
Ascorbate Plus Buformin in AML: A Metabolic Targeted Treatment

 , , , , , , and
, , , , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Patient Samples and Controls
2.2. Cell Lines and Cell Culture
2.3. Cell Viability
2.4. Colony Formation Unit Assay
2.5. Western Blot Analysis
2.6. Metabolic Assays
2.7. Cytofluorimetric Analysis
2.8. Statistical Analysis
3. Results
3.1. Metabolic Dependence of Primary AML Blasts
3.2. Metabolic Dependency in AML Cell Lines
3.3. Action of Buformin on OXPHOS Metabolism in AML Cells
3.4. Effect of Ascorbate on Glycolytic Metabolism in AML Cells
3.5. Metabolic Background Influences Apoptotic Response to Metabolic Treatments of AML Cell Lines
3.6. Ascorbate Plus Buformin Combination Treatment Effectively Induces Apoptosis in Primary AML Blasts
3.7. Ascorbate Plus Buformin Inhibits Glycolysis in AML Cells
3.8. Resistance to Ascorbate–Buformin Combined Treatment Depends on Metabolic Plasticity of AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [Green Version]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [Green Version]
- Noguera, N.I.; Hasan, S.K.; Ammatuna, E.; Venditti, A. Editorial: Metabolic Rewiring in Leukemias. Front. Oncol. 2021, 11, 775167. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Alexeev, E.; Nusbacher, N.; Minhajuddin, M.; Stevens, B.M.; Winters, A.C.; Lin, X.; Ashton, J.M.; et al. Subversion of Systemic Glucose Metabolism as a Mechanism to Support the Growth of Leukemia Cells. Cancer Cell 2018, 34, 659–673.e6. [Google Scholar] [CrossRef] [Green Version]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Culp-Hill, R.; Reisz, J.A.; Pei, S.; Gustafson, A.; Khan, N.; DeGregori, J.; Pollyea, D.A.; et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 2019, 134, 389–394. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.-R.; Song, H.-C.; Byun, B.H.; Oh, J.-R.; Kim, H.-S.; Hong, S.-P.; Kwon, S.Y.; Chong, A.; Kim, J.; Cho, S.-G.; et al. Intratumoral Metabolic Heterogeneity for Prediction of Disease Progression After Concurrent Chemoradiotherapy in Patients with Inoperable Stage III Non-Small-Cell Lung Cancer. Nucl. Med. Mol. Imaging 2014, 48, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 1. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic Regulation in Pluripotent Stem Cells during Reprogramming and Self-Renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Presti, C.L.; Fauvelle, F.; Jacob, M.-C.; Mondet, J.; Mossuz, P. The metabolic reprogramming in acute myeloid leukemia patients depends on their genotype and is a prognostic marker. Blood Adv. 2021, 5, 156–166. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [Green Version]
- Smith-Díaz, C.C.; Magon, N.J.; McKenzie, J.L.; Hampton, M.B.; Vissers, M.C.M.; Das, A.B. Ascorbate Inhibits Proliferation and Promotes Myeloid Differentiation in TP53-Mutant Leukemia. Front. Oncol. 2021, 11, 709543. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [Green Version]
- Granger, M.; Eck, P. Dietary Vitamin C in Human Health. Adv. Food Nutr. Res. 2018, 83, 281–310. [Google Scholar] [CrossRef]
- Mastrangelo, D.; Massai, L.; Fioritoni, G.; Coco, F.L.; Noguera, N.; Testa, U. High Doses of Vitamin C and Leukemia: In Vitro Update. In Myeloid Leukemia; Lasfar, A., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Noguera, N.I.; Pelosi, E.; Angelini, D.F.; Piredda, M.L.; Guerrera, G.; Piras, E.; Battistini, L.; Massai, L.; Berardi, A.; Catalano, G.; et al. High-dose ascorbate and arsenic trioxide selectively kill acute myeloid leukemia and acute promyelocytic leukemia blasts in vitro. Oncotarget 2017, 8, 32550–32565. [Google Scholar] [CrossRef] [Green Version]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, D.; Massai, L.; Coco, F.L.; Noguera, N.I.; Borgia, L.; Fioritoni, G.; Berardi, A.C.; Iacone, A.; Muscettola, M.; Pelosi, E.; et al. Cytotoxic effects of high concentrations of sodium ascorbate on human myeloid cell lines. Ann. Hematol. 2015, 94, 1807–1816. [Google Scholar] [CrossRef]
- Fiorani, M.; De Sanctis, R.; Scarlatti, F.; Vallorani, L.; De Bellis, R.; Serafini, G.; Bianchi, M.; Stocchi, V. Dehydroascorbic acid irreversibly inhibits hexokinase activity. Mol. Cell. Biochem. 2000, 209, 145–153. [Google Scholar] [CrossRef]
- Heneberg, P. Redox Regulation of Hexokinases. Antioxid. Redox Signal. 2019, 30, 415–442. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin Is an AMP Kinase–Dependent Growth Inhibitor for Breast Cancer Cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Wahdan-Alaswad, R.S.; Cochrane, D.R.; Spoelstra, N.S.; Howe, E.N.; Edgerton, S.M.; Anderson, S.M.; Thor, A.D.; Richer, J.K. Metformin-Induced Killing of Triple-Negative Breast Cancer Cells Is Mediated by Reduction in Fatty Acid Synthase via miRNA-193b. Horm. Cancer 2014, 5, 374–389. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, E.A.I.F.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.H.; Fortes, Z.B.; Khaper, N. Metformin Induces Apoptosis and Cell Cycle Arrest Mediated by Oxidative Stress, AMPK and FOXO3a in MCF-7 Breast Cancer Cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A.; et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int. J. Cancer 2015, 136, E534–E544. [Google Scholar] [CrossRef]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin Prevents Tobacco Carcinogen–Induced Lung Tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Liu, Z.; Jiang, L.; Liu, M.; Ma, J.; Yang, C.; Han, L.; Nan, K.; Liang, X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol. Med. Rep. 2016, 13, 2590–2596. [Google Scholar] [CrossRef] [Green Version]
- Tomic, T.; Botton, T.; Cerezo, M.; Robert, G.; Luciano, F.; Puissant, A.; Gounon, P.; Allegra, M.; Bertolotto, C.; Bereder, J.-M.; et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011, 2, e199. [Google Scholar] [CrossRef] [Green Version]
- Janjetovic, K.; Harhaji-Trajkovic, L.; Misirkic-Marjanovic, M.; Vucicevic, L.; Stevanovic, D.; Zogovic, N.; Sumarac-Dumanovic, M.; Micic, D.; Trajkovic, V. In vitro and in vivo anti-melanoma action of metformin. Eur. J. Pharmacol. 2011, 668, 373–382. [Google Scholar] [CrossRef]
- Bhat, M.; Yanagiya, A.; Graber, T.; Razumilava, N.; Bronk, S.; Zammit, D.; Zhao, Y.; Zakaria, C.; Metrakos, P.; Pollak, M.; et al. Metformin requires 4E-BPs to induce apoptosis and repress translation of Mcl-1 in hepatocellular carcinoma cells. Oncotarget 2017, 8, 50542–50556. [Google Scholar] [CrossRef] [Green Version]
- Kilgore, J.; Jackson, A.L.; Clark, L.H.; Guo, H.; Zhang, L.; Jones, H.M.; Gilliam, T.P.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. Buformin exhibits anti-proliferative and anti-invasive effects in endometrial cancer cells. Am. J. Transl. Res. 2016, 8, 2705–2715. [Google Scholar]
- Careccia, S.; Mainardi, S.; Pelosi, A.; Gurtner, A.; Diverio, D.; Riccioni, R.; Testa, U.; Pelosi, E.; Piaggio, G.; Sacchi, A.; et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 2009, 28, 4034–4040. [Google Scholar] [CrossRef] [Green Version]
- Banella, C.; Ginevrino, M.; Catalano, G.; Fabiani, E.; Falconi, G.; Divona, M.; Curzi, P.; Panetta, P.; Voso, M.T.; Noguera, N.I. Absence of FGFR3–TACC3 rearrangement in hematological malignancies with numerical chromosomal alteration. Hematol. Oncol. Stem Cell Ther. 2021, 14, 163–168. [Google Scholar] [CrossRef]
- Masciarelli, S.; Capuano, E.; Ottone, T.; Divona, M.; De Panfilis, S.; Banella, C.; Noguera, N.I.; Picardi, A.; Fontemaggi, G.; Blandino, G.; et al. Retinoic acid and arsenic trioxide sensitize acute promyelocytic leukemia cells to ER stress. Leukemia 2018, 32, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Noguera, N.I.; Piredda, M.L.; Taulli, R.; Catalano, G.; Angelini, G.; Gaur, G.; Nervi, C.; Voso, M.T.; Lunardi, A.; Pandolfi, P.P.; et al. PML/RARa inhibits PTEN expression in hematopoietic cells by competing with PU.1 transcriptional activity. Oncotarget 2016, 7, 66386–66397. [Google Scholar] [CrossRef] [Green Version]
- Quattrocchi, A.; Maiorca, C.; Billi, M.; Tomassini, S.; De Marinis, E.; Cenfra, N.; Equitani, F.; Gentile, M.; Ceccherelli, A.; Banella, C.; et al. Genetic lesions disrupting calreticulin 3′-untranslated region in JAK2 mutation-negative polycythemia vera. Am. J. Hematol. 2020, 95, E263–E267. [Google Scholar] [CrossRef]
- Banella, C.; Catalano, G.; Travaglini, S.; Divona, M.; Masciarelli, S.; Guerrera, G.; Fazi, F.; Lo-Coco, F.; Voso, M.T.; Noguera, N.I. PML/RARa Interferes with NRF2 Transcriptional Activity Increasing the Sensitivity to Ascorbate of Acute Promyelocytic Leukemia Cells. Cancers 2019, 12, 95. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, I.; Kohno, B. 18 FDG-PET/CT: 21st century approach to leukemic tumors in 124 cases. Am. J. Hematol. 2016, 91, 379–384. [Google Scholar] [CrossRef]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Sarrazy, V.; Viaud, M.; Westerterp, M.; Ivanov, S.; Giorgetti-Peraldi, S.; Guinamard, R.; Gautier, E.L.; Thorp, E.B.; De Vivo, D.C.; Yvan-Charvet, L. Disruption of Glut1 in Hematopoietic Stem Cells Prevents Myelopoiesis and Enhanced Glucose Flux in Atheromatous Plaques of ApoE(-/-) Mice. Circ. Res. 2016, 118, 1062–1077. [Google Scholar] [CrossRef] [Green Version]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.L.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Leukaemia: A pre-leukaemic reservoir. Nat. Rev. Cancer 2014, 14, 212. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Scotland, S.; Saland, E.; Skuli, N.; De Toni, F.; Boutzen, H.; Micklow, E.; Sénégas, I.; Peyraud, R.; Peyriga, L.; Theodoro, F.; et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013, 27, 2129–2138. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [Green Version]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Rampal, R.; Akalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA Hydroxymethylation Profiling Reveals that WT1 Mutations Result in Loss of TET2 Function in Acute Myeloid Leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk. Res. 2018, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
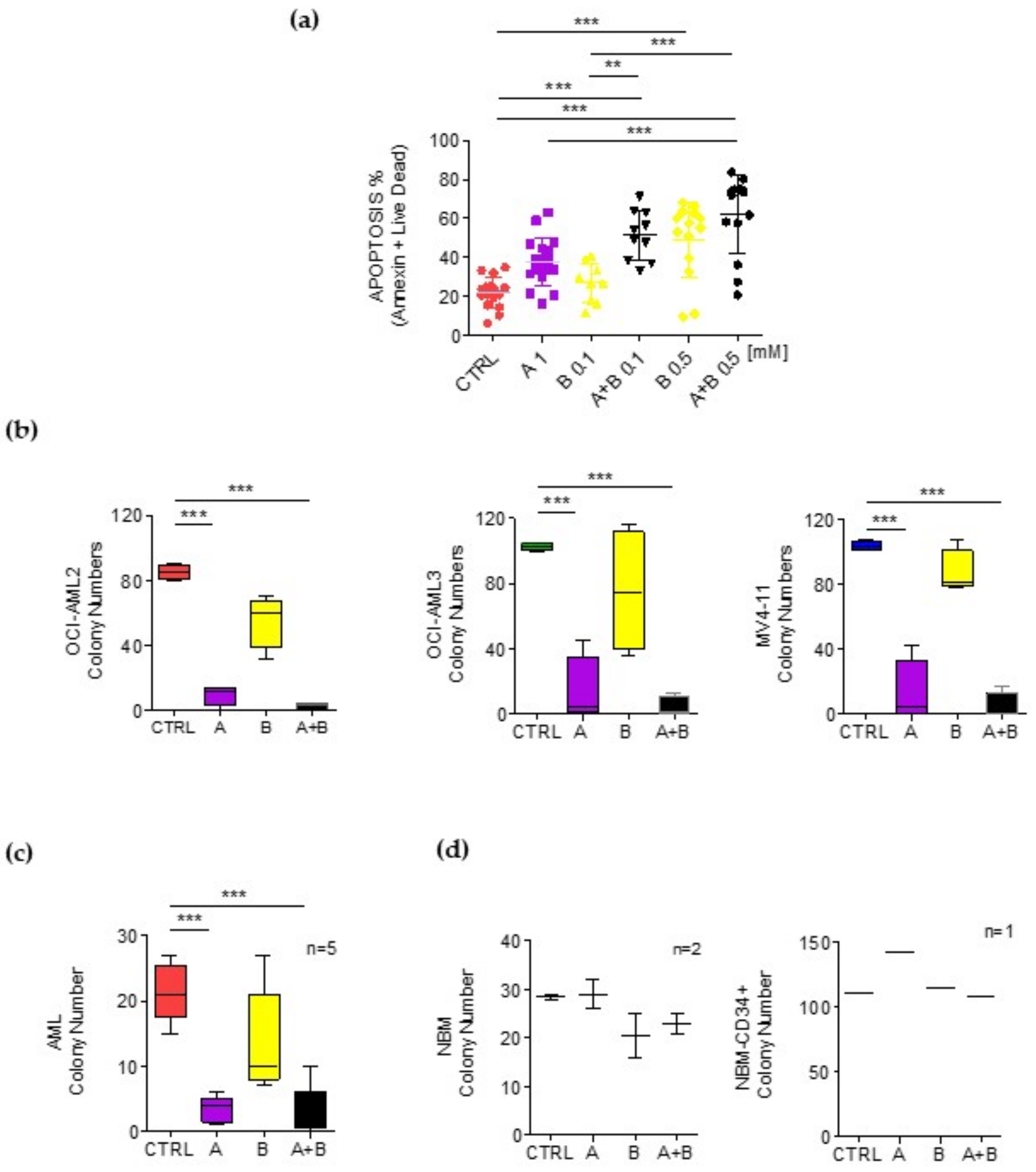{kind=link}
{kind=link}
| N° | Age | Sex | Molecular Biology | Cytogenetic |
|---|---|---|---|---|
| * 1 | 71 | F | Negative panel | 46, XX |
| 2 | 64 | F | NPM1; FLT3-ITD (R 0.36) | NA |
| 3 | 79 | F | NPM1 | FISH negative (chr 5, 7, 8, 11, 20) |
| 4 | 61 | M | Negative panel | 46, XY |
| 5 | 81 | F | NPM1; FLT3-ITD (R 0.33) | NA |
| 6 | 77 | M | PLZF/RARa | 46, XY, t (11; 17) (q23; q21) |
| 7 | 76 | F | NPM1; FLT3-ITD (R 0.22) | 46, XX |
| 8 | 75 | F | FLT3-TKD (AR:0.5) | NA |
| 9 | 74 | M | Negative panel | 46, XY |
| 10 | 51 | M | NPM1 | FISH negative (chr 5, 7, 8, 11, 20) |
| 11 | 69 | M | Negative panel | 46, XY, t (4; 16) |
| 12 | 60 | M | NPM1; FLT3-ITD (R 1.67) | 46, XY |
| 13 | 78 | M | NPM1; FLT3-ITD (R 0.58) | NA |
| 14 | 41 | F | MPM1; FLT3-ITD | NA |
| 15 | 53 | F | Negative panel | NA |
| 16 | 75 | F | NPM1; FLT3-ITD (R 0.67) | 46, XX |
| 17 | 45 | M | Negative panel | 46, XX |
| GlycolysisECAR (mpH/min/105 Cells) | AML | EP/P (N7) | EP/P (N13) | NBM | p-Value (AML vs. EP/P, N7) |
|---|---|---|---|---|---|
| Basal | 19 ± 19 | 37 ± 6 | 18 ± 4 | 3 ± 0 | - |
| Capacity | 22 ± 20 | 41 ± 10 | 28 ± 7 | 4 ± 1 | - |
| Reserve | 3 ± 2 | 10 ± 3 | 11 ± 5 | 0.5 ± 0.7 | 0.03 |
| Mitochondrial Respiration OCR (pmol/min/105 Cells) | AML | EP/P (N7) | EP/P (N13) | NBM | p-Value (AML vs. EP/P) |
|---|---|---|---|---|---|
| Basal | 40 ± 24 | 50 ± 12 | 24 ± 16 | 9 ± 1 | - |
| Spare Respiratory Capacity | 76 ± 25 | 153 ± 54 | 56 ± 56 | 23 ± 4 | 0.02 |
| Proton Leak | 18 ± 16 | 10 ± 2 | 5 ± 1 | 0.5 ± 0.7 | - |
| ATP | 23 ± 16 | 41 ± 10 | 20 ± 16 | 8 ± 1 | - |
| Glycolysis ECAR (mpH/min/105 Cells) | OCI- AML2 | OCI- AML3 | MV4-11 | p-Value | p-Value |
|---|---|---|---|---|---|
| Basal | 96 ± 10 | 104 ± 12 | 56 ± 5 | <0.5 (Oci2 vs. MV4-11) | <0.005 (OCI3 vs. MV4-11) |
| Capacity | 104 ± 31 | 146 ± 64 | 83 ± 10 | - | <0.005 (OCI3 vs. MV4-11) |
| Reserve | 9 ± 12 | 43 ± 32 | 27 ± 11 | <0.005 (OCI2 vs. OCI3) | <0.05 (OCI2 vs. MV4-11) |
| Mitochondrial Respiration OCR (pmol/min/105 Cells) | OCI- AML2 | OCI- AML3 | MV4-11 | p-Value | p-Value |
|---|---|---|---|---|---|
| Basal | 106 ± 31 | 97 ± 18 | 160 ± 27 | <0.05 MV4-11 vs. OCI3 | - |
| Spare Respiratory Capacity | 103 ± 21 | 180 ± 47 | 116 ± 15 | <0.005 OCI3 vs. OCI2 | - |
| Proton Leak | 19 ± 6 | 22 ± 5 | 34 ± 4 | <0.005 MV4-11 vs. OCI2 | <0.05 MV4-11 vs. OCI3 |
| ATP | 87 ± 26 | 75 ± 14 | 141 ± 13 | <0.05 MV4-11 vs. OCI2 | <0.005 MV4-11 vs. OCI3 |
| OCI-AML2 | OCI-AML3 | MV4-11 | p-Value (MV4-11 vs. Oci2) | p-Value (MV4-11 vs. Oci3) | |
|---|---|---|---|---|---|
| CT2 | 0.4 ± 0.2 | 0.5 ± 0.3 | 1.5 ± 0.6 | <0.0005 | <0.0005 |
| CPT1A | 0.5 ± 0.4 | 0.1 ± 0.03 | 2.2 ± 0.2 | <0.0005 | <0.0005 |
| OCI-AML2 | OCI-AML3 | MV4-11 | AML | |
|---|---|---|---|---|
| Ctrl | 3 ± 1 | 3 ± 1 | 6 ± 1 | 22 ± 8 |
| Ascorbate 1 mM | 6 ± 3 | 22 ± 13 | 8 ± 1 | 38 ± 12 |
| Buformin 0.1 mM | 11 ± 2 | 10 ± 1 | 14 ± 1 | 27 ± 10 |
| Ascorbate–buformin 0.1 mM | 18 ± 5 | 33 ± 9 | 14 ± 1 | 51 ± 13 |
| Buformin 0.5 mM | 7 ± 4 | 3 ± 2 | 27 ± 15 | 49 ± 19 |
| Ascorbate–buformin 0.5 mM | 14 ± 8 | 72 ± 6 | 28 ± 10 | 62 ± 20 |
| Ctrl | Ascorbate 1 mM | Buformin (0.1 mM) | Ascorbate–Buformin | |
|---|---|---|---|---|
| OCI-AML2 | 103 ± 3 | 11 ± 9 | 58 ± 16 | 2 ± 3 |
| OCI-AML3 | 102 ± 2 | 14 ± 21 | 75 ± 38 | 4 ± 6 |
| MV4-11 | 104 ± 2 | 13 ± 20 | 87 ± 14 | 4 ± 8 |
| AML blasts | 21 ± 5 | 3 ± 2 | 14 ± 8 | 3 ± 4 |
| N-BMC | 29 ± 4 | 29 ± 4 | 21 ± 6 | 23 ± 3 |
| CD34+ | 142 | 142 | 115 | 109 |
| Ctrl | Ascorbate 1 mM | Buformin (0.1 mM) | Ascorbate–Buformin 0.1 | Buformin (0.5 mM) | Ascorbate–Buformin 0.5 | |
|---|---|---|---|---|---|---|
| U937-Mock | 1 ± 0 | 0.81 ± 0.1 | 0.93 ± 0.06 | 0.97 ± 0.05 | 0.85 ± 0.06 | 0.78 ± 0.1 |
| U937-AETO | 1 ± 0 | 0.67 ± 0.1 | 0.84 ± 0.1 | 0.67 ± 0.1 | 0.80 ± 0.1 | 0.48 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banella, C.; Catalano, G.; Travaglini, S.; Pelosi, E.; Ottone, T.; Zaza, A.; Guerrera, G.; Angelini, D.F.; Niscola, P.; Divona, M.; et al. Ascorbate Plus Buformin in AML: A Metabolic Targeted Treatment. Cancers 2022, 14, 2565. https://doi.org/10.3390/cancers14102565
Banella C, Catalano G, Travaglini S, Pelosi E, Ottone T, Zaza A, Guerrera G, Angelini DF, Niscola P, Divona M, et al. Ascorbate Plus Buformin in AML: A Metabolic Targeted Treatment. Cancers. 2022; 14(10):2565. https://doi.org/10.3390/cancers14102565
Chicago/Turabian StyleBanella, Cristina, Gianfranco Catalano, Serena Travaglini, Elvira Pelosi, Tiziana Ottone, Alessandra Zaza, Gisella Guerrera, Daniela Francesca Angelini, Pasquale Niscola, Mariadomenica Divona, and et al. 2022. "Ascorbate Plus Buformin in AML: A Metabolic Targeted Treatment" Cancers 14, no. 10: 2565. https://doi.org/10.3390/cancers14102565