
BRAF Inhibitors in Non-Small Cell Lung Cancer
 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
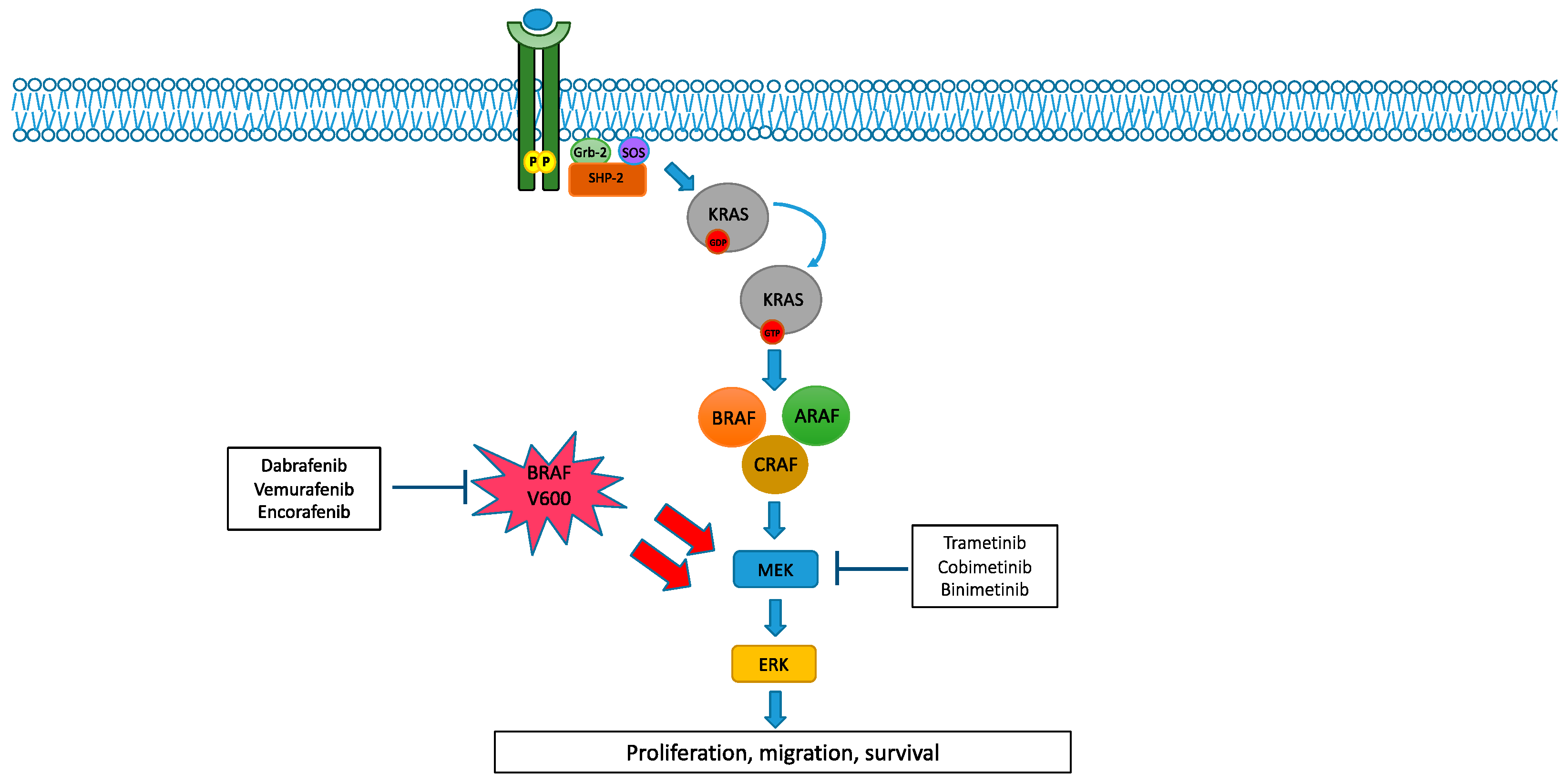
2. MAPK Pathway and Its Alterations
2.1. BRAF and MEK Alterations in NSCLC
2.2. BRAF and MEK Inhibitors in NSCLC
3. Discussion

{kind=link}
| Trial | Phase | Setting | Stage | Pts | Treatment | Primary Endpoints |
|---|---|---|---|---|---|---|
| NCT04526782 (ENCO-BRAF Trial) [59] | Phase 2 | Naïve or subsequent lines (two different arms) | Extensive stage | 119 | Encorafenib + Binimetinib | ORR |
| NCT03915951 [60] | Phase 2 | Any line of treatment | Extensive stage | 97 | Encorafenib + binimetinib | ORR |
| NCT04585815 (Landscape 1011 trial) [62] | Phase 1/2 | Any line of treatment | Extensive stage | 375 | Sasalnimab + encorafenib + binimetinib | Durable objective response rate |
| NCT04591431 (ROME trial) [63] | Phase 2 | Subsequent lines (no more than two treatments are allowed) | Extensive stage | 384 | Vemurafenib + cobimetinib vs. SOC * | ORR |
| NCT03178552 (BFAST Trial) [64] | Phase 2/3 | First line | Extensive stage | 1000 | Vemurafenib + Cobimetinib+ Atezolizumab | Time in response (TIR) |
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, L.K.; Hindley, A.D.; O’Neill, E.; Kolch, W. Regulation and Role of Raf-1/B-Raf Heterodimerization. Mol. Cell. Biol. 2006, 26, 2262–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; van Schil, P.E.; Hellmann, M.D.; et al. ESMO Guidelines Committee. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, J.R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Papint, C.; Denouel-Galy, A.; Laugier, D.; Caloty, G.; Eychene, A. Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B-Raf. J. Biol. Chem. 1998, 273, 24939–24947. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, J.R. MEK1/2 dual-specificity protein kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2012, 417, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, S.; Yang, S.; Wu, X.; Li, H.; Wang, Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF–MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Danker, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutation. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Alvarez, J.G.B.; Otterson, G.A. Agents to treat BRAF-mutant lung cancer. Drugs Context 2019, 8, 212566. [Google Scholar] [CrossRef]
- Negrao, M.V.; Raymond, V.M.; Lanman, R.B.; Robichaux, J.P.; He, J.; Nilsson, M.B.; Ng, P.K.S.; Amador, B.E.; Roarty, E.B.; Nagy, R.J.; et al. Molecular landscape of BRAF-Mutant NSCLC reveals an association between clonality and driver mutations and identifies targetable non-V600 driver mutations. J. Thorac. Oncol. 2020, 15, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardarella, S.; Ogino, A.; Nishino, M.; Butaney, M.; Shen, J.; Lydon, C.; Yeap, B.Y.; Sholl, L.M.; Johnson, B.E.; Janne, P.J. Clinical, pathological and biological features associated with BRAF mutations in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2013, 19, 4532–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustugun, O.T.; Khattak, A.M.; Tromborg, A.K.; Beigi, M.; Beiske, K.; Lund-Iversen, M.; Helland, A. BRAF-mutations in non-small cell lung cancer. Lung Cancer 2014, 84, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Facchinetti, F.; Rossi, G.; Minari, R.; Conti, A.; Friboulet, L.; Tiseo, M.; Planchard, D. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat. Rev. 2018, 66, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Felicioni, L.; Malatesta, S.; Sciarrotta, M.G.; Guetti, L.; Chella, A.; Viola, P.; Pullara, C.; Mucilli, F.; Buttitta, F. Clinical features and outcome of patients with Non–Small-Cell Lung Cancer harboring BRAF mutations. J. Clin. Oncol. 2011, 29, 3574–3579. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, L.Q.; Huang, J.F.; Liu, K.; Chuai, Z.R.; Yang, Z.; Wang, Y.X.; Shi, D.C.; Liu, Q.; Huang, Q.; et al. BRAF mutations in patients with Non-Small Cell Lung Cancer: A systematic review and meta-Analysis. PLoS ONE 2014, 9, e101354. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H. The association between BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J. Transl. Med. 2019, 17, 298. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, D.P.; Dagogo-Jack, I.; Chen, T.; Padole, A.; Shepherd, J.O.A.; Shaw, A.T.; Digumarthy, S.R. Imaging characteristics of BRAF-mutant non-small cell lung cancer by functional class. Lung Cancer 2019, 129, 80–84. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Martinez, P.; Yeap, B.Y.; Ambrogio, C.; Ferris, L.A.; Lydon, C.; Nguen, T.; Jessop, N.A.; Iafrate, A.J.; Johnson, B.E.; et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant Lung Cancer. Clin. Cancer Res. 2018, 25, 158–165. [Google Scholar] [CrossRef]
- Frisone, D.; Friedlaender, A.; Malapelle, U.; Banna, G.; Addeo, A. A BRAF new world. Crit. Rev. Oncol. Hematol. 2020, 152, 103008. [Google Scholar] [CrossRef]
- Passaro, A.; Attili, I.; Rappa, A.; Vacirca, D.; Ranghiero, A.; Fumagalli, C.; Guarize, J.; Spaggiari, L.; de Marinis, F.; Barberis, M.; et al. Genomic characterization of concurrent alterations in Non-Small Cell Lung Cancer (NSCLC) harboring actionable mutations. Cancers 2021, 13, 2172. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhao, C.; Li, J.; Su, C.; Chen, X.; Ren, S.; Li, X.; Zhou, C. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med. 2019, 8, 1858–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martorell, P.M.; Huerta, M.; Quilis, A.C.; Abellan, R.; Seda, E.; Blesa, S.; Chaves, F.J.; Beltran, D.D.; Keranen, S.R.; Franco, J.; et al. Coexistence of EGFR, KRAS, BRAF, and PIK3CA mutations and ALK rearrangement in a comprehensive cohort of 326 consecutive spanish non squamous NSCLC patients. Clin. Lung Cancer 2017, 18, e395–e402. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Yang, J.C.H. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thor. Oncol. 2016, 12, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Dudnik, E.; Peled, N.; Nechushtan, H.H.; Wollner, M.; Onn, A.; Agbarya, A.; Moskovitz, M.; Keren, S.; Popovits-Hadari, N.; Urban, D.; et al. BRAF mutant lung cancer: Programmed Death Ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitor. J. Thor. Oncol. 2018, 13, 1128–1137. [Google Scholar] [CrossRef] [Green Version]
- Rihawi, K.; Giannarelli, D.; Galetta, D.; Delmonte, A.; Giavarra, M.; Turci, D.; garassino, M.; Tiseo, M. BRAF Mutant NSCLC and immune checkpoint inhibitors: Results from a real-world experience. J. Thor. Oncol. 2019, 14, e57–e59. [Google Scholar] [CrossRef] [Green Version]
- Li, S.D.; martial, A.; Schrock, A.B.; Liu, J.J. Extraordinary clinical benefit to sequential treatment with targeted therapy and immunotherapy of a BRAF V600E and PD-L1 positive metastatic lung adenocarcinoma. Exp. Hematol. Oncol. 2017, 6, 29. [Google Scholar] [CrossRef]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Reddy, V.P.; Gay, L.M.; Elvin, J.A.; Vergilio, J.A.; Suh, J.; Ramkissoon, S.; Daniel, S.; Severson, E.A.; Ali, S.M.; Schrock, A.B.; et al. BRAF fusions in clinically advanced non-small cell lung cancer: An emerging target for anti-BRAF therapies. J. Clin. Oncol. 2017, 35, 9072. [Google Scholar] [CrossRef]
- Marks, J.L.; Gong, Y.; Chitale, D.; Golas, B.; McLellan, M.D.; Kasai, Y.; Ding, L.; Mardis, E.R.; Wilson, R.K.; Soliti, D.; et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008, 68, 5524–5528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcila, M.E.; Drilon, A.; Sylvester, B.E.; Lovly, C.M.; Borsu, L.; Reva, B.; Kris, M.; Solit, D.B.; Ladanyi, M. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin. Cancer Res. 2015, 21, 1935–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Gautschi, O.; Pauli, C.; Strobel, K.; Hirschmann, A.; Printzen, G.; Aebi, S.; Diebold, J. A Patient with BRAF V600E Lung Adenocarcinoma Responding to Vemurafenib. J. Thorac. Oncol. 2012, 7, e23–e24. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Michielin, O.; Zimmermann, S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J. Clin. Oncol. 2013, 31, e341–e344. [Google Scholar] [CrossRef]
- Gautschi, O.; Milia, J.; Cabarrou, B.; Bluthgen, M.V.; Besse, B.; Smit, E.F.; Wolf, J.; Peters, S.; Früh, M.; Koeberle, D.; et al. Targeted therapy for patients with BRAF-mutant lung cancer results from the European EURAF cohort. J. Thorac. Oncol. 2015, 10, 1451–1457. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Subbiah, V.; Gervais, R.; Riely, G.J.; Hollebecque, A.; Blay, J.-Y.; Felip, E.; Schuler, M.; Gonçalves, A.; Italiano, A.; Keedy, V.; et al. Efficacy of Vemurafenib in Patients with Non–Small-Cell Lung Cancer with BRAF V600 Mutation: An Open-Label, Single-Arm Cohort of the Histology-Independent VE-BASKET Study. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Cropet, C.; Montané, L.; Barlesi, F.; Souquet, P.; Quantin, X.; Dubos-Arvis, C.; Otto, J.; Favier, L.; Avrillon, V.; et al. Vemurafenib in non-small-cell lung cancer patients with BRAFV600 and BRAFnonV600 mutations. Ann. Oncol. 2020, 31, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.J.; Smit, E.F.; Groen, H.J.; Kelly, R.J.; et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: A single arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Hashemi, S.M.S.; Mazieres, J.; Kim, T.M.; Quoix, E.; Souquet, P.J.; Barlesi, F.; Baik, C.S.; et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients with BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022, 17, 103–115. [Google Scholar] [CrossRef]
- Robinson, S.D.; O’Shaughnessy, J.A.; Lance, C.C.; Konduri, K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer 2014, 85, 326–330. [Google Scholar] [CrossRef]
- Fernandes, M.G.; Costa, J.; Reis, J.; Jacob, M.; Moura, C.; Machado, J.; Hespanhol, V. OA08.07 BRAF-V600E Advanced lung adenocarcinoma with leptomeningeal (LM) disease treated with vemurafenib. J. Thorac. Oncol. 2017, 12, S274–S275. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role played by signalling pathways in overcoming BRAF inhibitor resistance in melanoma. Int. J. Mol. Sci. 2017, 18, 1527. [Google Scholar] [CrossRef]
- Oberholzer, P.A.; Kee, D.; Dziunycz, P.; Sucker, A.; Kamsukom, N.; Jones, R.; Roden, C.; Chalk, C.J.; Ardlie, K.; Palescandolo, E.; et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J. Clin. Oncol. 2012, 30, 316–321. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The Raf inhibitor paradox: Unexpected consequences of targeted drugs. Cancer Cell 2010, 17, 221–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Approved Drugs—FDA Grants Regular Approval to Dabrafenib and Trametinib Combination for Metastatic NSCLC with BRAF V600E Mutation 2017. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm564331.htm (accessed on 10 November 2017).
- Trametinib in Combination with Dabrafenib Is Indicated for the Treatment of Adult Patients with Advanced Non-Small Cell Lung Cancer with a BRAF V600 Mutation 2017. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/002643/WC500222159.pdf (accessed on 23 November 2017).
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04526782 (accessed on 4 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03915951 (accessed on 4 October 2022).
- Johnson, M.L.; Braiteh, F.; Grilley-Olson, J.E.; Chou, J.; Davda, J.; Forgie, A.; Li, R.; Jacobs, I.; Kazazi, F.; Hu-Lieskovan, S. Assessment of Subcutaneous vs Intravenous Administration of Anti-PD-1 Antibody PF-06801591 in Patients with Advanced Solid Tumors: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2019, 5, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04585815 (accessed on 4 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT04591431 (accessed on 4 October 2022).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03178552 (accessed on 4 October 2022).
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Hong, K.; Streit, M. Molecular characterization of acquired resistance to the BRAF Inhibitor dabrafenib in a patient With BRAF Mutant Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, e41–e42. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.; Lopes, F.; Preece, N.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Whittaker, S.; Saturno, G.; Viros, A.; Pedersen, M.; et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015, 27, 85–96. [Google Scholar] [CrossRef]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.; Rizvi, N.; Wolchok, J.D.; Chan, T.A. Genomic profile, smoking, and response to anti-PD-1 therapy in non-small cell lung carcinoma. Mol. Cell. Oncol. 2016, 3, e1048929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussiotis, V.A. Somatic mutations and immunotherapy outcome with CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2230–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guisier, F.; Dubos-Arvis, C.; Viñas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and Safety of Anti-PD-1 Immunotherapy in Patients with Advanced NSCLC With BRAF, HER2, or MET Mutations or RET Translocation: GFPC 01-2018. J. Thorac. Oncol. 2020, 15, 628–636. [Google Scholar] [CrossRef] [PubMed]

| Study | Author | Setting | Pts | Treatment | Response Rate (%) | Disease Control Rate (%) | Progression-Free Survival (Months) | Overall Survival (Months) | Toxicity |
|---|---|---|---|---|---|---|---|---|---|
| Phase 2 (VE-BASKET study) | Subbiah, 2019 [41] | Advanced solid tumours, Cohort NSCLC BRAFV600 | 62 | Vemurafenib | 37.1 | - | 6.5 | 15.4 | Nausea, hyperkeratosis, decreased appetite, arthralgia, cutaneous SCC |
| Phase 2 (AcSé-BASKET study) | Mazieres, 2020 [42] | Advanced solid tumours, Cohort NSCLC: BRAFnonV600 BRAFV600 | 115 15 100 | Vemurafenib | 0 44.8 | - - | 1.8 5.2 | 5.2 10 | Asthenia, decreased appetite, acneiform dermatitis, nausea and diarrhoea |
| Phase 2 (BRF113928) | Planchard 2016; 2022 [43,44,45] | BRAFV600E advanced NSCLC: Cohort A pretreated Cohort B pretreated Cohort C naïve | 171 78 57 36 | Dabrafenib Dabrafenib+ Trametinib Dabrafenib+ Trametinib | 33 68.4 63.9 | 58 80.7 75 | 5.5 10.2 10.8 | 12.7 18.2 17.3 | Pyrexia, asthenia, hyperkeratosis, decreased appetite, nausea |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sforza, V.; Palumbo, G.; Cascetta, P.; Carillio, G.; Manzo, A.; Montanino, A.; Sandomenico, C.; Costanzo, R.; Esposito, G.; Laudato, F.; et al. BRAF Inhibitors in Non-Small Cell Lung Cancer. Cancers 2022, 14, 4863. https://doi.org/10.3390/cancers14194863
Sforza V, Palumbo G, Cascetta P, Carillio G, Manzo A, Montanino A, Sandomenico C, Costanzo R, Esposito G, Laudato F, et al. BRAF Inhibitors in Non-Small Cell Lung Cancer. Cancers. 2022; 14(19):4863. https://doi.org/10.3390/cancers14194863
Chicago/Turabian StyleSforza, Vincenzo, Giuliano Palumbo, Priscilla Cascetta, Guido Carillio, Anna Manzo, Agnese Montanino, Claudia Sandomenico, Raffaele Costanzo, Giovanna Esposito, Francesca Laudato, and et al. 2022. "BRAF Inhibitors in Non-Small Cell Lung Cancer" Cancers 14, no. 19: 4863. https://doi.org/10.3390/cancers14194863