1. Introduction
Endometrial cancer (EC) is the most common gynecological cancer in western countries. It involves 90% of all uterine cancers. It accounts for 90,000 deaths worldwide per year [
1], whereas the five-year survival of patients diagnosed at advanced stages is only 20% [
2,
3]. Despite the recent increase in molecularly targeted therapies approved by the FDA, including immune checkpoint inhibitors (pembralizumab, dostarlimab), angiogenesis inhibitors (lenvatinib, bevacizumab) or mTOR inhibitors (everolimus, temsirolimus), the first line therapy remains still surgery, chemotherapy and radiotherapy [
4]. The modest response to conventional treatments, along with their high rates of resistance to chemotherapy, explains why current therapies are not successful in treating high-risk, recurrent or metastatic EC patients [
5].
Thus, there is an unmet medical need calling to develop new therapeutic approaches that improve current treatment, both by preventing metastatic development in high-risk EC patients and achieving a higher inhibition of metastatic dissemination, along with a significant reduction in its associated systemic toxicity and severe adverse effects [
6,
7]. Moreover, while selective therapeutic alternatives such as antibody–drug conjugates (ADC), consisting of a targeted monoclonal antibody chemically attached to a cytotoxic agent, have been approved for other cancer types [
8]; however there are none currently available to treat EC [
9].
The development of toxin-based therapies, and particularly the immunotoxins displaying antitumor activity, is a highly dynamic research field, which is being evaluated in clinical trials despite their low therapeutic window. Immunotoxins are similar to ADC since they pursue targeted drug delivery; thus, they consist of a targeting monoclonal antibody, or cytokine, chemically linked to a cytotoxic domain of bacterial, plant or animal toxins [
10]. Among the clinical trials that evaluated toxin-based therapies against cancer, however, the FDA approved only those to treat hematological neoplasias. In contrast, immunotoxins did not show sufficient activity against solid tumors because of poor specificity and severe adverse effects [
11].
To prevent these handicaps, we are developing novel nanoparticles displaying multivalent-CXCR4 targeting ligands. These nanoparticles are capable of selectively delivering conjugated drugs or cytotoxic protein domains to cancer cells that overexpress the CXCR4 receptor (CXCR4
+) in cancer tissues, avoiding their delivery in healthy organs because of the low or negligible CXCR4 expression level in normal cells [
12,
13]. Thus, our goal is to tackle the urgent need to develop therapeutic agents that could be exploited to achieve targeted delivery of bacterial exotoxins to CXCR4
+ tumor cells and thus reach personalized therapy that enhances the antineoplastic effect while reducing adverse effects in EC patients [
14].
CXCR4 is a chemokine receptor overexpressed in EC tumor cells both at mRNA and protein levels [
15], showing mostly membrane staining in cancer tissues of EC patients, which has been reported to promote metastases in our previous work [
16]. We also recently proved the role of CXCR4 in enhancing metastasis in the EC mouse model. Moreover, we demonstrated a CXCR4-dependent biodistribution for the T22-GFP-H6 nanocarrier, and its enhanced uptake in tumor tissue in a subcutaneous EC model. In this work, we selected the CXCR4
+ Luciferase
+ AN3CA cell line, among the tested HEC1A, ARK-2 and AN3CA cells, to generate the subcutaneous and metastatic EC models to be used in the evaluation of these novel therapeutic nanoparticles because of their higher internalization of our CXCR4-targeted nanocarrier in this cell line. Moreover, the orthotopic model derived from CXCR4
+ Luciferase
+ AN3CA yielded the highest metastatic and aggressive malignancy [
16].
In these models, two nanotoxins were tested: T22-DITOX-H6, which incorporates the deimmunized catalytic domain of the exotoxin from
Corynebacterium diphtheriae (DITOX) and T22-PE24-H6, which incorporates the
Pseudomonas aeruginosa (PE24) exotoxin domain as previously described [
17] (
Figure S1). Both nanotoxins maintained the same nanostructure as that described for the T22-GFP-H6 nanocarrier and, similarly, formed self-assembled protein-only nanoparticles that achieve a multivalent display of CXCR4-targeted T22 ligands. These nanotoxins had previously shown high preclinical therapeutic potential in head and neck and colorectal cancer models and in hematological cancer models [
18,
19,
20]. Additional therapeutic nanoparticles derived from the same carrier that include human proapoptotic domains, specifically BAK, PUMA or BAX pore-forming domains [
13], are here compared with the nanotoxins regarding their cytotoxic effect in vitro.
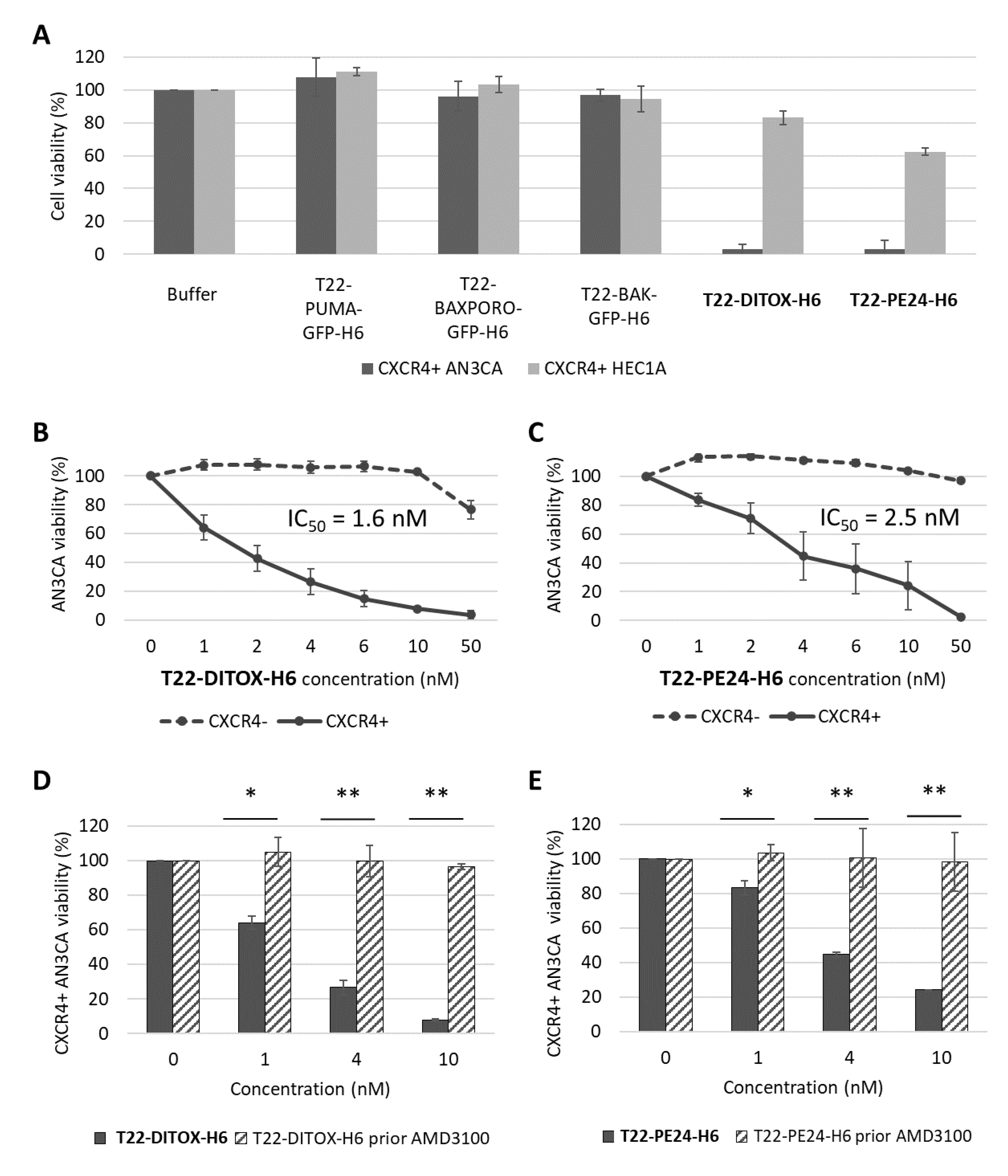
Thus, in this work, we performed a screening of nanotoxins and proapoptotic nanoparticles in terms of their anticancer effect in vitro, selecting only those with a higher effect, namely T22-DITOX-H6 and T22-PE24-H6. Then, we evaluated the antitumor and antimetastatic effect of the nanotoxins in aggressive subcutaneous and orthotopic EC models. Our results support the nanotoxins as alternative therapies that could improve the current treatment of high-risk EC because of their capacity to prevent metastasis development at the clinically relevant dissemination sites for EC using our recently developed orthotopic model of metastatic CXCR4+ EC.
2. Materials and Methods
2.1. Cell Culture
AN3CA cell line (ATCC) and transduced CXCR4
+ Luciferase
+ AN3CA were cultured in DMEM and F12 medium 1:1, while HEC1A (ATCC) and transduced CXCR4
+ Luciferase
+ HEC1A were cultured in high glucose DMEM. All cell lines were maintained in their medium supplemented with 10% fetal bovine serum and penicillin/streptomycin, at 37 °C and 5% CO
2. The CXCR4
+ Luciferase
+ AN3CA cell line was obtained previously by lentiviral transduction in a previous work by our group. CXCR4 cell expression was assessed before all in vitro and in vivo experiments by flow cytometry as previously described [
16].
2.1.1. Cytotoxic Effect
In vitro cytotoxicity was evaluated using XTT assay (Roche, Basel, Switzerland) following the manufacturer’s instructions; 25,000 cells per well were seeded in 96 well plates and exposed to therapeutic nanoparticles at different concentrations ranging from 1 nM to 1000 nM for 48 h. A competition assay was performed by preincubating the cells with 1 µM CXCR4 antagonist AMD3100 for 1 h, followed by nanoparticle exposure. Absorbance at 490 nm was read (FLUOstar OPTIMA, BMG Labtech, Ortenberg, Germany) and cell viability was quantified and expressed in relation to the viability of buffer-treated cells.
2.1.2. Apoptosis Assessment by Flow Cytometry
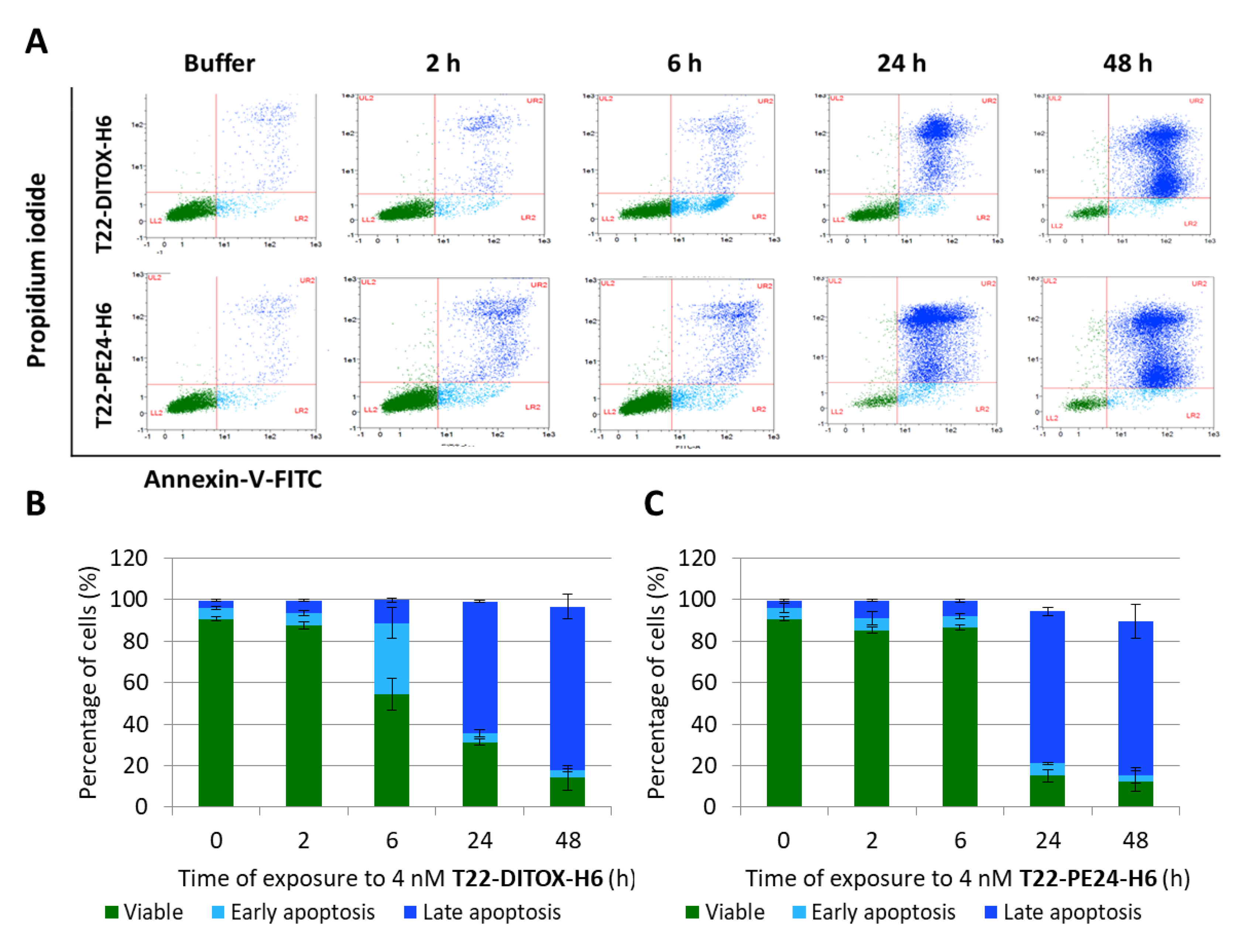
The percentage of cells undergoing early or late apoptosis was quantified using an Annexin V-FITC/propidium iodide (PI) detection kit (Sigma-Aldrich, St. Louis, MO, USA) following the manufacturer’s instructions. Data were analyzed using MACSQuant cytometer and MACS Quantify v2.3 software (Miltenyi Biotec, Bergisch Gladbach, Germany).
2.1.3. Paraffin-Embedded Cell Blocks
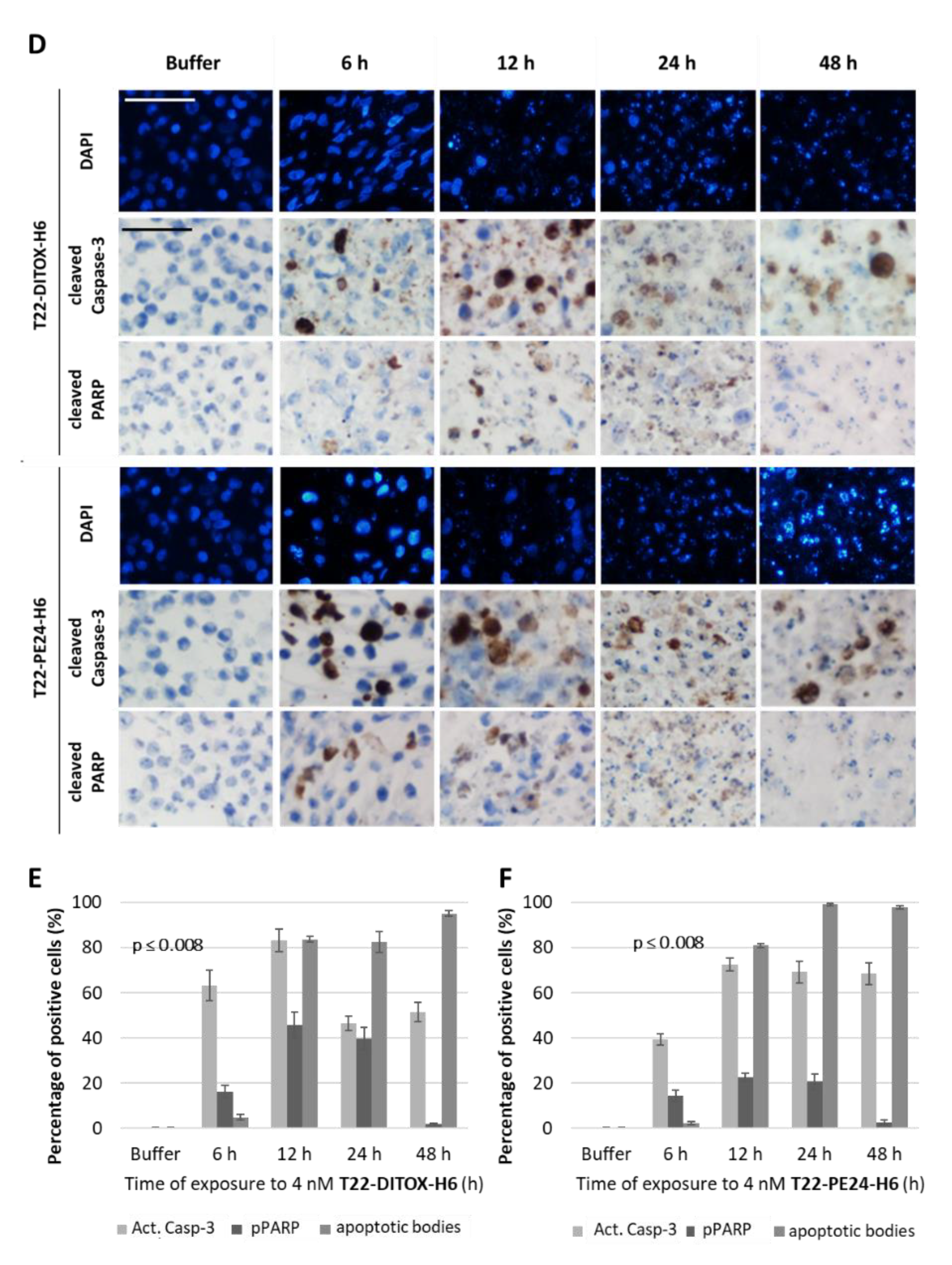
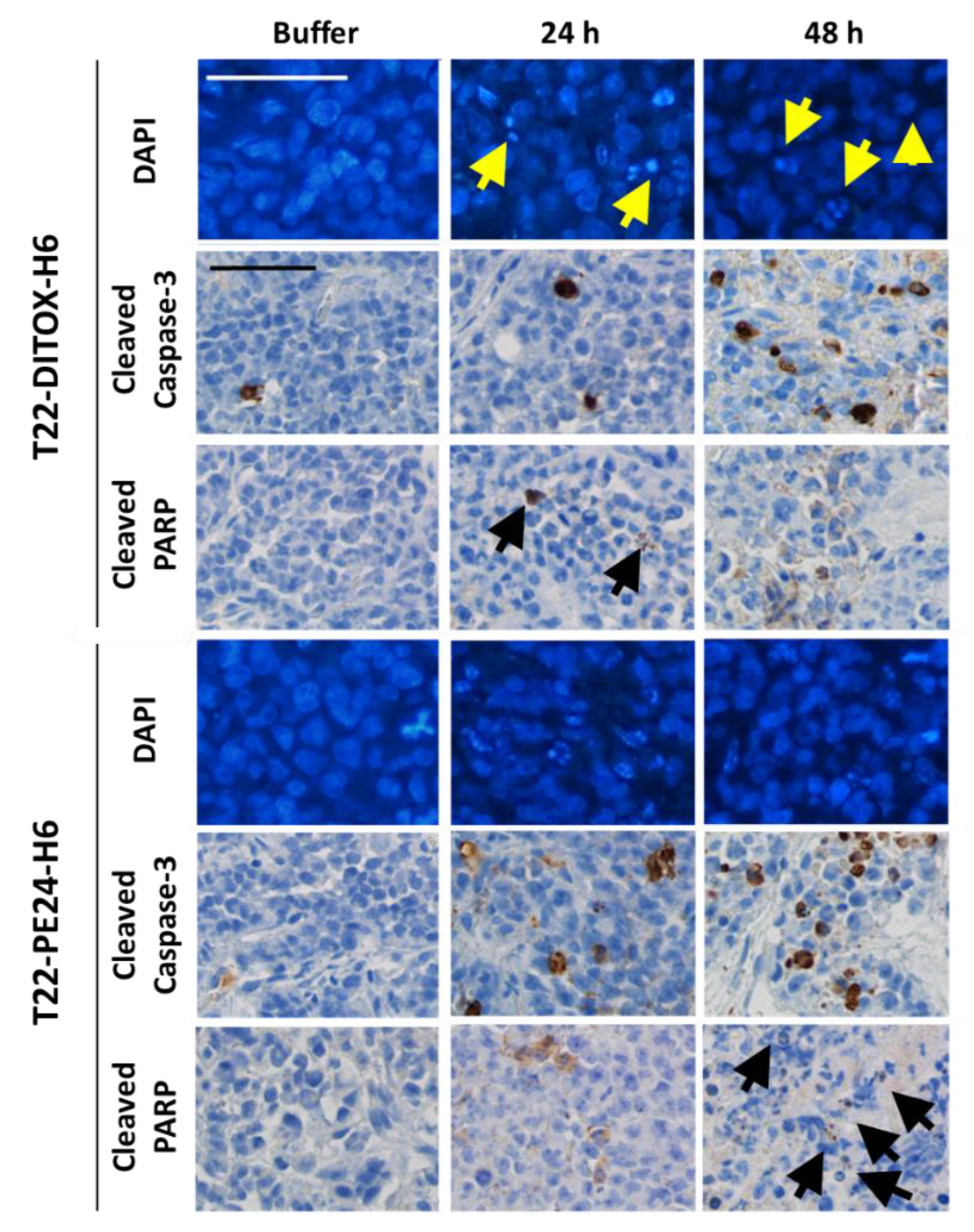
In total, 3.5 × 106 CXCR4+ Luciferase+ AN3CA cells were seeded in 150 cm2 cell culture flasks. Then, 24 h later, they were treated with either buffer or 4 nM of T22-DITOX-H6 or T22-PE24-H6 nanotoxins for 6, 12, 24 or 48 h. Supernatants were collected and attached cells were washed, trypsinized, collected in the supernatant and washed in PBS. Cell blocks were obtained by gently mixing 2 drops of human plasma and 2 drops of human thrombin (Roche). Cell blocks were fixed in paraformaldehyde 4% and embedded in paraffin for further immunocytochemistry or DAPI staining analysis. All slides were examined under an optic or fluorescence microscope (Olympus BX53, Olympus, Tokyo, Japan) and representative pictures were taken using an Olympus DP73 camera.
2.1.4. DAPI Fluorescent Staining
ProLong Gold Antifade Mountant with DAPI (Invitrogen, Waltham, MA, USA) was used to assess DNA condensation and apoptotic cell bodies in paraffin slides of cell blocks. 4 µM slides were incubated at 60 °C for 1 h and rehydrated with 3 × 5 min incubations in xylene and decreasing concentrations of alcohol to 76° to be stained with DAPI.
2.1.5. Immunocytochemistry
Anti-active caspase-3 (1:300; BD Pharmingen, 559565) and anti-cleaved poly (ADP-ribose) polymerase PARP p85 (1:300; Promega, G73411) were used to determine apoptosis induction in cell blocks. Staining was performed in a DAKO Autostainer Link48 (Agilent, Santa Clara, CA, USA) following the manufacturer’s instructions, using 4 μm paraffin sections of cell pellets. All slides were examined by a blind observer under an optic microscope and representative pictures were taken using an Olympus DP73 camera. Positively stained cells were identified using QuPath software v.0.2.3 (Queen’s University, Belfast, Northern Ireland, UK) [
21] at 400× magnification and compared to control samples.
2.2. Animals
Five-week-old female Swiss nude (Crl:NU(Ico)-Foxn1nu) and NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (Charles River, Écully, France) used for the experiments were maintained under specific pathogen-free conditions, being fed with irradiated food (14% protein, 4% fat, Teklad Global diet, ENVIGO) and reverse osmosis autoclaved water ad libitum. Their light/dark cycle was 12 h and they were housed in individually ventilated cage units (Sealsafe Plus GM500, Techniplast, Buguggiate, Italy) in groups of five. All procedures were performed using sterile material in a type II biosafety cabinet (BIO-II-A/P, Telstar, Terrassa, Spain). During surgery, eye dryness was avoided using a saline drip and body temperature was maintained using a heating blanket under a drape.
The correct implantation in vivo of EC cells, either in the subcutaneous or the orthotopic model, was assessed using firefly D-luciferin (Perkin Elmer, Waltham, MA, USA), a substrate for luciferase that is expressed in transduced AN3CA cells. Mice were injected intraperitoneally with 2.25 mg/mouse, and anesthetized with 3% isoflurane in oxygen to capture the intensity of the emitted bioluminescence (BLI) 5 min after injection. Images were analyzed using Living Image v.4.7.3. software (Perkin Elmer, Waltham, MA, USA).
The endpoint criteria included 10–20% body weight loss, signs of pain or distress, such as abnormal postures or development of ulcers, alopecia, ruffled fur, abnormal breathing, abnormal activity, coma, ataxia or tremors. All mice were euthanized by cervical dislocation at the experimental endpoint.
All the in vivo procedures were approved by the Hospital de la Santa Creu i de Sant Pau Animal Ethics Committee and Generalitat de Catalunya (FUE-2018-00819447, 24 April 2019) and performed according to European Council directives (CEA-OH/9721/2).
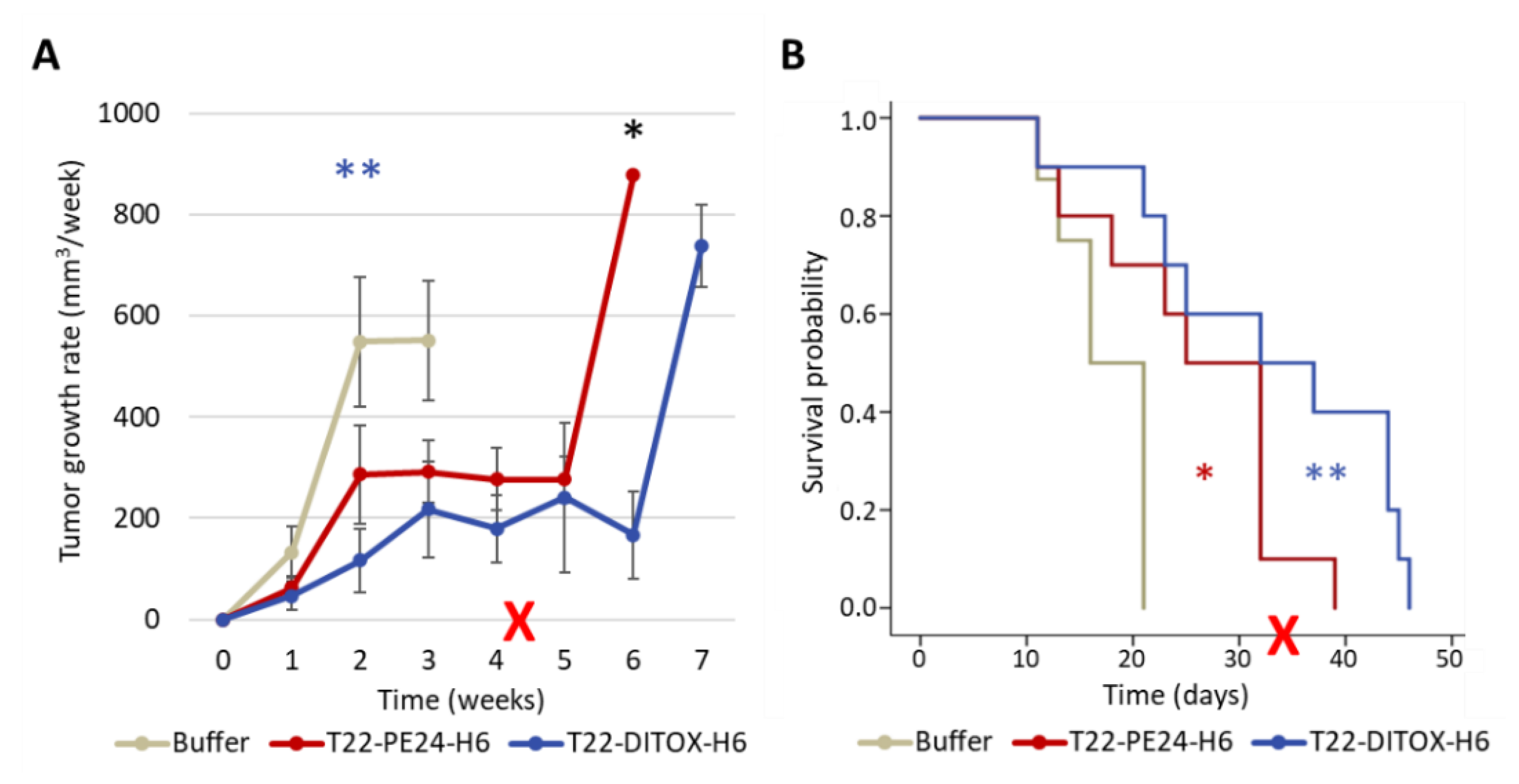
2.2.1. Antitumor Effect of Nanotoxins in a Subcutaneous CXCR4+ EC Model
In total, 40 Swiss nude mice were anesthetized with 2% isofluorane, and the back was swabbed with povidone-iodine. Then, 107 cells were inoculated subcutaneously (SC) in both flanks. Tumor growth was monitored using a caliper and the formula V = width2 × length/2, twice a week.
To assess the effect of each nanotoxin on tumor growth and mice survival, animals were recruited when tumors reached 30–70 mm
3. They were randomized into three groups (
n = 10/group), receiving either 10 µg T22-DITOX-H6, 10 µg T22-PE24-H6 or carbonate buffer as a control, three times a week for a maximum of 15 doses (
Figure S2).
The tumor size endpoint criterium was set at a volume of 900 mm3. In case mice did not reach this criterium after the administration of the 15th dose, they were left untreated until the endpoint.
To determine the activation of protein markers of apoptotic cell death, animals were recruited when tumors reached 150–200 mm3 to receive a single bolus of either T22-DITOX-H6 (30 μg), T22-PE24-H6 (100 μg) or carbonate buffer (n = 2 per group). Mice were euthanized at 24 or 48 h after treatment.
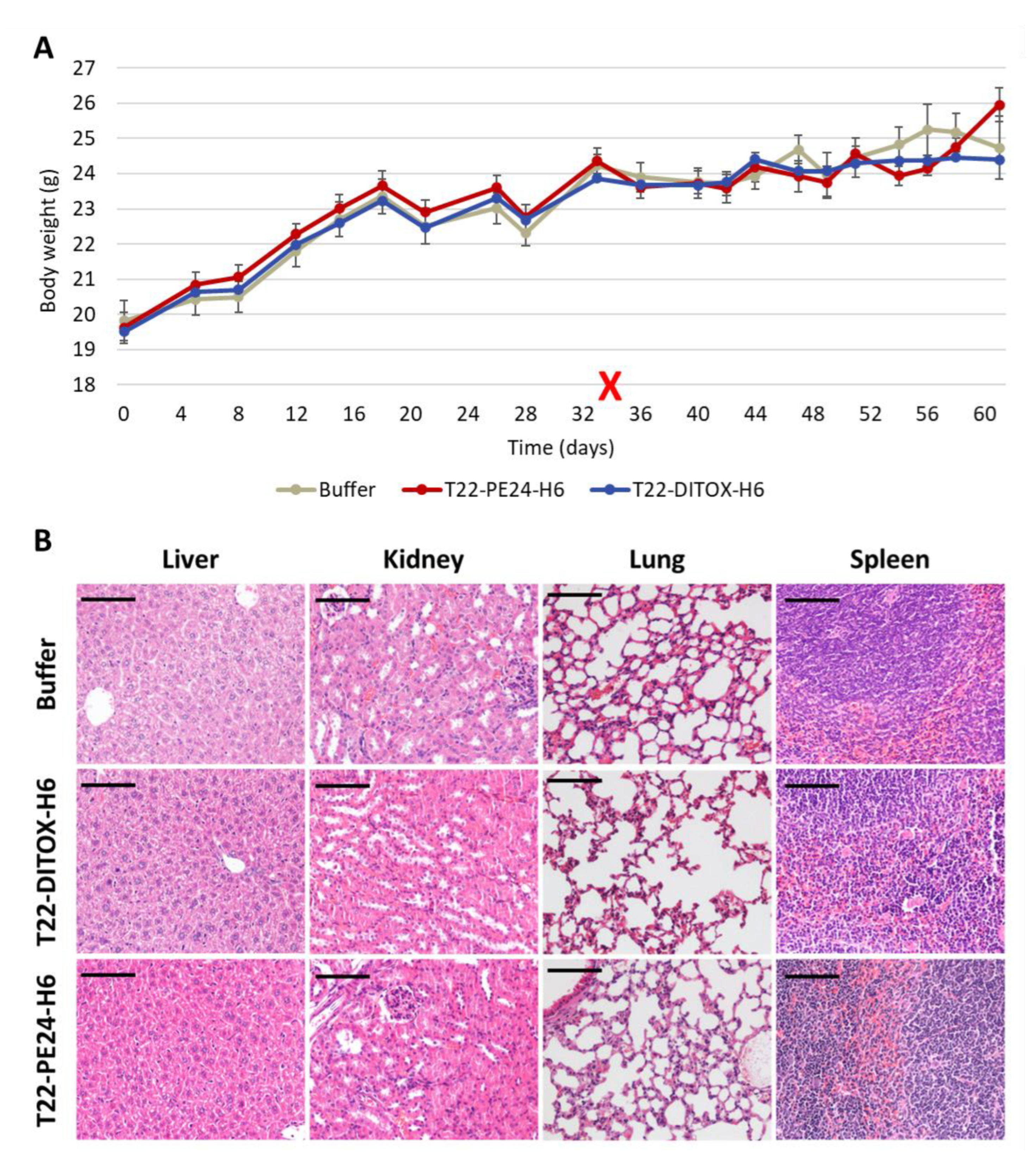
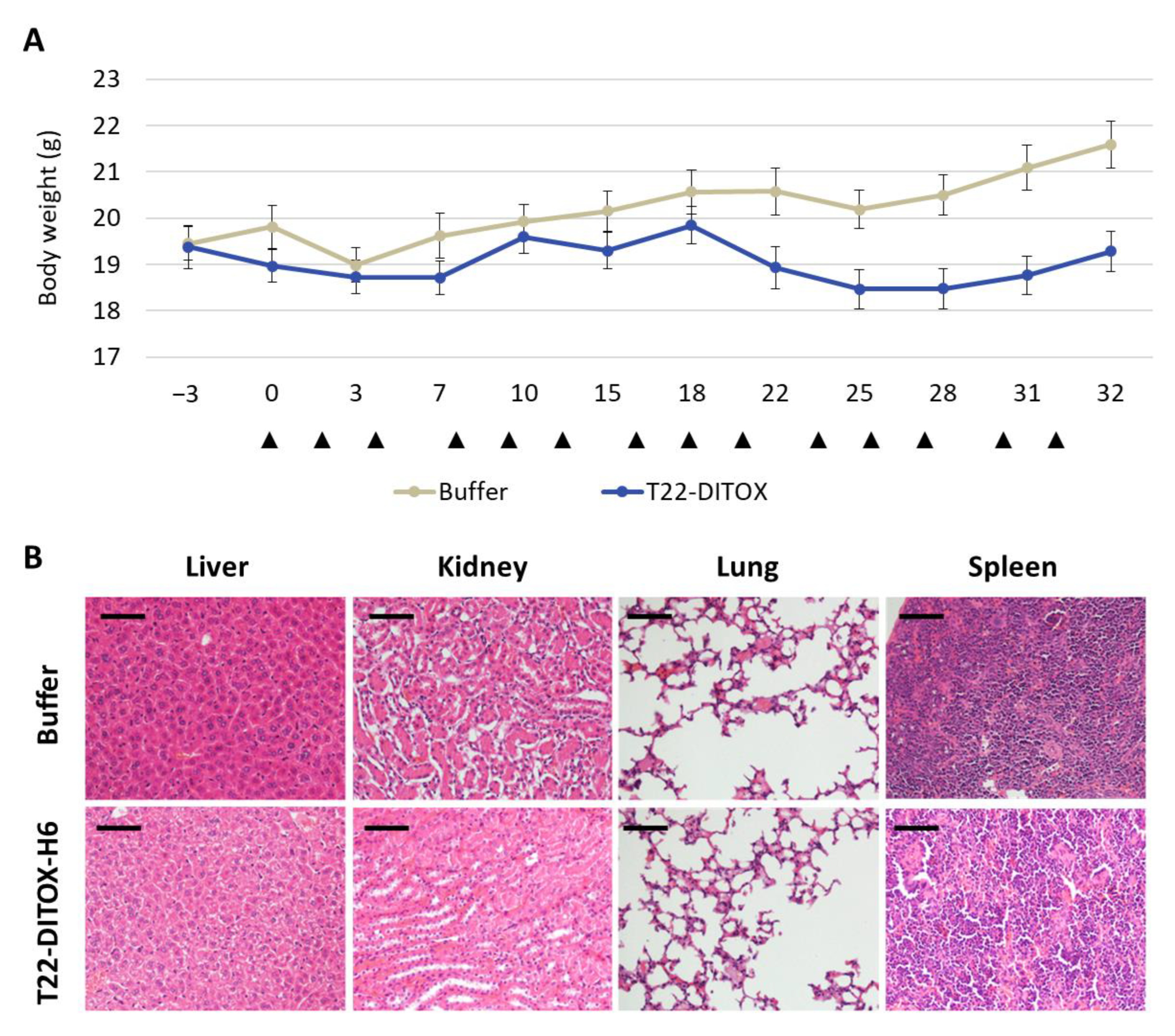
Finally, tumor and non-tumor organs (lung, kidneys, liver, spleen) were collected, fixed in 4% formaldehyde and paraffin-embedded for histopathological or IHC evaluation of metastasis, apoptotic markers and toxicity.
2.2.2. Antimetastatic Effect of T22-DITOX-H6 Using a Highly Metastatic Orthotopic CXCR4+ EC Model
In total, 22 NSG mice were anesthetized with 100 mg/kg ketamine (Ketolar, Pfizer, New York, NY, USA) and 10 mg/kg xylacyn (Rompun, Bayer, Barmen, Germany). Then, 10
6 CXCR4
+ Luciferase
+ AN3CA cells resuspended in 25 uL of culture medium were inoculated through the myometrium into the endometrial cavity using a 29G Hamilton syringe (Microliter Serie 800, Hamilton, Reno, NV, USA), according to the procedure described in [
16].
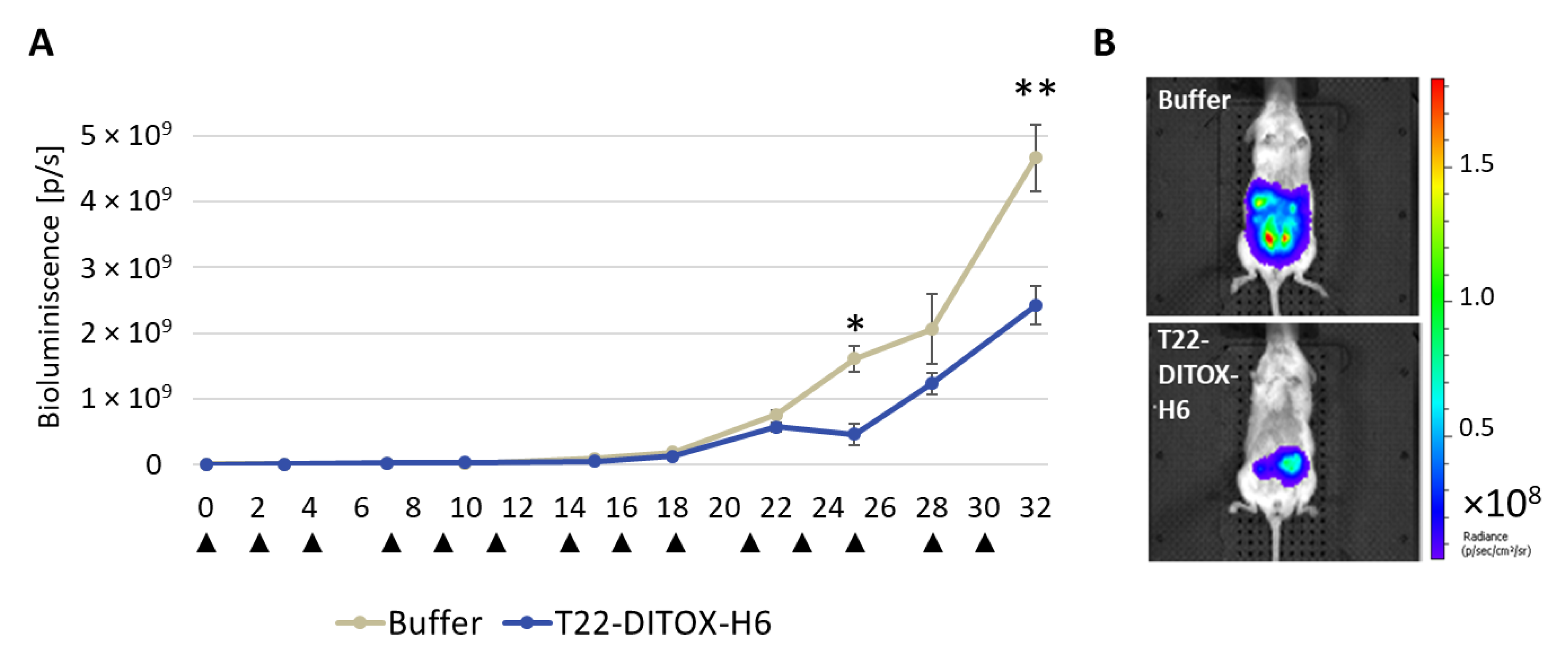
Three days after implantation, cell engraftment and the bioluminescence emission were assessed using IVIS Spectrum 200. Mice were then separated into two groups (
n = 10/group) to receive either 5 µg T22-DITOX-H6 or carbonate buffer as control, three times a week. Whole body BLI was determined twice a week (
Figure S3).
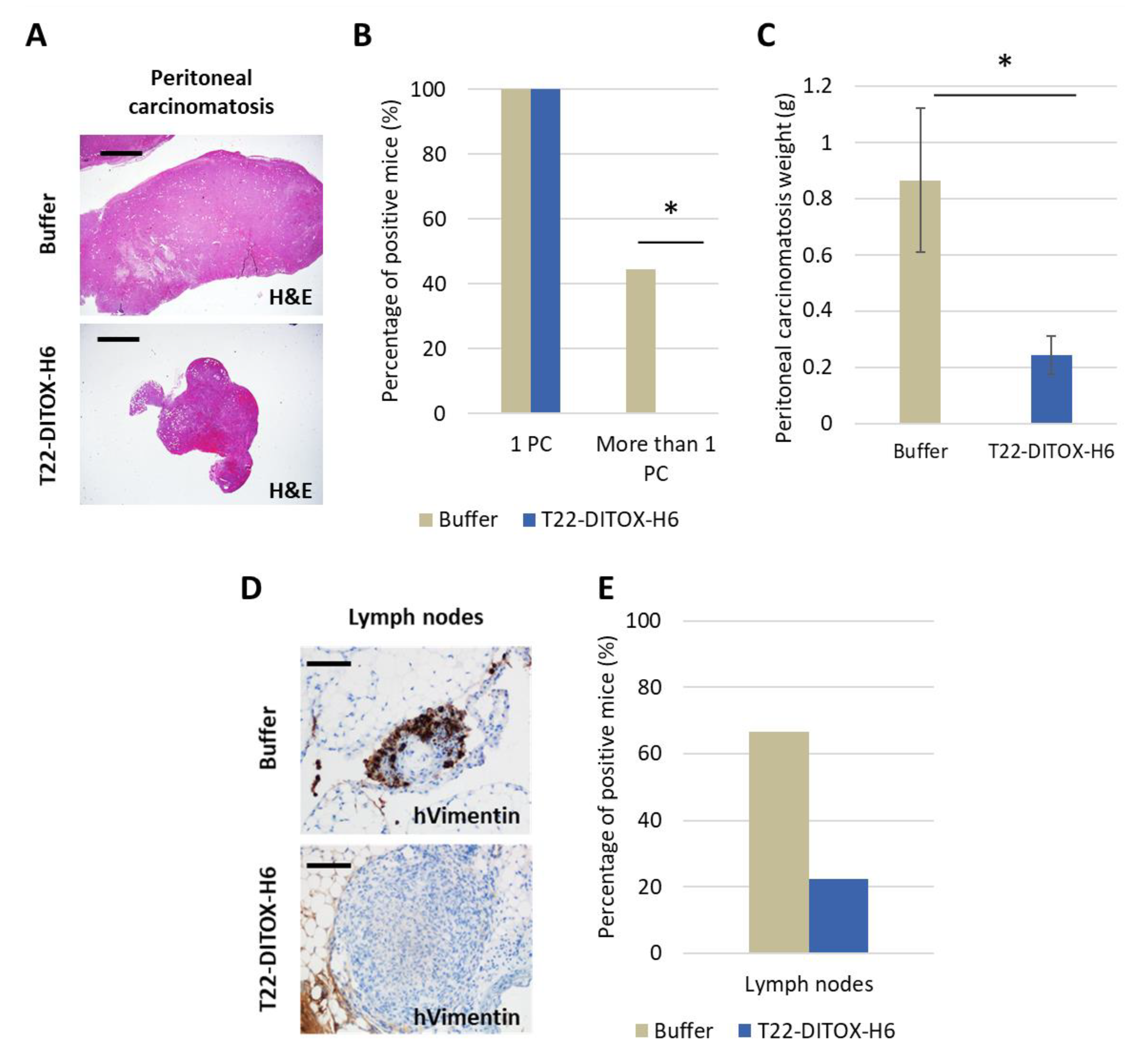
All mice were euthanized when at least one of them reached endpoint criteria, such as a high primary tumor or peritoneal carcinomatosis growth or abdominal distension, which happened two days after the 14th dose. All clinically relevant organs (uterus and ovaries, peritoneal carcinomatosis, lung, kidneys, liver, spleen, abdominal lymph nodes) were collected, fixed in 4% formaldehyde and paraffin-embedded for histopathological analysis, IHC evaluation of metastatic foci and possible toxicity of T22-DITOX-H6 on normal organs.
2.2.3. Histological Examination
Paraffin sections of all organs were stained with hematoxylin-eosin (H&E) and analyzed by two independent blind observers to assess the possible toxicity on non-tumor organs.
2.2.4. Immunohistochemistry
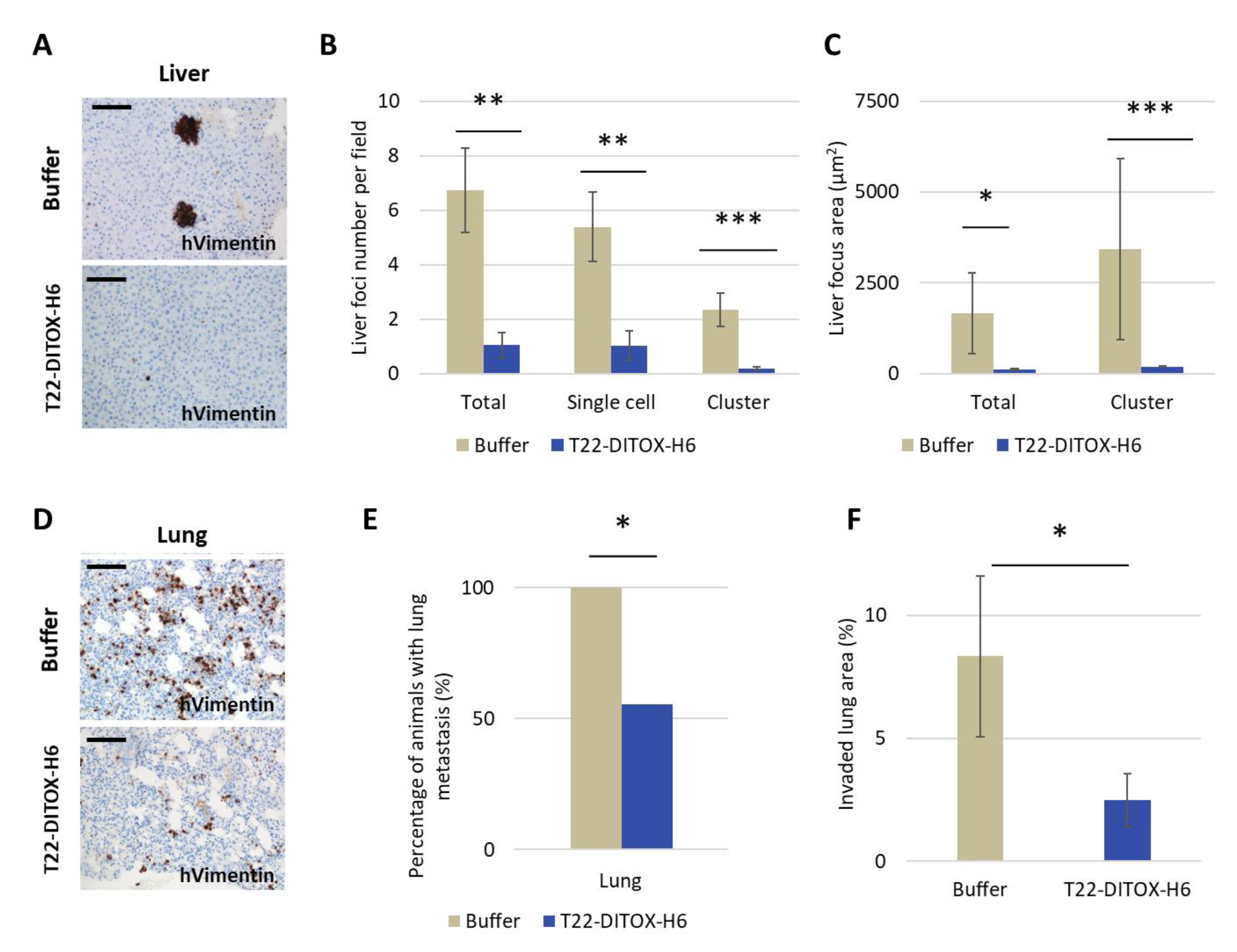
Immunohistochemical staining was performed in a DAKO Autostainer Link48 (Agilent) following the manufacturer’s instructions, using 4 μm paraffin sections of cell pellets or organs. Anti-human vimentin (Dako IGA63061-) was used to determine the presence of tumor cells in clinically relevant organs. Anti-active caspase-3 (1:300; BD Pharmingen, 559565) and anti-cleaved poly (ADP-ribose) polymerase PARP p85 (1:300; Promega, G73411) were used to determine apoptosis induction in vivo.
All slides were examined under an optic microscope and representative pictures were taken using an Olympus DP73 camera. Liver sections were processed with cellSens software (Olympus), while lung sections were analyzed with QuPath, both at 200× magnifications.
2.3. Statistical Analysis
All in vitro experiments were performed in biological triplicates. Mice number for in vivo experiments was defined in preliminary experiments to measure the interindividual variability among mice in terms of tumor growth and metastatic load. Randomizations of animals in experimental and control groups were performed using a dice. Two mice with undetectable orthotopic cell engraftment assessed by bioluminescence were excluded from the experiment. The investigator was not blinded to group allocation during in vivo manipulation of animals; nevertheless, she was blinded for the histological analysis. All results were expressed as mean ± standard error (SE). Differences between groups were analyzed using either the Kruskal–Wallis test, Student T-test or Mann–Whitney test, after the determination of normality by the Shapiro–Wilk test. Differences were considered statistically significant at p ≤ 0.05. Statistical calculations were performed using SPSS software v.23 (IBM, Armonk, NY, USA).
4. Discussion
Women bearing EC at an advanced stage or at high risk of developing locoregional or distal metastasis lack effective treatments, and currently approved drugs lead to poor outcomes and severe adverse effects for EC patients.
We, here, first demonstrated in vitro that our nanotoxins exert a potent and CXCR4-dependent anticancer effect, in CXCR4
+ AN3CA cells (IC
50 = low nanomolar range), as compared to a moderate effect on HEC1A cells showing lower CXCR4 membrane expression. Moreover, both T22-PE24-H6 and T22-DITOX-H6 nanotoxins induced apoptosis, as detected by Annexin V expression and DNA condensation in apoptotic bodies. Interestingly, T22-DITOX-H6 shows higher early apoptosis compared to T22-PE24-H6, a result that is consistent with the faster DITOX path to reach the cytosol, opening a pore in the endosome, whereas PE24 needs to translocate from the endosome to Golgi and endoplasmic reticulum prior to reaching the cytoplasm. Finally, both toxins trigger cell death by blocking protein synthesis through eEF-2 inhibition [
12,
22].
Next, we used two previously developed models by our group, derived from CXCR4
+ EC AN3CA cells, a subcutaneous (SC) and a highly metastatic EC model, which allows the detection of single EC cells invading distant organs because of their expression of human Vimentin [
16]. Thus, repeated intravenous administration of each nanotoxin in the SC EC model showed a highly significant antineoplastic effect, also mediated by apoptosis with DNA condensation and Caspase-3 and PARP cleavage. Moreover, this treatment increased survival compared to control mice and lacked on-target or off-target systemic toxicity; therefore, implying a wide therapeutic window in terms of harm–benefit balance for both nanotoxins.
In addition, repeated intravenous T22-DITOX-H6 administration in the CXCR4+ EC AN3CA orthotopic model induced a potent antimetastatic effect by displaying a reduction in primary tumor growth, and a blockage of metastasis development, again without systemic toxicity. Furthermore, peritoneal metastases were reduced by 50%, while lymph node metastases were diminished by 60%; however, the study of their significance was limited by the direct contact and tissue merging between lymph nodes and peritoneal metastases. On the other hand, T22-DITOX-H6 shrinks 5-fold the number of liver metastases and 10-fold their size compared to control mice. Finally, nanotoxin treatment significantly reduced 8-fold the total area of the lung invaded by metastatic cancer cells. Thus, it is highly likely that this potent antimetastatic effect occurs through the elimination of CXCR4+ cells while they migrate, invade or grow at the colonized organs.
The relevance of our original approach relies on its high therapeutic potency in the absence of systemic toxicity. In contrast, several immunotoxins, in which antibodies targeting HER-2 are bound to PE24 or DITOX, are being tested in early clinical trials that include EC patients; however, they still develop severe side effects [
23]. Moreover, in 2004, a molecular therapy targeting HER-2, incorporating active caspase domains and the cytotoxic domain of P. aeruginosa (the same employed in T22-PE24-H6) was successful in preclinical experiments but, as far as we know, it did not reach clinical trials [
24,
25,
26]. Recently, antitumor the effect of angubindin-1 (derived from
C. perfingens iota toxin) has been evaluated in vitro, but no preclinical in vivo or clinical results have been published to date [
27,
28].
On the other hand, and as previously explained, immunotoxins have been approved only for leukemia or lymphoma, while they have not been effective enough on solid tumors, due to limitations such as low accessibility to tumor cells and their toxicity and immunogenicity [
11]. In contrast, several antibody-drug conjugates (ADCs) are currently being tested in clinical trials for EC to target the Folate receptor-α Tissue factor, HER-2 or Trop-2, which are conjugated to microtubule inhibitors (DM-4, MMAE) or genotoxic agents (deruxetan, SN-38 or Duocarmycin) [
8]. It is relevant to indicate that all of these approaches clearly differ regarding the target cancer cells since our nanotoxins target the CXCR4 receptor.
It is equally important to remark that, instead of delivering microtubule inhibitors or genotoxic agents to the cytosol in the case of ADCs, the T22-DITOX-H6 nanotoxin delivers the exotoxin domain to the cytosol to activate an alternative mechanism of action that consists of the inhibition of eEF-2. This molecular inhibition blocks protein translation to induce cancer cell death not only in dividing tumor cells but also in cancer cells in the Go phase of the cell cycle. This mechanism, never studied in EC patients, could have a clinical impact because of its capacity to prevent metastasis development. Furthermore, no immunotoxins are being tested in EC patients, while the ADCs being tested for EC are still in early clinical phases (I or II), and there are no significant results so far in EC to determine whether or not they are showing activity. However, severe adverse effects that limit the dosage have been reported in these trials [
29,
30,
31], which are consistent with the reports on ADCs that deliver microtubule inhibitors or genotoxic agents and also the fact that only ~0.1% of the ADC administered drug reaches cancer tissues [
32]. All these limitations seem to be overcome by the use of our multivalent display of multiple ligands for the CXCR4 receptor in our nanotoxin, which makes for a highly selective delivery of the bacterial cytotoxin domain to CXCR4
+ EC cells. Therefore, their highly selective delivery is likely to obtain higher cancer tissue uptake and achieve an effective control of tumor growth or dissemination, without on-target or off-target toxicity in normal organs.
In addition, we believe that our nanotoxins could be combined with other drugs already explored for EC therapy, which would include a wider range of candidate patients for treatment. They are PI3K/Akt inhibitors (perifosin, BKM120, temsirolimus or everolimus) or immune checkpoint inhibitors (pembrolizumab, dostarlimab), which have been FDA-approved for microsatellite instability-high and mismatch repair deficient patients; thus, their combination could enhance the current success rate by lowering their dosages and reducing adverse effects. Of note, for the in vivo assessment of nanotoxins, we have used CXCR4
+ mice models derived from the AN3CA cell line, which has described both alterations of the PI3K/Akt pathway and high microsatellite instability [
33]. Furthermore, this is a novel and highly metastatic model that overcomes the low metastatic rates shown in previous models; previously developed anticancer drugs that are currently applied were never tested in EC disseminated models. One major limitation of immunotoxin development is its severe adverse effects. They could be due, in part, to the targeting antibodies that the immunotoxins carry, which display only two binding sites, whereas our nanotoxins have a significantly higher multivalency since they are estimated to reach as far as eleven T22-ligands for the target CXCR4 receptor. On the other hand, being aware of the immunogenicity associated with immunotoxicity treatment, we plan to carry out further in vivo experiments using immunocompetent mice strains to better evaluate if the nanotoxins induce the activation of the immune system to increase the antitumor effect and whether they trigger antibodies against the toxin that could neutralize their effectiveness, before the initiation of their clinical translation. In summary, and based on all described results, we propose that the nanotoxin T22-DITOX-H6, and also most likely T22-PE24-H6, emerge as promising therapeutic candidates for their further preclinical and regulatory development to be performed before entering clinical trials in advanced CXCR4
+ EC patients, which could improve the low selectivity of current treatments for this aggressive illness.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









