iizuka, T.; Yin, P.; Zuberi, A.; Kujawa, S.; Coon, J.S.; Björvang, R.D.; Damdimopoulou, P.; Pacyga, D.C.; Strakovsky, R.S.; Flaws, J.A.; et al. Mono-(2-ethyl-5-hydroxyhexyl) phthalate promotes uterine leiomyoma cell survival through tryptophan-kynurenine-AHR pathway activation. Proc. Natl. Acad. Sci. USA 2022, 119, e2208886119. [Google Scholar] [CrossRef]
- Ye, X.; Li, H.; Anjum, K.; Zhong, X.; Miao, S.; Zheng, G.; Liu, W.; Li, L. Dual Role of Indoles Derived From Intestinal Microbiota on Human Health. Front. Immunol. 2022, 13, 903526. [Google Scholar] [CrossRef]
- Xu, Y.; Zheng, X.; Qiu, Y.; Jia, W.; Wang, J.; Yin, S. Distinct Metabolomic Profiles of Papillary Thyroid Carcinoma and Benign Thyroid Adenoma. J. Proteome Res. 2015, 14, 3315–3321. [Google Scholar] [CrossRef]
- Du, Y.; Fan, P.; Zou, L.; Jiang, Y.; Gu, X.; Yu, J.; Zhang, C. Serum Metabolomics Study of Papillary Thyroid Carcinoma Based on HPLC-Q-TOF-MS/MS. Front. Cell Dev. Biol. 2021, 9, 593510. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ying, T.; Wang, X.; Yao, Y.; Yuan, J.; Chen, S.; Wen, L.; Chen, Z.; Wang, X.; Luo, C.; Sheng, J.; et al. Integrative Methylome and Transcriptome Characterization Identifies SERINC2 as a Tumor-Driven Gene for Papillary Thyroid Carcinoma. Cancers 2023, 15, 243. https://doi.org/10.3390/cancers15010243
Ying T, Wang X, Yao Y, Yuan J, Chen S, Wen L, Chen Z, Wang X, Luo C, Sheng J, et al. Integrative Methylome and Transcriptome Characterization Identifies SERINC2 as a Tumor-Driven Gene for Papillary Thyroid Carcinoma. Cancers. 2023; 15(1):243. https://doi.org/10.3390/cancers15010243
Chicago/Turabian StyleYing, Tianxing, Xumeng Wang, Yunjin Yao, Jimeng Yuan, Shitu Chen, Liping Wen, Zhijian Chen, Xiaofeng Wang, Chi Luo, Jinghao Sheng, and et al. 2023. "Integrative Methylome and Transcriptome Characterization Identifies SERINC2 as a Tumor-Driven Gene for Papillary Thyroid Carcinoma" Cancers 15, no. 1: 243. https://doi.org/10.3390/cancers15010243