
Regulation of NKG2D Stress Ligands and Its Relevance in Cancer Progression
by
 , and
, and
Amber B. Jones
1, 
Abbey Rocco
2,
Lawrence S. Lamb
3,
Gregory K. Friedman
2 and
Anita B. Hjelmeland
1,* 1
Department of Cell, Developmental and Integrative Biology, University of Alabama at Birmingham, Birmingham, AL 35233, USA
2
Department of Pediatrics, Division of Pediatric Hematology and Oncology, University of Alabama at Birmingham, Birmingham, AL 35233, USA
3
IN8bio Inc., New York, NY 10016, USA
*
Author to whom correspondence should be addressed.
Cancers 2022, 14(9), 2339; https://doi.org/10.3390/cancers14092339
Submission received: 18 March 2022
/
Revised: 22 April 2022
/
Accepted: 26 April 2022
/
Published: 9 May 2022
(This article belongs to the Special Issue Advances in Immuno-Oncology Research)
Abstract
:Simple Summary
Immune surveillance during periods of cellular stress is necessary to maintain homeostasis. In distressed cells, the expression of stress (natural killer group 2 member D (NKG2D)) ligands is induced on cells to promote recognition by cytotoxic immune populations. This process is exemplified during cancer initiation, progression, and maintenance. Unfortunately, cancer cell adaptation yields multiple mechanisms to evade immune cell recognition. Therefore, extensive efforts have been investigated to induce stress ligands on tumor cells to complement immunotherapy efforts. In this review, we provide updates on the current regulatory mechanisms involved with both stress ligand induction and repression and offer areas of consideration as research on this topic progresses.
Abstract
Under cellular distress, multiple facets of normal homeostatic signaling are altered or disrupted. In the context of the immune landscape, external and internal stressors normally promote the expression of natural killer group 2 member D (NKG2D) ligands that allow for the targeted recognition and killing of cells by NKG2D receptor-bearing effector populations. The presence or absence of NKG2D ligands can heavily influence disease progression and impact the accessibility of immunotherapy options. In cancer, tumor cells are known to have distinct regulatory mechanisms for NKG2D ligands that are directly associated with tumor progression and maintenance. Therefore, understanding the regulation of NKG2D ligands in cancer will allow for targeted therapeutic endeavors aimed at exploiting the stress response pathway. In this review, we summarize the current understanding of regulatory mechanisms controlling the induction and repression of NKG2D ligands in cancer. Additionally, we highlight current therapeutic endeavors targeting NKG2D ligand expression and offer our perspective on considerations to further enhance the field of NKG2D ligand biology.
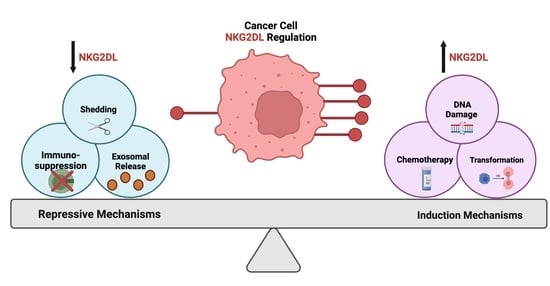
1. Introduction
To maintain normal homeostatic conditions, immune surveillance in the body is necessary to ensure the clearance of damaged, infected, or neoplastically transformed cells [1,2,3,4]. However, dysregulated immune surveillance can strongly contribute to disease etiology and progression [5,6,7]. Specifically in cancer, there are a host of mechanisms that these cells implement to evade immune recognition and ensure sustained survival [8,9,10,11]. Many of these adaptive mechanisms are implemented to mitigate both intrinsic (excessive proliferation, oxidative stress, etc.) and extrinsic cellular (chemo-, radiotherapy, etc.) stress, which can negatively impact immune cell activity [12,13,14,15,16,17]. Adverse immune reactions in response to stress are evident in the natural killer group 2 member D (NKG2D) stress response pathway. After exposure to stress-inducing stimuli, there is a consequent induction of NKG2D ligands on target cells. NKG2D ligands are then recognized by NKG2D receptors on subsets of neighboring cytotoxic immune cells [18,19,20]. However, in many cancers, there is a dysregulation of the stress response pathway resulting in decreased levels of NKG2D ligands, which interferes with immune recognition by effector cell populations [21,22,23]. Mechanisms of NKG2D pathway regulation have been investigated by multiple groups, and much emphasis has been placed on the activity of NKG2D receptor bearing immune cell populations [19,24,25,26,27]. However, the mechanisms through which the NKG2D ligands are regulated have not been fully elucidated, and further investigation is warranted to provide additional molecular insight into this clinically relevant immune pathway. In this review, we examine the current understanding of the regulatory mechanisms that govern NKG2D ligands in cancer, and we provide insight into therapeutic modulation of the stress response pathway for anti-cancer treatments.
2. NKG2D Pathway Overview
2.1. NKG2D Receptor
As a master regulator of immune surveillance, NKG2D receptor activation is essential for downstream signal transduction by cytotoxic effector cell populations (Figure 1) [28,29]. The NKG2D receptor, encoded by Killer Cell Lectin Like Receptor K1 (KLRK1), is found on natural killer (NK) cells, natural killer T (NKT) cells, subsets of gamma delta T cells, and small populations of CD4+ T cells [30,31,32,33,34]. Distinct polymorphisms of the NKG2D receptor have been described and have varying roles within disease etiology [25,35]. Polymorphisms in the receptor have been shown to correlate with susceptibility of chronic hepatitis B [36] and protection against systemic lupus erythematosus [37]. In cancer, a guanine to alanine mutation (Thr72Alanine) displayed increased expression in control samples in a cervical cancer study, suggesting a protective effect against cancer progression through the modulation of NK and T cell activity [38]. Functionally, the transmembrane homo-dimeric protein structure of the NKG2D receptor allows for signal transduction via the utilization of adapter molecules, as the intracellular domains of this receptor lack signaling capacity [39,40,41]. Humans are thought to exclusively utilize the DNAX activation protein 10 (DAP10) adaptor molecule, as there has only been one identified isoform of NKG2D in humans [42,43]. However, alternative splicing of the KLRK1 gene in mice results in two distinct isoforms of the NKG2D receptor, which warrants the utilization of two adaptor molecules, DAP10 and DNAX activation protein 12 (DAP12) [44,45]. Upon activation in humans, DAP10 recruits intracellular signal transducers, phosphatidylinositol-3-kinase (PI3K) and growth factor receptor-bound protein 2 (Grb2), to sustain the proliferation and survival of NK cells and to potentiate their cytotoxic effect [46,47]. Activation of the NKG2D receptor is induced by external changes to neighboring cells that promote signaling cascades that cause either an inhibition of homeostatic maintenance and/or induction of cellular stress (Figure 1) [19,48]. NKG2D receptor activity is further enhanced through additional immune-modulating stimuli, such as exposure to pro-inflammatory cytokines interleukin (IL)-15 and IL-2 [49,50,51,52]. Specifically, IL-15 promotes the phosphorylation of the DAP10 adaptor molecule to further support downstream signaling via the NKG2D receptor [53]. In contrast to NK receptors aimed at recognizing major histocompatibility complex (MHC) I molecules (HLA-A, HLA-B, HLA-C), the NKG2D receptor preferentially recognizes non-MHC I molecules known as NKG2D ligands [54,55,56].
2.2. NKG2D Ligands
There are a variety of NKG2D ligands, and the regulatory mechanisms that control their expression vary contextually [57,58,59,60]. NKG2D ligands have been identified only in placental and marsupial mammals [61]. Humans express two distinct categories of NKG2D ligands with a large degree of polymorphism that have developed over time, possibly in response to various microbial evasions [62]. One family consists of MHC Class I Polypeptide-related sequence A and B (MICA and MICB, respectively), while the other family of NKG2D ligands consists of a six-member glycoprotein family of UL16-binding proteins (ULBP1–6) [34,63,64,65,66]. Compared to humans, NKG2D ligands in mice consist of retinoic acid early inducible-1 (RAE-1) with five distinct isoforms (alpha-epsilon), three isoforms of histocompatibility H60 (a, b, c), and one homologous murine UL-16 binding protein (MULT-1) [67,68,69,70,71]. Further contributing to the diversity of NKG2D ligands, there is a high degree of polymorphic expression [24,72]. For example, there are over 60 distinct allelic sequence of MICA and 20 of MICB, and this, coupled with differential receptor binding affinity, can significantly impact immune effector-mediated cytotoxicity [73,74,75,76,77,78]. The varying binding affinity of the respective ligands to the NKG2D receptor is particularly high for immune reactions in terms of dissociation constant (KD) ranges [68,79,80,81]. In addition to the canonical NKG2D ligands, novel non-canonical ligands have been discovered that activate the NKG2D receptor (i.e., LETAL, MUTS) [82,83]. While it is important to recognize there are alternative molecules that can potentiate this pathway, this review will focus primarily on the foundational NKG2D ligands and their regulation by distinct stimuli with known roles in cancer development and progression.
2.3. Cell Type Specific Expression
As the NKG2D pathway can be modulated to advance anti-cancer therapies, there is a need to understand which cell populations are more resistant or susceptible to its targeting. The expression of NKG2DLs in healthy human tissues is generally low or restricted [63]: however, there can be the heterogeneous expression of each ligand based on its specific role, tissue type, regulation, and exposure to certain inducers [63,84]. For example, ULBP1 is found in human B cells and monocytes and may aid in hematopoiesis [85], whereas MICA, ULBP1, and ULBP3 are present in human bone marrow and early progenitor cells [86]. In addition to varying baseline expression, the ligands have differing responses to various stressors. For example, in hematopoietic tissues, stimulation with alloantigen or superantigen upregulates NKG2DLs in T cells [87], and myelomonocytic differentiation leads to an increase in ULBP1 [85]. Stimulation with various growth factors induces ULBPs in CD14+ monocytes, and both IFN-α and IL-15 can upregulate MICA expression on mature dendritic cells [88]. In gut epithelial cells, MICA may be present and increased in response to changes or disruptions in gut flora [63]. Lastly, in airway epithelial cells, the upregulation of surface NKG2DLs can occur in response to oxidative stress [89].
Off-target effects of any therapy can limit clinical implementation; therefore, identifying potential risks to the healthy surrounding tissue is critical [90,91]. Although studies demonstrated NKG2D ligand expression on normal tissue, other reports have emphasized the semi-restricted tumor-targeting potential of effector populations. For example, a 2015 study by Pereboeva et al. showed that stress-inducing stimuli, such as irradiation and chemotherapy, did indeed induce both MICA and ULBP2 ligands on both glioblastoma tumor cells and human astrocytes [92]. However, despite these ligands being expressed on the astrocytes, these cells were significantly spared from the cytotoxic effect of ex vivo expanded gamma-delta T cells in comparison to their tumor cell counterparts [92]. Additionally, work from Lamb et al. utilized normal human astrocytes as an experimental control for the cytotoxic potential of temozolomide-resistant gamma delta T cells toward glioblastoma cells, and no toxicity to the astrocytes was reported [93]. Importantly, these findings provide preclinical evidence supporting the safe implementation of NKG2D-based therapies without risk to the surrounding non-tumor tissue.
In addition to understanding the impact that NKG2D ligands have on normal tissue in response to cancer therapy, it is also important to acknowledge that NKG2D ligands do play homeostatic roles. The expression of NKG2D ligands serves as a vital signal to the immune surveillance system where NKG2D receptor-bearing cells promote the clearance of damaged tissue. For instance, in response to events such chronic inflammation, burn injury, and obesity, NKG2D ligands are induced: this results in the recruitment of effector cells to rid the affected area of damaged cells to maintain tissue health [94,95,96]. Since the NKG2D ligands are stress responsive, a process such as tumorigenesis can have an overlap of stimuli that regulate NKG2D ligand expression. For example, infection with the bacteria Helicobacter pylori (H. pylori) is an established risk factor associated with the onset of gastric adenocarcinoma [97]. A recent study by Hernandez et al. demonstrated that exposure to H. pylori induced MICA and ULBP4 in gastric adenocarcinoma cell lines, which promoted NK cell cytotoxic activation [97]. In addition, stress to the endoplasmic reticulum induced MULT-1 expression in human endothelial cells is also linked to tumorigenesis [98]. A functionally intact NKG2D system also had a protective effect against tumor initiation as evident in both a fibrosarcoma and ovarian cancer model, respectively [99,100]. With the relatively harsh conditions that tumors maintain, the exploitation of NKG2D ligands to enhance immunotherapies is a promising approach.
Another important cell-specific consideration for therapeutic intervention is the ability of certain malignant cell populations to evade this pathway. For example, chemo- and radiotherapy-resistant tumor-initiating cell or cancer stem cell (CSC) populations may have relatively lower levels of NKG2D ligands [101,102,103]. For example, acute myeloid leukemia (AML) stem cells evaded killing by NK cells through increased expression of the DNA repair enzyme poly-ADP-ribose-polymerase 1 (PARP1), which caused a repression of NKG2D ligands [104]. Similarly, stem cells in solid tumors such as glioblastoma have also been shown to evade immune system detection via dysregulation of NKG2D ligand expression. Zhang et al. determined that epigenetic silencing resulted in ULBP1 and ULBP3 ligand repression in glioblastoma [105]. Following treatment with the hypomethylating agent decitabine, ligand repression was reversed, and NK-mediated killing increased [105]. However, other reports using glioblastoma cells have noted the increased expression of stress ligands in cancer stem cell populations, supporting a heterogeneous expression pattern that is not yet fully understood [106,107]. Substantial efforts have been dedicated to manipulating and targeting CSC populations to make them more susceptible to cytotoxic killing by NKG2D receptor-bearing effector populations [108,109,110]. However, it is important to note that other cell populations critical for tumor biology also express NKG2D ligands: this pathway can influence the more differentiated, non-CSC tumor cells as well as macrophages, dendritic cells, B-cells, and T-cells [69,111,112]. Despite the expression of NKG2D ligands being necessary for immunoreactivity and immune surveillance, sustained expression can be detrimental to pathway activity [113]. Constitutive MICA and Rae-1ε expression in a cutaneous carcinoma model was associated with the downregulation of NKG2D receptor expression and diminished NK-mediated cytotoxicity via receptor internalization and lysosomal-mediated degradation [114,115]. Adding to the complexity of NKG2D pathway regulation, there are also heterogeneous induction mechanisms for stress ligand expression that are relevant for cancer etiology.
3. Positive Regulators of NKG2D Ligand Expression
The regulation and activation of the NKG2D receptor has been extensively reviewed (see Refs. [19,24,25,26,27]); therefore, we have focused on the cancer-relevant regulation of NKG2D ligands and the potential for developing novel, targeted cancer therapeutics related to this pathway.
3.1. DNA Damage
Many current cancer treatment strategies rely on initiating DNA damage in malignant cells: DNA damage has an important role in immunoreactivity, specifically as it pertains to NKG2D ligand expression [116]. In transduced ovarian cancer cells, DNA damage from ionizing radiation and certain chemotherapies induced both human (MICA/B, ULBP1-3) and mouse (RAE-1) NKG2D ligands [117]. Along with ligand induction, there was increased activity of DNA damage response (DDR) pathway regulators, such as ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and RAD3 related (ATR): their activity was supported by an increased phosphorylation of downstream pathway mediators Chk1 and Chk2 and the tumor-suppressor protein p53 [117]. Other cancer types, such as multiple myeloma and leukemia, also have a dependency on ATM and ATR for NKG2D ligand induction [118,119].
Along with the direct influence that the DNA damage response has on NKG2D ligand expression, there is also evidence that indirect mechanisms can contribute to ligand induction [120]. For example, as DNA damage occurs, there is an increased presence of cytosolic DNA that activates corresponding sensory pathways. These pathways can further activate the stimulator of interferon (IFN) genes (STING) pathway, which is an established immune regulatory pathway [121]. Lam et al. determined that RAE-1 expression in lymphoma cell lines was dependent on the activation of IFN regulatory factor-3 (IRF3), whose phosphorylation was regulated by DNA sensor pathway activation [122].
In addition to cytosolic sensory pathways, downstream molecular targets of DNA damage, such as the transcription factor and tumor suppressor protein p53, have also been shown to influence the expression of NKG2D ligands [122]. In lung cancer models, p53 mutational status influenced the expression of ULBP1 and ULBP2, as these ligands are direct transcriptional targets of p53 [123]. Cancer cells transduced with wild-type p53 resulted in a significant upregulation of the ULBP1 and ULBP2 ligands as compared to those with mutant p53 [123]. In the context of DNA damage, p53 is a downstream target of activated response elements, which promotes ligand expression, suggesting its necessity [117,124,125].
3.2. Cell Cycle Alterations
While DNA damage is one of the more established mechanisms that regulates NKG2D ligand expression, other phenotypes associated with cancer initiation and maintenance have also been implicated in ligand regulation. Malignant cells commonly have alterations in their cell cycle, and investigators are working to elucidate mechanistic links between cell cycle regulators and immune surveillance. An established family of cell cycle regulating transcription factors, the E2 factor family (E2F), is a direct regulator of NKG2D ligands. Specifically, the Rae family of NKG2D ligands are direct transcriptional targets of E2F, and increased overall E2F family (1,2,3) transcriptional activity leads to increased proliferation and Rae family expression [126]. E2F is also a direct phosphorylation target of ATM and ATR kinase activity. Therefore, E2F provides further regulation of NKG2D ligands by the DNA damage response pathway.
In addition to pathway regulators, the behavior of malignant cells has been linked to the expression of NKG2D ligands. The induction of hyperploidy in various cancer cell lines by cytochalasin D, a microfilament polymerization inhibitor, resulted in a subsequent upregulation of MICA and an increased NK cell cytotoxic response [127]. In multiple myeloma cells, a high-self renewal capacity resulted in increased NKG2D ligand expression and preferential targeting by NK cells [128]. As energetic output is often associated with increased cell division, some studies have determined that nutrient availability influences ligand expression. For example, enhanced mitochondrial citrate metabolism, glycolysis, and fatty acid metabolism promoted either ligand expression (MICA/B) or enhanced NK cell functionality in their respective tumor models, which supports the potential targeting of metabolically active cell populations [129,130,131,132].
3.3. Oncogenic Transformation
The acquired genetic changes during cancer can also influence NKG2D ligand expression. During oncogenesis, there is an immune landscape shift that contributes to disease progression [133,134,135]. Genetic alterations commonly associated with tumor formation have been reported to regulate NKG2D ligand expression. Unni et al. demonstrated a novel link between NKG2D ligands and genetic transformation: the combination of increased oncogenic Myc activity and diminished tumor-suppressor activity of either p53 or Arf resulted in an increased surface expression of Rae-1ε during lymphomagenesis [136]. Furthermore, Schuster et al. determined that not only did p53 need to be lost but the anti-apoptotic protein BCL2 also needed to be overexpressed to induce expression of the murine NKG2D ligand, MULT-1, in a Eµ-myc model of lymphoma [137]. These multiple oncogenic “hits” were necessary to facilitate NK cell-mediated killing of the lymphoma cells. An important distinction to note is that diminished tumor suppressor activity of p53 can regulate ligand expression independent of the DNA damage response cascade.
The phosphatidylinositol-3-kinase (PI3K) pathway is also commonly altered during transformation and implicated in NKG2D signaling. In a model of cellular stress in mouse fibroblast infected with cytomegalovirus, the PI3K catalytic subunit, p110𝛼, induced RAE-1 family ligands. Inhibition of this subunit caused a reduction in expression of RAE-1 ligands [138]. PI3K-mediated regulation of ligands has also been demonstrated in human breast cancer cell lines. Specifically, the dimerization of epidermal growth factor family members, HER2 and HER3, upregulated MICA and MICB expression via the PI3K axis, which increased NK cell recognition and killing of tumor cells [139].
Upstream of both Myc and PI3K, the oncogene RAS also influences stress ligand expression. Constitutive activation of mutant RAS in a human breast cancer model resulted in an increase in downstream pathway mediators such as mitogen-activated protein kinase (MAPK) and PI3K and in murine RAET1 ligand expression in mouse breast cancer cell lines [140]. Although established as a regulator of stress ligand expression, there was no dependency on enhanced ATM or ATR expression to substantiate NKG2D ligand expression in this model. This supports the heterogeneous regulation of NKG2D ligand expression independent of established pathways, such as that of the DNA damage response pathway [140].
Another common occurrence during oncogenesis is epithelial-to-mesenchymal transition, which has been shown to repress ULBP1 ligand expression in a gastric cancer model and further contribute to the immunosuppressive tumor microenvironment [141]. In additional studies related to cancer stressors, there is an increased expression of heat shock proteins, which facilitate the restoration of homeostatic signaling through refolding of denatured proteins and are known to be both upregulated under malignant conditions and regulate the expression of NKG2D ligands [142,143,144,145]. As illustrated above, increasing evidence suggests a relationship between NKG2D ligands and tumor initiation/maintenance. However, the impact of carcinogenesis on NKG2D ligands also depends on the immune cell influenced by the ligand expression. A study by Strid et al., using the epidermis as a model, reported that in response to local Rae-1 induction, certain immune cell populations exuded differing effects on carcinogenesis [146]. For example, with Rae-1 expression, gamma delta cells were able to protect the tissue from carcinogenesis, while Langerhans cells promoted it [146]. The immune cell responsive to NKG2D ligand induction could be one explanation for the diversity in cancer-mediated regulation of this pathway. Understanding the heterogeneous induction of this immune surveillance pathway is extremely important for targeting cancer cell vulnerabilities (Figure 2): however, there is also a necessity to understand compensatory mechanisms that cancer cells may employ to diminish pathway activity.
4. Biological Repression of NKG2D Ligands in Cancer
Mechanisms to combat NKG2D ligand-mediated recognition are commonly implored in cancer cells. Due to their high degree of plasticity and adaptability, cancer cells can manipulate their biological activity to evade the NKG2D immune surveillance pathway. Next, we highlight the established immunosuppressive mechanisms utilized by cancer cells to downregulate NKG2D ligands and avoid NKG2D-mediated killing (Figure 3).
4.1. Shedding
A regulatory mechanism of the immune surveillance pathway is the proteolytic cleavage of cell surface bound NKG2D ligands, which is also known as shedding. Two primary families of proteases associated with this shedding phenotype include matrix metalloproteinases (MMPs) and disintegrin and metalloproteinases (ADAMs). First established using epithelial-derived tumors, MICA was shown to be actively released by tumor cells as a means of immunosuppression [147]. Inhibition of metalloproteinases reversed the shedding phenotype and promoted the accumulation of MICA on the tumor cell surface [147]. NKG2D ligand shedding occurs in many other tumor types and has been validated in analyses from cancer patient serum [148,149,150,151,152]. Further supporting the complexity of the immune landscape, immune regulatory cytokines also have multi-faceted roles in NKG2D ligand expression, which is mediated partly by MMP cleavage. For example, interferon alpha (IFN-𝛼) has been shown to enhance MICA expression on tumor cells through increased promoter activity. Alternatively, interferon-gamma has been shown to enhance the cleavage of MICA via MMP9 [153]. As mentioned earlier, ADAM proteases are also responsible for the proteolytic shedding of NKG2D ligands. Using a standard model of glioblastoma, ADAM17 was shown to promote MICB solubilization through specific active sites at detergent-resistant membrane microdomains (DRMs) [154]. In addition to ADAM17, ADAM10 and ADAM9 have both been implicated in the shedding of NKG2D ligands [155,156,157,158]. Of note, hypoxia is a potent negative regular of NKG2D ligand expression: hypoxia directly influences both exosome release and the shedding of ligands [159]. Additionally, hypoxia diminishes the effector functionality of the NKG2D receptor expressing immune populations [160,161,162]. Relevant to clinical implications, investigators have sought to target hypoxia-induced immunosuppression in the hopes of enhancing immunotherapies [159,163,164,165,166,167].
4.2. Exosomes
One mechanism cancer cells use to regulate NKG2D ligands is their extracellular release through vesicular transport via exosomes [168]. In response to different stimuli, exosomes are intracellularly produced during endo-lysosomal processing and then released into the external microenvironment. Multiple tumor models including ovarian, mesothelioma, and cervical cancer have displayed the regulation of ligands through enhanced exosome production and release [169,170,171]. Specifically in leukemia cells, tumor-specific stressors, such as thermal and oxidative stress, promoted the release of exosomes containing NKG2D ligands that subsequently diminished NK cell cytotoxicity [172]. This exosome-mediated immunosuppressive phenotype has also been observed in prostate cancer cells where cancer cell-derived exosomes containing MICA/B, ULBP1, or ULBP2 led to the downregulation of NKG2D receptor expression on effector populations [173]. Elegantly proving this mechanism, the supplementation of tumor-derived exosomes to healthy lymphocytes led to the subsequent downregulation of NKG2D receptor expression [173]. In the broader context of the tumor microenvironment, exosomes derived from other cell populations, such as dendritic cells, regulate immune reactivity. Interestingly, dendritic cell-derived exosomes containing NKG2D ligands were able to enhance the proliferation and activity of NK cells through an IL-15Ralpha-mediated mechanism [174]. The exact mechanisms that regulate ligand secretion via exosomes remains to be elucidated, as exosomes are an evolving area of study.
4.3. Immunosuppressive Signaling Regulators
Many signaling pathways are aberrantly active under malignant conditions, and some of these may repress the expression and activity of NKG2D pathway intermediates. For example, transforming growth factor beta (TGF-β) has a multi-faceted role in cancer progression, where its effects on the tumor microenvironment can dampen immune cell activity [175]. In a lung cancer model, Lee et al. recently determined that TGF- β negatively regulated NGK2D ligand expression through the upregulation of MMP activity [176]. Similarly, TGF-β-suppressed NKG2D ligands MICA, ULBP2, and ULBP4 via MMP activity in a glioma model [177]. Importantly, in addition to repressing ligand expression, TGF- β has also diminishes the effector activity of NKG2D-bearing T-cells [178,179,180].
Another pathway implicated in the negative regulation of NKG2D ligand expression is the Janus Kinase and Signal Transducer and Activator of Transcription (JAK/STAT) pathway. In melanoma, MICA expression was negatively regulated by the production of interferon-gamma mediated by the activity of STAT1 [181]. In a castration-resistant pancreatic cancer model, the JAK/STAT signaling axis also regulated NKG2D ligand levels [182]. Specifically, IL-6 mediated the activation of JAK-STAT3, which led to a repression of multiple NKG2D ligands (MICA, MICB, ULBP1, ULBP2, and ULBP3) and evasion from NK cell killing [182]. Interestingly, this study also determined that the IL-6/JAK/STAT3 signaling cascade regulated the expression of the immune cell inhibitory molecule Programmed Death Receptor Ligand 1 (PD-L1), which is known inhibit effector cell activity [182,183,184]. Other studies have also demonstrated the negative regulation of NKG2D ligand expression via the PDL1/2 axis. For example, Okita et al. determined that MICA, ULBP2, ULBP5, and ULBP6 expression was induced in response to cisplatin [185]. However, along with this increase in NKG2D ligands, there was also an induction of PDL-1, diminishing the potential of NK cell-mediated killing [185]. A recent study by Lee et al. suggested that radiotherapy could induce expression of MICA, MICB, ULBP1, ULBP2, and ULBP3 in melanoma cells, but the concurrent induction of PD-L1 on the cells diminished the potential cytotoxic effect of the NK-92 cell line [186].
There is also increasing evidence that the epigenetic landscape regulates the expression patterns of NKG2D ligands. For example, histone deacetylases suppress NKG2D ligand expression. In epithelial tumor cells, both pharmacologic and genetic inhibition of histone deacetylase 3 (HDAC3) were able to rescue the suppressed expression of ULBP1-3. Mechanistically, it was determined that in response to stress, HDAC3 was recruited to the promoter regions of ULBP1-3, where it mediated the direct repression of these ligands. Recently, Histone Deacetylase 8 (HDAC8) was shown to modulate glioma immune responses through regulation of NKG2D ligands. Following pharmacologic inhibition of HDAC8, chromatin immunoprecipitation sequencing (ChIP) analysis identified a direct regulation of MULT-1, Rae-1, and H60 murine NKG2D ligands [187]. Collectively, the diverse regulatory mechanisms for NKG2D ligand expression provides promising therapeutic targets.
5. Therapeutic Approaches Utilizing NKG2D Ligand Expression
Identifying treatments that induce NKG2D ligands to enhance immune cell-mediated killing is a promising immunotherapy approach (Table 1). While they have off-target effects, standard of care radio- and chemotherapy can alter the NKG2D pathway [188,189,190]. In glioblastoma, radiation and the DNA alkylating agent, temozolomide, were shown to induce MICA, MICB, ULBP2, and ULBP3 ligand expression in vitro and in vivo, which conferred higher immunoreactivity [191]. Additionally, temozolomide was able to induce multiple NKG2D ligands (MICA/B, ULBP1-3) in a glioblastoma stem cell model [106]. These findings support previous work performed by Lamb et al. where a transient induction of MICA/B, ULBP1-ULBP3 ligands was observed in standard models of glioblastoma after treatment with temozolomide [93]. Interestingly, the addition of gamma delta T-cells engineered to be temozolomide-resistant (through overexpression of the DNA repair enzyme methylguanine DNA methyltransferase (MGMT)) to temozolomide treatment increased overall survival in mice bearing glioblastoma xenografts [192,193]. Based on these results, temozolomide-resistant gamma delta T cells combined with standard of care temozolomide are being investigated in a phase 1 clinical trial as upfront maintenance therapy in adults with glioblastoma (NCT04165941).
The heterogeneous regulation of NKG2D ligands provides multiple potential avenues for immunotherapies that enhance ligand expression. As illustrated in Figure 2, TGF- β is a negative regulator of NKG2D ligand expression. Either genetic or pharmacologic inhibition of this pathway has shown promise at restoring ligand expression and effector cell activity [194,195]. Recently, in a preclinical breast cancer model, Liu et al. observed therapeutic efficacy of an encapsulated combination of TGF-β inhibitors with selenocystine. The combination enhanced NK cell-mediated tumor cell death through the induction of MICA, MICB and ULBP1-4 [196]. In addition, HDAC inhibitors are a promising approach and have proven successful for enhancing NKG2D ligand expression in a variety of cancer models [197,198,199,200,201,202]. Another approach being explored is inhibiting MMP activity to decrease the shedding activity that may limit cytotoxic effects [136,137,138]. In addition to targeting the negative regulators of NKG2D ligands, activating the positive regulators of NKG2D ligands has proven successful in a variety of cancer types. For example, DDR activation through small molecule inhibition can increase NKG2D ligands. Furthermore, heat shock protein inhibitors (i.e., HSP90), inhibitors of apoptosis (IAP), and proteosome inhibitors have been shown to enhance the expression of a variety of NKG2D ligands in different cancer types via the activation of DDR pathway mediators [203,204,205].
Another promising cancer therapeutic that may be responsive to NKG2D pathway manipulation is oncolytic virotherapy [206,207,208]. As a well-established stressor to induce NKG2D ligand expression, viral infection targeting malignant cells may result in ligand induction to potentiate the cytotoxic effect of NKG2D bearing immune populations [209,210,211,212,213]. As cancer cells are highly adaptable, there are mechanisms to evade viral-induced NKG2D ligand expression. Therefore, strategic manipulation is necessary to maximize therapeutic effect. Table 1 summarizes current clinical trials utilizing NKG2D pathway manipulation (Table 1). As a by-product of ligand induction, an added advantage may be the identification of predictive biomarkers to support NKG2D-driven immunotherapy [214,215,216,217].
Although this notion of NKG2D ligand induction for immunotherapies is exciting, the heterogeneity of tumors across tissue types, and even the intratumoral heterogeneity amongst patients, makes universal prognostic indicators extremely difficult. In certain instances, there is a benefit to having NKG2D ligands robustly induced and expressed. However, in other tumors, sustained NKG2D ligand overexpression is a negative indicator of overall patient survival. For example, from an immunohistology screen, MICA/B and ULBP1 were shown to be both correlated and good prognostic indicators for cervical cancer patient survival [215]. On the other hand, a recent bioinformatic analysis of ULBP1 expression in colon adenocarcinoma patients revealed ULBP1 as a negative indicator of overall patient survival [218]. Interestingly, in colorectal cancer, MICA and ULBP5 (RAET1G) co-expression was shown to positively influence overall patient survival [219]. However, their expression was most abundant in stage 1 metastatic tumor nodes, while expression decreased as the tumor stage increased [219]. The tissue and patient-specific reasoning behind differential survival responses to NKG2D ligand expression has not been clearly elucidated, and further research investigating the regulation of this pathway that carefully considers heterogeneity, including in preclinical models, is needed.
6. Conclusions
Employing the NKG2D stress response pathway for therapeutic benefit is an exciting immunotherapy strategy. While significant strides have been made in the foundational understanding of these immune-modulating ligands, there is still ample room to increase our understanding to utilize this pathway more precisely for clinical benefit. The diverse regulation of NKG2D ligands provides numerous therapeutic opportunities. There remains a need for further investigation of the mechanistic role that oxidative stress has on NKG2D ligand expression. In addition, a better understanding of the tumor microenvironment is critical. The nutrient availability, oxygen tension, and overall acidity of the tumor microenvironment largely affect signaling cascades. The large-scale implications of these environmental conditions have not been well established in the context of NKG2D ligand expression and may provide needed opportunities to enhance the effectiveness of immunotherapies. Additionally, further investigation into tumor-specific niches and regulation is necessary.
As evidenced by the diverse clinical responses amongst different tumor types, variability in NKG2D ligand-based immunotherapy efficiency is apparent: this warrants further investigation to identify strategies that may need to be disease-specific to overcome this differential response. For example, blood-borne (liquid) malignancies have often been utilized as models for the foundational mechanistic studies for NKG2D ligand biology, and these tumor types have shown significant success this specific type of immunotherapy [109,119,152,220]. However, due to the cellular heterogeneity and physiologic complexity that many solid tumors possess, targeting solid tumors with NKG2D ligand induction may have more therapeutic hurdles. Resulting from the diverse nature of solid tumors, the expression of NKG2D ligands can also be heterogeneously induced depending on the stimuli. As many cells within the tumor microenvironment differ in both therapeutic sensitivity and antigen generation/presentation, there is a benefit to utilizing stress-responsive NKG2D ligands. Under stressful stimuli, multiple ligands may be induced on differing cell populations within the tumor. These stimuli may arise from a variety of sources such as the different chemotherapeutic agents commonly employed as first-line treatments to newly diagnosed cancers. Despite the potential NKG2D ligand induction being diverse, with ligands displaying varying induction time/stability, intratumoral heterogeneity, and abundance, there is still the potential therapeutic benefit as the tumor becomes more recognized by the immune system. This enhanced recognition by effector cells may allow for the better implementation of cell-based immunotherapies by mechanistically making historically immunogenically cold tumors (inactive) turn hot (active). To illustrate this point further, some chemotherapies that have been investigated for NKG2D ligand induction in cancer are summarized below (Table 2). In summation, targeting the NKG2D stress response pathway has shown extraordinary promise in preclinical settings for treatment of cancer, and with further research, there is an opportunity to implement these strategies to maximize therapeutic efficacy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers14092339/s1, File S1: Figures S1–S3 and Graphical Abstract Publication License.
Author Contributions
Original draft preparation, A.B.J.; reviewing and editing, A.R., A.B.H., G.K.F. and L.S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NINDS (5R01NS104339-05), NINDS (1R03NS125506), UAB Start Up Funds (NS104339), CDIB Pilot Grant, and In8Bio Inc.—A.B.H. G.K.F. is supported by the Rally Foundation, Cure Search for Children’s Cancer, The V Foundation, Hyundai Hope on Wheels, Andrew McDonough B+ Foundation, the Pediatric Cancer Research Foundation, and the National Pediatric Cancer Foundation.
Acknowledgments
We would like to thank Kate Rochlin, the chief operating officer of In8bio, for her thoughtful comments and suggestions that improved the review. Additionally, we would like to thank the members of the Hjelmeland laboratory for their review of this manuscript: Kaysaw Tuy, Sajina GC, Cyntanna Hawkins, and Sarah Williford. All images were generated using Biorender.com (accessed on 22 April 2022), and licensure certificates are provided in the Supplemental Materials.
Conflicts of Interest
The authors declare no conflict of interest for this review. Lawrence Lamb is the executive vice president, chief scientific officer, and co-founder of In8bio whose clinical trial is mentioned in Table 1. The Hjelmeland lab has financial support from In8bio for academic research.
References
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaço, H.G.; Moita, L.F. Initiation of innate immune responses by surveillance of homeostasis perturbations. FEBS J. 2016, 283, 2448–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, O.P.; Lichtnekert, J.; Anders, H.J.; Mulay, S.R. The Immune System in Tissue Environments Regaining Homeostasis after Injury: Is “Inflammation” Always Inflammation? Mediat. Inflamm. 2016, 2016, 2856213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.I.; Lee, L.; Schwarz, E.; Groh, V.; Spies, T.; Ebert, E.C.; Jabri, B. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J. Immunol. 2001, 167, 5527–5530. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D. The concept of immune surveillance against tumors. The first theories. Oncotarget 2017, 8, 7175–7180. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Steinert, G.; Schölch, S.; Niemietz, T.; Iwata, N.; García, S.A.; Behrens, B.; Voigt, A.; Kloor, M.; Benner, A.; Bork, U.; et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014, 74, 1694–1704. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.C.; Zhang, X.F.; Peng, J.; Li, X.F.; Wang, A.L.; Bie, Y.Q.; Shi, L.H.; Lin, M.B.; Zhang, X.F. Survival Mechanisms and Influence Factors of Circulating Tumor Cells. BioMed Res. Int. 2018, 2018, 6304701. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genese Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Mercer, S.J.; Knight, L.A.; Gabriel, F.G.; Whitehouse, P.A.; Sharma, S.; Fernando, A.; Glaysher, S.; Di Palma, S.; Johnson, P.; et al. Cancer cell adaptation to chemotherapy. BMC Cancer 2005, 5, 78. [Google Scholar] [CrossRef] [Green Version]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunology 2013, 2, e26097. [Google Scholar] [CrossRef] [Green Version]
- Wensveen, F.M.; Jelenčić, V.; Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- López-Soto, A.; Huergo-Zapico, L.; Acebes-Huerta, A.; Villa-Alvarez, M.; Gonzalez, S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer 2015, 136, 1741–1750. [Google Scholar] [CrossRef]
- Raffaghello, L.; Prigione, I.; Airoldi, I.; Camoriano, M.; Levreri, I.; Gambini, C.; Pende, D.; Steinle, A.; Ferrone, S.; Pistoia, V. Downregulation and/or release of NKG2D ligands as immune evasion strategy of human neuroblastoma. Neoplasia 2004, 6, 558–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemann, K.; Mittrücker, H.W.; Feger, U.; Welte, S.A.; Yokoyama, W.M.; Spies, T.; Rammensee, H.G.; Steinle, A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J. Immunol. 2005, 175, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- López-Larrea, C.; Suárez-Alvarez, B.; López-Soto, A.; López-Vázquez, A.; Gonzalez, S. The NKG2D receptor: Sensing stressed cells. Trends Mol. Med. 2008, 14, 179–189. [Google Scholar] [CrossRef]
- Coudert, J.D.; Held, W. The role of the NKG2D receptor for tumor immunity. Semin. Cancer Biol. 2006, 16, 333–343. [Google Scholar] [CrossRef]
- Zafirova, B.; Wensveen, F.M.; Gulin, M.; Polić, B. Regulation of immune cell function and differentiation by the NKG2D receptor. Cell Mol. Life Sci. 2011, 68, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Burgess, S.J.; Maasho, K.; Masilamani, M.; Narayanan, S.; Borrego, F.; Coligan, J.E. The NKG2D receptor: Immunobiology and clinical implications. Immunol. Res. 2008, 40, 18–34. [Google Scholar] [CrossRef]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Miyokawa, N.; Bach, F.H. Isolation and characterization of NK cell or NK/T cell-specific cDNA clones. J. Mol. Cell Immunol. 1990, 4, 295–304, discussion 296–305. [Google Scholar] [PubMed]
- Groh, V.; Steinle, A.; Bauer, S.; Spies, T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science 1998, 279, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Machuldova, A.; Holubova, M.; Caputo, V.S.; Cedikova, M.; Jindra, P.; Houdova, L.; Pitule, P. Role of Polymorphisms of NKG2D Receptor and Its Ligands in Acute Myeloid Leukemia and Human Stem Cell Transplantation. Front. Immunol. 2021, 12, 651751. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, X.; Wu, X.; Li, J.; Zhu, X.; Li, Z.; Li, J.; Pan, L.; Li, T.; Li, H.; et al. Association of NKG2D genetic polymorphism with susceptibility to chronic hepatitis B in a Han Chinese population. J. Med. Virol. 2010, 82, 1501–1507. [Google Scholar] [CrossRef]
- Piotrowski, P.; Lianeri, M.; Olesińska, M.; Jagodziński, P.P. Prevalence of the NKG2D Thr72Ala polymorphism in patients with systemic lupus erythematosus. Mol. Biol. Rep. 2012, 39, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Roszak, A.; Lianeri, M.; Jagodziński, P.P. Prevalence of the NKG2D Thr72Ala polymorphism in patients with cervical carcinoma. Genet. Test. Mol. Biomark. 2012, 16, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Cao, T.M.; Baker, J.A.; Verneris, M.R.; Soares, L.; Negrin, R.S. Silencing human NKG2D, DAP10, and DAP12 reduces cytotoxicity of activated CD8+ T cells and NK cells. J. Immunol. 2005, 175, 7819–7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Cherwinski, H.; Spies, T.; Phillips, J.H.; Lanier, L.L. DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J. Exp. Med. 2000, 192, 1059–1068. [Google Scholar] [CrossRef]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.B.; Araki, M.; Hamerman, J.A.; Chen, T.; Yamamura, T.; Lanier, L.L. A Structural basis for the association of DAP12 with mouse, but not human, NKG2D. J. Immunol. 2004, 173, 2470–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilfillan, S.; Ho, E.L.; Cella, M.; Yokoyama, W.M.; Colonna, M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 2002, 3, 1150–1155. [Google Scholar] [CrossRef]
- Roda-Navarro, P.; Reyburn, H.T. The traffic of the NKG2D/Dap10 receptor complex during natural killer (NK) cell activation. J. Biol. Chem. 2009, 284, 16463–16472. [Google Scholar] [CrossRef] [Green Version]
- Garrity, D.; Call, M.E.; Feng, J.; Wucherpfennig, K.W. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc. Natl. Acad. Sci. USA 2005, 102, 7641–7646. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Tomasello, E.; Lucas, M.; Jamieson, A.M.; Hsia, J.K.; Vivier, E.; Raulet, D.H. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 2002, 3, 1142–1149. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Ruck, T.; Bittner, S.; Afzali, A.M.; Göbel, K.; Glumm, S.; Kraft, P.; Sommer, C.; Kleinschnitz, C.; Preuße, C.; Stenzel, W.; et al. The NKG2D-IL-15 signaling pathway contributes to T-cell mediated pathology in inflammatory myopathies. Oncotarget 2015, 6, 43230–43243. [Google Scholar] [CrossRef]
- Kang, T.H.; Mao, C.P.; He, L.; Tsai, Y.C.; Liu, K.; La, V.; Wu, T.C.; Hung, C.F. Tumor-targeted delivery of IL-2 by NKG2D leads to accumulation of antigen-specific CD8+ T cells in the tumor loci and enhanced anti-tumor effects. PLoS ONE 2012, 7, e35141. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ronson, B.; Guo, M.; Liu, S.; Bishop, J.S.; Van Echo, D.A.; O’Malley, B.W., Jr. Interleukin 2 gene transfer prevents NKG2D suppression and enhances antitumor efficacy in combination with cisplatin for head and neck squamous cell cancer. Cancer Res. 2002, 62, 4023–4028. [Google Scholar] [PubMed]
- Horng, T.; Bezbradica, J.S.; Medzhitov, R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat. Immunol. 2007, 8, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.L.; Chalupny, N.J.; Cosman, D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 2001, 181, 185–192. [Google Scholar] [CrossRef]
- Eagle, R.A.; Trowsdale, J. Promiscuity and the single receptor: NKG2D. Nat. Rev. Immunol. 2007, 7, 737–744. [Google Scholar] [CrossRef]
- Cerwenka, A.; Lanier, L.L. NKG2D ligands: Unconventional MHC class I-like molecules exploited by viruses and cancer. Tissue Antigens 2003, 61, 335–343. [Google Scholar] [CrossRef]
- Spear, P.; Wu, M.R.; Sentman, M.L.; Sentman, C.L. NKG2D ligands as therapeutic targets. Cancer Immun. Arch. 2013, 13, 8. [Google Scholar]
- Heyward, C.Y.; Patel, R.; Mace, E.M.; Grier, J.T.; Guan, H.; Makrigiannis, A.P.; Orange, J.S.; Ricciardi, R.P. Tumorigenic adenovirus 12 cells evade NK cell lysis by reducing the expression of NKG2D ligands. Immunol. Lett. 2012, 144, 16–23. [Google Scholar] [CrossRef]
- Qian, M.; Geng, J.; Luo, K.; Huang, Z.; Zhang, Q.; Zhang, J.A.; Ji, L.; Wu, J. BCL11B regulates MICA/B-mediated immune response by acting as a competitive endogenous RNA. Oncogene 2020, 39, 1514–1526. [Google Scholar] [CrossRef]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef]
- Kondo, M.; Maruoka, T.; Otsuka, N.; Kasamatsu, J.; Fugo, K.; Hanzawa, N.; Kasahara, M. Comparative genomic analysis of mammalian NKG2D ligand family genes provides insights into their origin and evolution. Immunogenetics 2010, 62, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Bahram, S. MIC genes: From genetics to biology. Adv. Immunol. 2000, 76, 1–60. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bahram, S.; Bauer, S.; Herman, A.; Beauchamp, M.; Spies, T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. USA 1996, 93, 12445–12450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Randolph-Habecker, J.; Topp, M.S.; Riddell, S.R.; Spies, T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2001, 2, 255–260. [Google Scholar] [CrossRef]
- Eagle, R.A.; Traherne, J.A.; Hair, J.R.; Jafferji, I.; Trowsdale, J. ULBP6/RAET1L is an additional human NKG2D ligand. Eur. J. Immunol. 2009, 39, 3207–3216. [Google Scholar] [CrossRef]
- Cerwenka, A.; Bakker, A.B.; McClanahan, T.; Wagner, J.; Wu, J.; Phillips, J.H.; Lanier, L.L. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 2000, 12, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Carayannopoulos, L.N.; Naidenko, O.V.; Fremont, D.H.; Yokoyama, W.M. Cutting edge: Murine UL16-binding protein-like transcript 1: A newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 2002, 169, 4079–4083. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Jamieson, A.M.; Liu, S.D.; Shastri, N.; Raulet, D.H. Ligands for the murine NKG2D receptor: Expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 2000, 1, 119–126. [Google Scholar] [CrossRef]
- Malarkannan, S.; Shih, P.P.; Eden, P.A.; Horng, T.; Zuberi, A.R.; Christianson, G.; Roopenian, D.; Shastri, N. The molecular and functional characterization of a dominant minor H antigen, H60. J. Immunol. 1998, 161, 3501–3509. [Google Scholar]
- Zou, Z.; Nomura, M.; Takihara, Y.; Yasunaga, T.; Shimada, K. Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: A novel cDNA family encodes cell surface proteins sharing partial homology with MHC class I molecules. J. Biochem. 1996, 119, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Mizuki, N.; Ota, M.; Yamazaki, M.; Ohno, S.; Goto, K.; Miyata, Y.; Wakisaka, K.; Bahram, S.; Inoko, H. Allelic variants of the human MHC class I chain-related B gene (MICB). Immunogenetics 1997, 46, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gyllensten, U. MICA polymorphism: Biology and importance in cancer. Carcinogenesis 2014, 35, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
- Stephens, H.A. MICA and MICB genes: Can the enigma of their polymorphism be resolved? Trends Immunol. 2001, 22, 378–385. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.; Corell, A.; Argüello, J.R.; Cox, S.T.; McWhinnie, A.; Marsh, S.G.; Madrigal, J.A. A new MICA allele with ten alanine residues in the exon 5 microsatellite. Tissue Antigens 2000, 55, 162–165. [Google Scholar] [CrossRef]
- Gambelunghe, G.; Brozzetti, A.L.; Ghaderi, M.; Tortoioli, C.; Falorni, A. MICA A8: A new allele within MHC class I chain-related A transmembrane region with eight GCT repeats. Hum. Immunol. 2006, 67, 1005–1007. [Google Scholar] [CrossRef]
- Choy, M.K.; Phipps, M.E. MICA polymorphism: Biology and importance in immunity and disease. Trends Mol. Med. 2010, 16, 97–106. [Google Scholar] [CrossRef]
- Goto, K.; Kato, N. MICA SNPs and the NKG2D system in virus-induced HCC. J. Gastroenterol. 2015, 50, 261–272. [Google Scholar] [CrossRef]
- O’Callaghan, C.A.; Cerwenka, A.; Willcox, B.E.; Lanier, L.L.; Bjorkman, P.J. Molecular competition for NKG2D: H60 and RAE1 compete unequally for NKG2D with dominance of H60. Immunity 2001, 15, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Strong, R.K. Asymmetric ligand recognition by the activating natural killer cell receptor NKG2D, a symmetric homodimer. Mol. Immunol. 2002, 38, 1029–1037. [Google Scholar] [CrossRef]
- Mistry, A.R.; O’Callaghan, C.A. Regulation of ligands for the activating receptor NKG2D. Immunology 2007, 121, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.C.; Khang, E.; Zhang, L.; Mohamed-Hadley, A.; Vinocur, J.M.; Buckanovich, R.J.; Thompson, C.B.; Levine, B.; et al. Letal, A tumor-associated NKG2D immunoreceptor ligand, induces activation and expansion of effector immune cells. Cancer Biol. Ther. 2003, 2, 446–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Chen, H.; Mo, C.; Cui, L.; He, W. Ectopically expressed human tumor biomarker MutS homologue 2 is a novel endogenous ligand that is recognized by human γδ T cells to induce innate anti-tumor/virus immunity. J. Biol. Chem. 2012, 287, 16812–16819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Mohamed, R.H.; Kajikawa, M.; Koizumi, J.; Tanaka, M.; Fugo, K.; Otsuka, N.; Maenaka, K.; Yagita, H.; Chiba, H.; et al. Involvement of an NKG2D ligand H60c in epidermal dendritic T cell-mediated wound repair. J. Immunol. 2012, 188, 3972–3979. [Google Scholar] [CrossRef] [Green Version]
- Nowbakht, P.; Ionescu, M.C.; Rohner, A.; Kalberer, C.P.; Rossy, E.; Mori, L.; Cosman, D.; De Libero, G.; Wodnar-Filipowicz, A. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood 2005, 105, 3615–3622. [Google Scholar] [CrossRef]
- Poggi, A.; Prevosto, C.; Massaro, A.M.; Negrini, S.; Urbani, S.; Pierri, I.; Saccardi, R.; Gobbi, M.; Zocchi, M.R. Interaction between human NK cells and bone marrow stromal cells induces NK cell triggering: Role of NKp30 and NKG2D receptors. J. Immunol. 2005, 175, 6352–6360. [Google Scholar] [CrossRef] [Green Version]
- Cerboni, C.; Zingoni, A.; Cippitelli, M.; Piccoli, M.; Frati, L.; Santoni, A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood 2007, 110, 606–615. [Google Scholar] [CrossRef]
- Jinushi, M.; Takehara, T.; Kanto, T.; Tatsumi, T.; Groh, V.; Spies, T.; Miyagi, T.; Suzuki, T.; Sasaki, Y.; Hayashi, N. Critical role of MHC class I-related chain A and B expression on IFN-alpha-stimulated dendritic cells in NK cell activation: Impairment in chronic hepatitis C virus infection. J. Immunol. 2003, 170, 1249–1256. [Google Scholar] [CrossRef]
- Borchers, M.T.; Harris, N.L.; Wesselkamper, S.C.; Vitucci, M.; Cosman, D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 291, L222–L231. [Google Scholar] [CrossRef]
- Schrambach, S.; Ardizzone, M.; Leymarie, V.; Sibilia, J.; Bahram, S. In vivo expression pattern of MICA and MICB and its relevance to auto-immunity and cancer. PLoS ONE 2007, 2, e518. [Google Scholar] [CrossRef]
- Eagle, R.A.; Jafferji, I.; Barrow, A.D. Beyond Stressed Self: Evidence for NKG2D Ligand Expression on Healthy Cells. Curr. Immunol. Rev. 2009, 5, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereboeva, L.; Harkins, L.; Wong, S.; Lamb, L.S. The safety of allogeneic innate lymphocyte therapy for glioma patients with prior cranial irradiation. Cancer Immunol. Immunother. 2015, 64, 551–562. [Google Scholar] [CrossRef]
- Lamb, L.S., Jr.; Bowersock, J.; Dasgupta, A.; Gillespie, G.Y.; Su, Y.; Johnson, A.; Spencer, H.T. Engineered drug resistant γδ T cells kill glioblastoma cell lines during a chemotherapy challenge: A strategy for combining chemo- and immunotherapy. PLoS ONE 2013, 8, e51805. [Google Scholar] [CrossRef]
- Antonangeli, F.; Soriani, A.; Cerboni, C.; Sciumè, G.; Santoni, A. How Mucosal Epithelia Deal with Stress: Role of NKG2D/NKG2D Ligands during Inflammation. Front. Immunol. 2017, 8, 1583. [Google Scholar] [CrossRef] [Green Version]
- Babic, M.; Romagnani, C. The Role of Natural Killer Group 2, Member D in Chronic Inflammation and Autoimmunity. Front. Immunol. 2018, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.J.; Markiewicz, M.A.; Polić, B.; Shaw, A.S. Role of NKG2D in obesity-induced adipose tissue inflammation and insulin resistance. PLoS ONE 2014, 9, e110108. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Toledo-Stuardo, K.; García-González, P.; Garrido-Tapia, M.; Kramm, K.; Rodríguez-Siza, J.A.; Hermoso, M.; Ribeiro, C.H.; Molina, M.C. Heat-killed Helicobacter pylori upregulates NKG2D ligands expression on gastric adenocarcinoma cells via Toll-like receptor 4. Helicobacter 2021, 26, e12812. [Google Scholar] [CrossRef]
- Hosomi, S.; Grootjans, J.; Tschurtschenthaler, M.; Krupka, N.; Matute, J.D.; Flak, M.B.; Martinez-Naves, E.; Gomez Del Moral, M.; Glickman, J.N.; Ohira, M.; et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J. Exp. Med. 2017, 214, 2985–2997. [Google Scholar] [CrossRef]
- Smyth, M.J.; Swann, J.; Cretney, E.; Zerafa, N.; Yokoyama, W.M.; Hayakawa, Y. NKG2D function protects the host from tumor initiation. J. Exp. Med. 2005, 202, 583–588. [Google Scholar] [CrossRef]
- Cai, X.; Caballero-Benitez, A.; Gewe, M.M.; Jenkins, I.C.; Drescher, C.W.; Strong, R.K.; Spies, T.; Groh, V. Control of Tumor Initiation by NKG2D Naturally Expressed on Ovarian Cancer Cells. Neoplasia 2017, 19, 471–482. [Google Scholar] [CrossRef]
- Zhou, H.M.; Zhang, J.G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore) 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro. Oncol. 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Flüh, C.; Chitadze, G.; Adamski, V.; Hattermann, K.; Synowitz, M.; Kabelitz, D.; Held-Feindt, J. NKG2D ligands in glioma stem-like cells: Expression in situ and in vitro. Histochem. Cell Biol. 2018, 149, 219–233. [Google Scholar] [CrossRef]
- Oh, S.J.; Yang, J.I.; Kim, O.; Ahn, E.J.; Kang, W.D.; Lee, J.H.; Moon, K.S.; Lee, K.H.; Cho, D. Human U87 glioblastoma cells with stemness features display enhanced sensitivity to natural killer cell cytotoxicity through altered expression of NKG2D ligand. Cancer Cell Int. 2017, 17, 22. [Google Scholar] [CrossRef] [Green Version]
- Skov, S.; Pedersen, M.T.; Andresen, L.; Straten, P.T.; Woetmann, A.; Odum, N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005, 65, 11136–11145. [Google Scholar] [CrossRef] [Green Version]
- Diermayr, S.; Himmelreich, H.; Durovic, B.; Mathys-Schneeberger, A.; Siegler, U.; Langenkamp, U.; Hofsteenge, J.; Gratwohl, A.; Tichelli, A.; Paluszewska, M.; et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood 2008, 111, 1428–1436. [Google Scholar] [CrossRef]
- Langenkamp, U.; Siegler, U.; Jörger, S.; Diermayr, S.; Gratwohl, A.; Kalberer, C.P.; Wodnar-Filipowicz, A. Human acute myeloid leukemia CD34+CD38- stem cells are susceptible to allorecognition and lysis by single KIR-expressing natural killer cells. Haematologica 2009, 94, 1590–1594. [Google Scholar] [CrossRef] [Green Version]
- Trembath, A.P.; Markiewicz, M.A. More than Decoration: Roles for Natural Killer Group 2 Member D Ligand Expression by Immune Cells. Front. Immunol. 2018, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohner, A.; Langenkamp, U.; Siegler, U.; Kalberer, C.P.; Wodnar-Filipowicz, A. Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk. Res. 2007, 31, 1393–1402. [Google Scholar] [CrossRef]
- Molfetta, R.; Quatrini, L.; Zitti, B.; Capuano, C.; Galandrini, R.; Santoni, A.; Paolini, R. Regulation of NKG2D Expression and Signaling by Endocytosis. Trends Immunol. 2016, 37, 790–802. [Google Scholar] [CrossRef]
- Oppenheim, D.E.; Roberts, S.J.; Clarke, S.L.; Filler, R.; Lewis, J.M.; Tigelaar, R.E.; Girardi, M.; Hayday, A.C. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat. Immunol. 2005, 6, 928–937. [Google Scholar] [CrossRef]
- Coudert, J.D.; Scarpellino, L.; Gros, F.; Vivier, E.; Held, W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood 2008, 111, 3571–3578. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Sun, Y.; Song, Y.; Jiao, J.; Shen, B.; Li, W.; Jiang, C.; Li, Y.; Zhang, X.; Yu, J.; et al. ATM kinase regulates tumor immunoreactions in lymphocyte-predominant breast cancer through modulation of NKG2D ligand and TNF cytokines on tumor cells. Med. Mol. Morphol. 2020, 53, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [Green Version]
- Luis Espinoza, J.; Takami, A.; Trung, L.Q.; Nakao, S. Ataxia-telangiectasia mutated kinase-mediated upregulation of NKG2D ligands on leukemia cells by resveratrol results in enhanced natural killer cell susceptibility. Cancer Sci. 2013, 104, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Cecere, F.; Vulpis, E.; Fionda, C.; Molfetta, R.; Soriani, A.; Petrucci, M.T.; Ricciardi, M.R.; Fuerst, D.; Amendola, M.G.; et al. Genotoxic Stress Induces Senescence-Associated ADAM10-Dependent Release of NKG2D MIC Ligands in Multiple Myeloma Cells. J. Immunol. 2015, 195, 736–748. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.R.; Bert, N.L.; Ho, S.S.; Shen, Y.J.; Tang, L.F.; Xiong, G.M.; Croxford, J.L.; Koo, C.X.; Ishii, K.J.; Akira, S.; et al. RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res. 2014, 74, 2193–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef] [Green Version]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [Green Version]
- Cerboni, C.; Fionda, C.; Soriani, A.; Zingoni, A.; Doria, M.; Cippitelli, M.; Santoni, A. The DNA Damage Response: A Common Pathway in the Regulation of NKG2D and DNAM-1 Ligand Expression in Normal, Infected, and Cancer Cells. Front. Immunol. 2014, 4, 508. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Hsiung, B.; Pestal, K.; Procyk, E.; Raulet, D.H. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J. Exp. Med. 2012, 209, 2409–2422. [Google Scholar] [CrossRef] [Green Version]
- Acebes-Huerta, A.; Lorenzo-Herrero, S.; Folgueras, A.R.; Huergo-Zapico, L.; Lopez-Larrea, C.; López-Soto, A.; Gonzalez, S. Drug-induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM-1 receptors. Oncoimmunology 2016, 5, e1074378. [Google Scholar] [CrossRef]
- Leivas, A.; Risueño, R.M.; Guzmán, A.; Sánchez-Vega, L.; Pérez, M.; Megías, D.; Fernández, L.; Alonso, R.; Pérez-Martínez, A.; Rapado, I.; et al. Natural killer cells efficiently target multiple myeloma clonogenic tumor cells. Cancer Immunol. Immunother. 2021, 70, 2911–2924. [Google Scholar] [CrossRef]
- Høgh, R.I.; Møller, S.H.; Jepsen, S.D.; Mellergaard, M.; Lund, A.; Pejtersen, M.; Fitzner, E.; Andresen, L.; Skov, S. Metabolism of short-chain fatty acid propionate induces surface expression of NKG2D ligands on cancer cells. FASEB J. 2020, 34, 15531–15546. [Google Scholar] [CrossRef]
- Andresen, L.; Skovbakke, S.L.; Persson, G.; Hagemann-Jensen, M.; Hansen, K.A.; Jensen, H.; Skov, S. 2-deoxy D-glucose prevents cell surface expression of NKG2D ligands through inhibition of N-linked glycosylation. J. Immunol. 2012, 188, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Guan, D.; Wang, S.; Chai, L.Y.A.; Xu, S.; Lam, K.P. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front. Immunol. 2020, 11, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, S.H.; Mellergaard, M.; Madsen, M.; Bermejo, A.V.; Jepsen, S.D.; Hansen, M.H.; Høgh, R.I.; Aldana, B.I.; Desler, C.; Rasmussen, L.J.; et al. Cytoplasmic Citrate Flux Modulates the Immune Stimulatory NKG2D Ligand MICA in Cancer Cells. Front. Immunol. 2020, 11, 1968. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Unni, A.M.; Bondar, T.; Medzhitov, R. Intrinsic sensor of oncogenic transformation induces a signal for innate immunosurveillance. Proc. Natl. Acad. Sci. USA 2008, 105, 1686–1691. [Google Scholar] [CrossRef] [Green Version]
- Schuster, C.; Berger, A.; Hoelzl, M.A.; Putz, E.M.; Frenzel, A.; Simma, O.; Moritz, N.; Hoelbl, A.; Kovacic, B.; Freissmuth, M.; et al. The cooperating mutation or “second hit” determines the immunologic visibility toward MYC-induced murine lymphomas. Blood 2011, 118, 4635–4645. [Google Scholar] [CrossRef] [Green Version]
- Tokuyama, M.; Lorin, C.; Delebecque, F.; Jung, H.; Raulet, D.H.; Coscoy, L. Expression of the RAE-1 family of stimulatory NK-cell ligands requires activation of the PI3K pathway during viral infection and transformation. PLoS Pathog. 2011, 7, e1002265. [Google Scholar] [CrossRef]
- Okita, R.; Mougiakakos, D.; Ando, T.; Mao, Y.; Sarhan, D.; Wennerberg, E.; Seliger, B.; Lundqvist, A.; Mimura, K.; Kiessling, R. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J. Immunol. 2012, 188, 2136–2145. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.V.; Ho, S.S.; Tan, J.J.; Kamran, N.; Gasser, S. Ras activation induces expression of Raet1 family NK receptor ligands. J. Immunol. 2012, 189, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Tian, X.; Li, Y.; Liu, Y.; Yang, T.; Han, Z.; An, J.; Kong, L.; Li, Y. Epithelial-mesenchymal transition may be involved in the immune evasion of circulating gastric tumor cells via downregulation of ULBP1. Cancer Med. 2020, 9, 2686–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.H.; Kim, J.Y.; Kim, M.J.; Chang, S.H.; Park, Y.S.; Son, C.H.; Park, S.J.; Chung, J.S.; Lee, E.Y.; Kim, S.H.; et al. Quercetin enhances susceptibility to NK cell-mediated lysis of tumor cells through induction of NKG2D ligands and suppression of HSP70. J. Immunother. 2010, 33, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Hromadnikova, I.; Volchenkov, R.; Sedlackova, L.; Spacek, M.; Kozak, T. Expression of heat shock protein 70 and NKG2D ligands in acute myeloid leukemia cell lines. J. Recept. Signal Transduct. 2010, 30, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Fionda, C.; Soriani, A.; Malgarini, G.; Iannitto, M.L.; Santoni, A.; Cippitelli, M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J. Immunol. 2009, 183, 4385–4394. [Google Scholar] [CrossRef] [Green Version]
- Elsner, L.; Muppala, V.; Gehrmann, M.; Lozano, J.; Malzahn, D.; Bickeböller, H.; Brunner, E.; Zientkowska, M.; Herrmann, T.; Walter, L.; et al. The heat shock protein HSP70 promotes mouse NK cell activity against tumors that express inducible NKG2D ligands. J. Immunol. 2007, 179, 5523–5533. [Google Scholar] [CrossRef]
- Strid, J.; Roberts, S.J.; Filler, R.B.; Lewis, J.M.; Kwong, B.Y.; Schpero, W.; Kaplan, D.H.; Hayday, A.C.; Girardi, M. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat. Immunol. 2008, 9, 146–154. [Google Scholar] [CrossRef]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [Green Version]
- Salih, H.R.; Goehlsdorf, D.; Steinle, A. Release of MICB molecules by tumor cells: Mechanism and soluble MICB in sera of cancer patients. Hum. Immunol. 2006, 67, 188–195. [Google Scholar] [CrossRef]
- Waldhauer, I.; Steinle, A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 2006, 66, 2520–2526. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Wang, X.; Zhang, H.; Deng, L.; Zhang, Y. MMP9 mediates MICA shedding in human osteosarcomas. Cell Biol. Int. 2011, 35, 569–574. [Google Scholar] [CrossRef]
- Doubrovina, E.S.; Doubrovin, M.M.; Vider, E.; Sisson, R.B.; O’Reilly, R.J.; Dupont, B.; Vyas, Y.M. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J. Immunol. 2003, 171, 6891–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilpert, J.; Grosse-Hovest, L.; Grünebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Niu, J.; Zhang, J.; Wang, Y.; Zhou, Z.; Zhang, J.; Tian, Z. Opposing effects of interferon-alpha and interferon-gamma on the expression of major histocompatibility complex class I chain-related A in tumors. Cancer Sci. 2008, 99, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Boutet, P.; Agüera-González, S.; Atkinson, S.; Pennington, C.J.; Edwards, D.R.; Murphy, G.; Reyburn, H.T.; Valés-Gómez, M. Cutting edge: The metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J. Immunol. 2009, 182, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Kohga, K.; Takehara, T.; Tatsumi, T.; Miyagi, T.; Ishida, H.; Ohkawa, K.; Kanto, T.; Hiramatsu, N.; Hayashi, N. Anticancer chemotherapy inhibits MHC class I-related chain a ectodomain shedding by downregulating ADAM10 expression in hepatocellular carcinoma. Cancer Res. 2009, 69, 8050–8057. [Google Scholar] [CrossRef] [Green Version]
- Zocchi, M.R.; Catellani, S.; Canevali, P.; Tavella, S.; Garuti, A.; Villaggio, B.; Zunino, A.; Gobbi, M.; Fraternali-Orcioni, G.; Kunkl, A.; et al. High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas. Blood 2012, 119, 1479–1489. [Google Scholar] [CrossRef]
- Oh, S.; Park, Y.; Lee, H.J.; Lee, J.; Lee, S.H.; Baek, Y.S.; Chun, S.K.; Lee, S.M.; Kim, M.; Chon, Y.E.; et al. A Disintegrin and Metalloproteinase 9 (ADAM9) in Advanced Hepatocellular Carcinoma and Their Role as a Biomarker During Hepatocellular Carcinoma Immunotherapy. Cancers 2020, 12, 745. [Google Scholar] [CrossRef] [Green Version]
- Chitadze, G.; Lettau, M.; Luecke, S.; Wang, T.; Janssen, O.; Fürst, D.; Mytilineos, J.; Wesch, D.; Oberg, H.H.; Held-Feindt, J.; et al. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human γδ T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology 2016, 5, e1093276. [Google Scholar] [CrossRef]
- Yamada, N.; Yamanegi, K.; Ohyama, H.; Hata, M.; Nakasho, K.; Futani, H.; Okamura, H.; Terada, N. Hypoxia downregulates the expression of cell surface MICA without increasing soluble MICA in osteosarcoma cells in a HIF-1α-dependent manner. Int. J. Oncol. 2012, 41, 2005–2012. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Kim, H.J.; Kim, C.W.; Kim, H.C.; Jung, Y.; Lee, H.S.; Lee, Y.; Ju, Y.S.; Oh, J.E.; Park, S.H.; et al. Tumor hypoxia represses γδ T cell-mediated antitumor immunity against brain tumors. Nat. Immunol. 2021, 22, 336–346. [Google Scholar] [CrossRef]
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.; Wang, Y.; Lv, N.; Zhang, D.; Williamson, R.A.; Lei, L.; Chen, P.; Lei, L.; Wang, B.; Fu, J.; et al. Hypoxia Impairs NK Cell Cytotoxicity through SHP-1-Mediated Attenuation of STAT3 and ERK Signaling Pathways. J. Immunol. Res. 2020, 2020, 4598476. [Google Scholar] [CrossRef] [PubMed]
- Siegers, G.M.; Dutta, I.; Lai, R.; Postovit, L.M. Functional Plasticity of Gamma Delta T Cells and Breast Tumor Targets in Hypoxia. Front. Immunol. 2018, 9, 1367. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Kato-Kogoe, N.; Yamanegi, K.; Nishiura, H.; Fujihara, Y.; Fukunishi, S.; Okamura, H.; Terada, N.; Nakasho, K. Inhibition of Asparagine-linked Glycosylation Participates in Hypoxia-induced Down-regulation of Cell-surface MICA Expression. Anticancer Res. 2018, 38, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Siemens, D.R.; Hu, N.; Sheikhi, A.K.; Chung, E.; Frederiksen, L.J.; Pross, H.; Graham, C.H. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: Role of nitric oxide. Cancer Res. 2008, 68, 4746–4753. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Germeraad, W.T.; Rouschop, K.M.; Steeghs, E.M.; van Gelder, M.; Bos, G.M.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Israelsson, P.; Ottander, U.; Lundin, E.; Nagaev, I.; Nagaeva, O.; Dehlin, E.; Baranov, V.; Mincheva-Nilsson, L. Differential expression of ligands for NKG2D and DNAM-1 receptors by epithelial ovarian cancer-derived exosomes and its influence on NK cell cytotoxicity. Tumour Biol. 2016, 37, 5455–5466. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [Green Version]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; André, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Lee, Y.S.; Choi, H.; Cho, H.R.; Son, W.C.; Park, Y.S.; Kang, C.D.; Bae, J. Downregulation of NKG2DLs by TGF-β in human lung cancer cells. BMC Immunol. 2021, 22, 44. [Google Scholar] [CrossRef]
- Eisele, G.; Wischhusen, J.; Mittelbronn, M.; Meyermann, R.; Waldhauer, I.; Steinle, A.; Weller, M.; Friese, M.A. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 2006, 129, 2416–2425. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro. Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W., Jr.; Chatterjee, S.K. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J. Immunol. 2005, 175, 5541–5550. [Google Scholar] [CrossRef]
- Trinh, T.L.; Kandell, W.M.; Donatelli, S.S.; Tu, N.; Tejera, M.M.; Gilvary, D.L.; Eksioglu, E.A.; Burnette, A.; Adams, W.A.; Liu, J.; et al. Immune evasion by TGFβ-induced miR-183 repression of MICA/B expression in human lung tumor cells. Oncoimmunology 2019, 8, e1557372. [Google Scholar] [CrossRef] [Green Version]
- Schwinn, N.; Vokhminova, D.; Sucker, A.; Textor, S.; Striegel, S.; Moll, I.; Nausch, N.; Tuettenberg, J.; Steinle, A.; Cerwenka, A.; et al. Interferon-gamma down-regulates NKG2D ligand expression and impairs the NKG2D-mediated cytolysis of MHC class I-deficient melanoma by natural killer cells. Int. J. Cancer 2009, 124, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, X.; Shen, M.; Yang, D.R.; Fang, L.; Weng, G.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol. Oncol. 2018, 12, 269–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.J.; Xu, L.J.; Yang, L.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; Chen, Y. Radiation alters PD-L1/NKG2D ligand levels in lung cancer cells and leads to immune escape from NK cell cytotoxicity via IL-6-MEK/Erk signaling pathway. Oncotarget 2017, 8, 80506–80520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.J.; Ma, Q.; Zhu, J.; Li, J.; Xue, B.X.; Gao, J.; Sun, C.Y.; Zang, Y.C.; Zhou, Y.B.; Yang, D.R.; et al. Combined inhibition of JAK1,2/Stat3-PD-L1 signaling pathway suppresses the immune escape of castration-resistant prostate cancer to NK cells in hypoxia. Mol. Med. Rep. 2018, 17, 8111–8120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, R.; Maeda, A.; Shimizu, K.; Nojima, Y.; Saisho, S.; Nakata, M. Effect of platinum-based chemotherapy on the expression of natural killer group 2 member D ligands, programmed cell death-1 ligand 1 and HLA class I in non-small cell lung cancer. Oncol. Rep. 2019, 42, 839–848. [Google Scholar] [CrossRef]
- Lee, Y.S.; Heo, W.; Choi, H.J.; Cho, H.R.; Nam, J.H.; Ki, Y.G.; Lee, H.R.; Son, W.C.; Park, Y.S.; Kang, C.D.; et al. An inhibitor of programmed death ligand 1 enhances natural killer cell-mediated immunity against malignant melanoma cells. PLoS ONE 2021, 16, e0248870. [Google Scholar] [CrossRef]
- Mormino, A.; Cocozza, G.; Fontemaggi, G.; Valente, S.; Esposito, V.; Santoro, A.; Bernardini, G.; Santoni, A.; Fazi, F.; Mai, A.; et al. Histone-deacetylase 8 drives the immune response and the growth of glioma. Glia 2021, 69, 2682–2698. [Google Scholar] [CrossRef]
- Kim, J.Y.; Son, Y.O.; Park, S.W.; Bae, J.H.; Chung, J.S.; Kim, H.H.; Chung, B.S.; Kim, S.H.; Kang, C.D. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp. Mol. Med. 2006, 38, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Rosental, B.; Appel, M.Y.; Yossef, R.; Hadad, U.; Brusilovsky, M.; Porgador, A. The effect of chemotherapy/radiotherapy on cancerous pattern recognition by NK cells. Curr. Med. Chem. 2012, 19, 1780–1791. [Google Scholar] [CrossRef]
- Cao, G.; Wang, J.; Zheng, X.; Wei, H.; Tian, Z.; Sun, R. Tumor Therapeutics Work as Stress Inducers to Enhance Tumor Sensitivity to Natural Killer (NK) Cell Cytolysis by Up-regulating NKp30 Ligand B7-H6. J. Biol. Chem. 2015, 290, 29964–29973. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Schneider, H.; Silginer, M.; Steinle, A.; Pruschy, M.; Polić, B.; Weller, M.; Roth, P. NKG2D-Dependent Antitumor Effects of Chemotherapy and Radiotherapy against Glioblastoma. Clin. Cancer Res. 2018, 24, 882–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, L.S.; Pereboeva, L.; Youngblood, S.; Gillespie, G.Y.; Nabors, L.B.; Markert, J.M.; Dasgupta, A.; Langford, C.; Spencer, H.T. A combined treatment regimen of MGMT-modified γδ T cells and temozolomide chemotherapy is effective against primary high grade gliomas. Sci. Rep. 2021, 11, 21133. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.C.; Wan, Z.; Sheard, M.A.; Sun, J.; Jackson, J.R.; Malvar, J.; Xu, Y.; Wang, L.; Sposto, R.; Kim, E.S.; et al. TGFβR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) with Natural Killer Cells. Clin. Cancer Res. 2017, 23, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Lai, H.; Chen, T. Boosting Natural Killer Cell-Based Cancer Immunotherapy with Selenocystine/Transforming Growth Factor-Beta Inhibitor-Encapsulated Nanoemulsion. ACS Nano 2020, 14, 11067–11082. [Google Scholar] [CrossRef] [PubMed]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Imai, Y.; Kaito, Y.; Murayama, T.; Sato, K.; Ishida, T.; Yamamoto, J.; Ito, T.; Futami, M.; Ri, M.; et al. Small-molecule HDAC and Akt inhibitors suppress tumor growth and enhance immunotherapy in multiple myeloma. J. Exp. Clin. Cancer Res. 2021, 40, 110. [Google Scholar] [CrossRef]
- Bhat, J.; Dubin, S.; Dananberg, A.; Quabius, E.S.; Fritsch, J.; Dowds, C.M.; Saxena, A.; Chitadze, G.; Lettau, M.; Kabelitz, D. Histone Deacetylase Inhibitor Modulates NKG2D Receptor Expression and Memory Phenotype of Human Gamma/Delta T Cells Upon Interaction With Tumor Cells. Front. Immunol. 2019, 10, 569. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, D.; Schilham, M.W.; Vos, H.I.; Santos, S.J.; Kloess, S.; Buddingh, E.P.; Egeler, R.M.; Hogendoorn, P.C.; Lankester, A.C. Histone deacetylase inhibitors enhance expression of NKG2D ligands in Ewing sarcoma and sensitize for natural killer cell-mediated cytolysis. Clin. Sarcoma Res. 2012, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Son, C.H.; Keum, J.H.; Yang, K.; Nam, J.; Kim, M.J.; Kim, S.H.; Kang, C.D.; Oh, S.O.; Kim, C.D.; Park, Y.S.; et al. Synergistic enhancement of NK cell-mediated cytotoxicity by combination of histone deacetylase inhibitor and ionizing radiation. Radiat. Oncol. 2014, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Cobo, S.; Pieper, N.; Campos-Silva, C.; García-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Valés-Gómez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology 2018, 7, e1392426. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.; Reiners, K.S.; Hansen, H.P.; Engert, A.; Gasser, S.; von Strandmann, E.P. Induction of the DNA damage response by IAP inhibition triggers natural immunity via upregulation of NKG2D ligands in Hodgkin lymphoma in vitro. Biol. Chem. 2013, 394, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Dong, X.W.; Yan, B.; Liu, M.; Xue, T.H.; Liu, H.; You, J.H.; Li, F.; Wang, Z.L.; Chen, Z.N. MG132 selectively upregulates MICB through the DNA damage response pathway in A549 cells. Mol. Med. Rep. 2019, 19, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Böll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.J.; Lundgren, K.; Hansen, H.P.; Engert, A.; et al. Heat shock protein 90 inhibitor BIIB021 (CNF2024) depletes NF-kappaB and sensitizes Hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. 2009, 15, 5108–5116. [Google Scholar] [CrossRef] [Green Version]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Santos, J.M.; Havunen, R.; Hemminki, A. Modulation of the tumor microenvironment with an oncolytic adenovirus for effective T-cell therapy and checkpoint inhibition. Methods Enzymol 2020, 635, 205–230. [Google Scholar] [CrossRef]
- Lodoen, M.B.; Lanier, L.L. Viral modulation of NK cell immunity. Nat. Rev. Microbiol. 2005, 3, 59–69. [Google Scholar] [CrossRef]
- Draghi, M.; Pashine, A.; Sanjanwala, B.; Gendzekhadze, K.; Cantoni, C.; Cosman, D.; Moretta, A.; Valiante, N.M.; Parham, P. NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J. Immunol. 2007, 178, 2688–2698. [Google Scholar] [CrossRef] [Green Version]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McSharry, B.P.; Burgert, H.G.; Owen, D.P.; Stanton, R.J.; Prod’homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; et al. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.; Bonaparte, M.; Sacks, J.; Guterman, J.; Fogli, M.; Mavilio, D.; Barker, E. HIV modulates the expression of ligands important in triggering natural killer cell cytotoxic responses on infected primary T-cell blasts. Blood 2007, 110, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Easom, N.J.W.; Marks, M.; Jobe, D.; Gillmore, R.; Meyer, T.; Maini, M.K.; Njie, R. ULBP1 Is Elevated in Human Hepatocellular Carcinoma and Predicts Outcome. Front. Oncol. 2020, 10, 971. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Chung, J.Y.; Kim, S.; Braunschweig, T.; Kang, T.H.; Kim, J.; Chung, E.J.; Hewitt, S.M.; Kim, J.H. MICA/B and ULBP1 NKG2D ligands are independent predictors of good prognosis in cervical cancer. BMC Cancer 2014, 14, 957. [Google Scholar] [CrossRef]
- Watson, N.F.; Spendlove, I.; Madjd, Z.; McGilvray, R.; Green, A.R.; Ellis, I.O.; Scholefield, J.H.; Durrant, L.G. Expression of the stress-related MHC class I chain-related protein MICA is an indicator of good prognosis in colorectal cancer patients. Int. J. Cancer 2006, 118, 1445–1452. [Google Scholar] [CrossRef]
- Paschen, A.; Sucker, A.; Hill, B.; Moll, I.; Zapatka, M.; Nguyen, X.D.; Sim, G.C.; Gutmann, I.; Hassel, J.; Becker, J.C.; et al. Differential clinical significance of individual NKG2D ligands in melanoma: Soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin. Cancer Res. 2009, 15, 5208–5215. [Google Scholar] [CrossRef] [Green Version]
- Ruan, G.T.; Xie, H.L.; Zhu, L.C.; Ge, Y.Z.; Yan, L.; Liao, C.; Gong, Y.Z.; Shi, H.P. Immune ULBP1 is Elevated in Colon Adenocarcinoma and Predicts Prognosis. Front. Genet. 2022, 13, 762514. [Google Scholar] [CrossRef]
- McGilvray, R.W.; Eagle, R.A.; Watson, N.F.; Al-Attar, A.; Ball, G.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin. Cancer Res. 2009, 15, 6993–7002. [Google Scholar] [CrossRef] [Green Version]
- Boissel, N.; Rea, D.; Tieng, V.; Dulphy, N.; Brun, M.; Cayuela, J.M.; Rousselot, P.; Tamouza, R.; Le Bouteiller, P.; Mahon, F.X.; et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J. Immunol. 2006, 176, 5108–5116. [Google Scholar] [CrossRef]
- Okita, R.; Yukawa, T.; Nojima, Y.; Maeda, A.; Saisho, S.; Shimizu, K.; Nakata, M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2016, 65, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Heo, W.; Nam, J.; Jeung, Y.H.; Bae, J. The combination of ionizing radiation and proteasomal inhibition by bortezomib enhances the expression of NKG2D ligands in multiple myeloma cells. J. Radiat. Res. 2018, 59, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, C.; Jin, H.; Li, M.; Zhu, S.; Zhou, L.; Jin, F.; Zhou, Y.; Xu, D.; Xu, J.; Zhao, L.; et al. Low-dose bortezomib increases the expression of NKG2D and DNAM-1 ligands and enhances induced NK and γδ T cell-mediated lysis in multiple myeloma. Oncotarget 2017, 8, 5954–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Story, J.Y.; Zoine, J.T.; Burnham, R.E.; Hamilton, J.A.G.; Spencer, H.T.; Doering, C.B.; Raikar, S.S. Bortezomib enhances cytotoxicity of ex vivo-expanded gamma delta T cells against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Cytotherapy 2021, 23, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Gravett, A.M.; Dalgleish, A.G.; Copier, J. In vitro culture with gemcitabine augments death receptor and NKG2D ligand expression on tumour cells. Sci. Rep. 2019, 9, 1544. [Google Scholar] [CrossRef] [Green Version]
- Okita, R.; Wolf, D.; Yasuda, K.; Maeda, A.; Yukawa, T.; Saisho, S.; Shimizu, K.; Yamaguchi, Y.; Oka, M.; Nakayama, E.; et al. Contrasting Effects of the Cytotoxic Anticancer Drug Gemcitabine and the EGFR Tyrosine Kinase Inhibitor Gefitinib on NK Cell-Mediated Cytotoxicity via Regulation of NKG2D Ligand in Non-Small-Cell Lung Cancer Cells. PLoS ONE 2015, 10, e0139809. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, D.; Li, Z.; Jiao, D.; Jin, L.; Cong, J.; Zheng, X.; Xu, L. Low-Dose Gemcitabine Treatment Enhances Immunogenicity and Natural Killer Cell-Driven Tumor Immunity in Lung Cancer. Front. Immunol. 2020, 11, 331. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, W.J.; Zhang, J.N.; Zhang, X.Y. 5-Fluorouracil and interleukin-2 immunochemotherapy enhances immunogenicity of non-small cell lung cancer A549 cells through upregulation of NKG2D ligands. Asian Pac. J. Cancer Prev. 2014, 15, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Khallouf, H.; Märten, A.; Serba, S.; Teichgräber, V.; Büchler, M.W.; Jäger, D.; Schmidt, J. 5-Fluorouracil and interferon-α immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class I. J. Immunother. 2012, 35, 245–253. [Google Scholar] [CrossRef]
- Okimoto, T.; Kotani, H.; Iida, Y.; Koyanagi, A.; Tanino, R.; Tsubata, Y.; Isobe, T.; Harada, M. Pemetrexed sensitizes human lung cancer cells to cytotoxic immune cells. Cancer Sci. 2020, 111, 1910–1920. [Google Scholar] [CrossRef]
- Frazao, A.; Rethacker, L.; Jeudy, G.; Colombo, M.; Pasmant, E.; Avril, M.F.; Toubert, A.; Moins-Teisserenc, H.; Roelens, M.; Dalac, S.; et al. BRAF inhibitor resistance of melanoma cells triggers increased susceptibility to natural killer cell-mediated lysis. J. Immunother. Cancer 2020, 8, e000275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Li, S.; Li, B.; Sun, L.; Li, H.; Lin, P.; Wang, S.; Teng, W.; Zhou, X.; et al. Decitabine Enhances Vγ9Vδ2 T Cell-Mediated Cytotoxic Effects on Osteosarcoma Cells via the NKG2DL-NKG2D Axis. Front. Immunol. 2018, 9, 1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Kim, W.J.; Rao, A.V.; Jaman, E.; Deibert, C.P.; Sandlesh, P.; Krueger, K.; Allen, J.C.; Amankulor, N.M. In vivo efficacy of decitabine as a natural killer cell-mediated immunotherapy against isocitrate dehydrogenase mutant gliomas. Neurosurg. Focus 2022, 52, E3. [Google Scholar] [CrossRef] [PubMed]
- Allende-Vega, N.; Marco Brualla, J.; Falvo, P.; Alexia, C.; Constantinides, M.; de Maudave, A.F.; Coenon, L.; Gitenay, D.; Mitola, G.; Massa, P.; et al. Metformin sensitizes leukemic cells to cytotoxic lymphocytes by increasing expression of intercellular adhesion molecule-1 (ICAM-1). Sci. Rep. 2022, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Yin, T.; Li, D.; Gao, X.; Wan, Y.; Ma, X.; Ye, T.; Guo, F.; Sun, J.; Lin, Z.; et al. Enhanced interaction between natural killer cells and lung cancer cells: Involvement in gefitinib-mediated immunoregulation. J. Transl. Med. 2013, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.Z.; Liu, G.J.; Zhang, X.J.; Zhao, J.Z.; Feng, R.T. Erlotinib enhances the CIK cell-killing sensitivity of lung adenocarcinoma A549 cells. Genet. Mol. Res. 2015, 14, 3082–3089. [Google Scholar] [CrossRef]
- Hervieu, A.; Rébé, C.; Végran, F.; Chalmin, F.; Bruchard, M.; Vabres, P.; Apetoh, L.; Ghiringhelli, F.; Mignot, G. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J. Investig. Dermatol. 2013, 133, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Hervieu, A.; Mignot, G.; Ghiringhelli, F. Dacarbazine mediate antimelanoma effects via NK cells. Oncoimmunology 2013, 2, e23714. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wang, Y.; Li, Y.; Guo, K.; He, Y. Role of sorafenib and sunitinib in the induction of expressions of NKG2D ligands in nasopharyngeal carcinoma with high expression of ABCG2. J. Cancer Res. Clin. Oncol. 2011, 137, 829–837. [Google Scholar] [CrossRef]
- Huang, Y.X.; Chen, X.T.; Guo, K.Y.; Li, Y.H.; Wu, B.Y.; Song, C.Y.; He, Y.J. Sunitinib Induces NK-κB-dependent NKG2D Ligand Expression in Nasopharyngeal Carcinoma and Hepatoma Cells. J. Immunother. 2017, 40, 164–174. [Google Scholar] [CrossRef]
- Cucè, M.; Gallo Cantafio, M.E.; Siciliano, M.A.; Riillo, C.; Caracciolo, D.; Scionti, F.; Staropoli, N.; Zuccalà, V.; Maltese, L.; Di Vito, A.; et al. Trabectedin triggers direct and NK-mediated cytotoxicity in multiple myeloma. J. Hematol. Oncol. 2019, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Amin, P.J.; Shankar, B.S. Sulforaphane induces ROS mediated induction of NKG2D ligands in human cancer cell lines and enhances susceptibility to NK cell mediated lysis. Life Sci. 2015, 126, 19–27. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
NKG2D ligand (NKG2DL) binding activates the cytotoxic potential of effector cells. In response to stressors, human or murine NKG2D ligands are expressed on the surface of target cells. NKG2D ligands bind to the respective NKG2D receptor found on cells such as natural killer (NK) cells or gamma-delta T cells. Utilizing adaptor molecules DAP10 or DAP12 (murine), the signal transducers, PI3K and Grb2, are recruited to the NKG2D-binding domain. Through intracellular signaling cascades, transcriptional activation of target genes associated with effector cell maintenance, including those regulating proliferation, cytotoxicity, and cytokine release, is initiated. Image generated using Biorender.com (accessed on 22 April 2022), licensure certificate available as File S1.
Figure 1.
NKG2D ligand (NKG2DL) binding activates the cytotoxic potential of effector cells. In response to stressors, human or murine NKG2D ligands are expressed on the surface of target cells. NKG2D ligands bind to the respective NKG2D receptor found on cells such as natural killer (NK) cells or gamma-delta T cells. Utilizing adaptor molecules DAP10 or DAP12 (murine), the signal transducers, PI3K and Grb2, are recruited to the NKG2D-binding domain. Through intracellular signaling cascades, transcriptional activation of target genes associated with effector cell maintenance, including those regulating proliferation, cytotoxicity, and cytokine release, is initiated. Image generated using Biorender.com (accessed on 22 April 2022), licensure certificate available as File S1.

Figure 2.
Mechanisms associated with NKG2D ligand induction. Commonly activated pathways associated with cancer progression may influence stress ligand expression. The DNA damage response pathway, which is activated during cancer treatments, increases NKG2D ligand expression via activation of signaling kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and RAD3 related (ATR). ATM/ATR promote the activation of downstream targets that increase and maintain ligand expression. Genetic alterations, such as the oncogenic hits that promote tumorigenesis and behavioral phenotypes, including excessive proliferation and “epithelial-to-mesenchymal” transition, associated with tumor biology are also important regulators of NKG2D ligand expression. Image generated using Biorender.com (accessed on 16 March 2022), licensure certificate available as File S1.
Figure 2.
Mechanisms associated with NKG2D ligand induction. Commonly activated pathways associated with cancer progression may influence stress ligand expression. The DNA damage response pathway, which is activated during cancer treatments, increases NKG2D ligand expression via activation of signaling kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and RAD3 related (ATR). ATM/ATR promote the activation of downstream targets that increase and maintain ligand expression. Genetic alterations, such as the oncogenic hits that promote tumorigenesis and behavioral phenotypes, including excessive proliferation and “epithelial-to-mesenchymal” transition, associated with tumor biology are also important regulators of NKG2D ligand expression. Image generated using Biorender.com (accessed on 16 March 2022), licensure certificate available as File S1.

Figure 3.
Repression mechanisms of NKG2D ligands in cancer. Multiple mechanisms have been identified in cancer cells to evade immune cell recognition. Generation and release of NKG2D ligands into the extracellular space via exosomes is enhanced and stimulated by the hypoxic tumor microenvironment. Similarly, enhanced proteolytic cleavage activity of matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase (ADAM) family members is regulated by the hypoxic tumor microenvironment. Immunosuppressive and inhibitory mediators (Transforming Growth Factor Beta (TGF-β), Janus Kinase and Signal Transducer and Activator of Transcription (JAK/STAT), and Programmed Death Receptor Ligand 1 and 2 (PDL1/2)) also collectively diminish NKG2D ligand expression by potentiating the immunosuppressive landscape in cancer. Image generated using Biorender.com (accessed on 16 March 2022), licensure certificate available as File S1.
Figure 3.
Repression mechanisms of NKG2D ligands in cancer. Multiple mechanisms have been identified in cancer cells to evade immune cell recognition. Generation and release of NKG2D ligands into the extracellular space via exosomes is enhanced and stimulated by the hypoxic tumor microenvironment. Similarly, enhanced proteolytic cleavage activity of matrix metalloproteinase (MMP) and a disintegrin and metalloproteinase (ADAM) family members is regulated by the hypoxic tumor microenvironment. Immunosuppressive and inhibitory mediators (Transforming Growth Factor Beta (TGF-β), Janus Kinase and Signal Transducer and Activator of Transcription (JAK/STAT), and Programmed Death Receptor Ligand 1 and 2 (PDL1/2)) also collectively diminish NKG2D ligand expression by potentiating the immunosuppressive landscape in cancer. Image generated using Biorender.com (accessed on 16 March 2022), licensure certificate available as File S1.


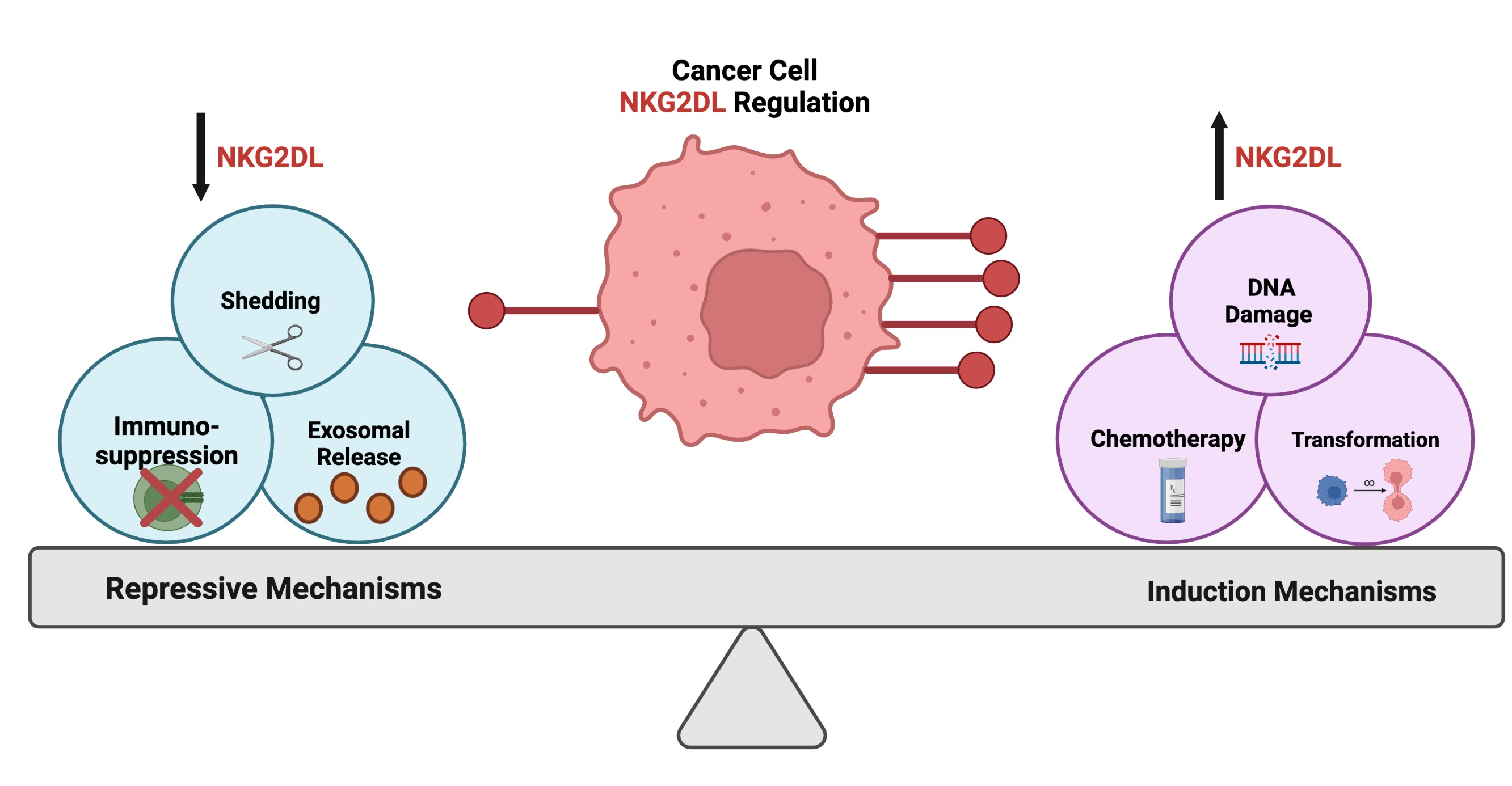{kind=link}
{kind=link}
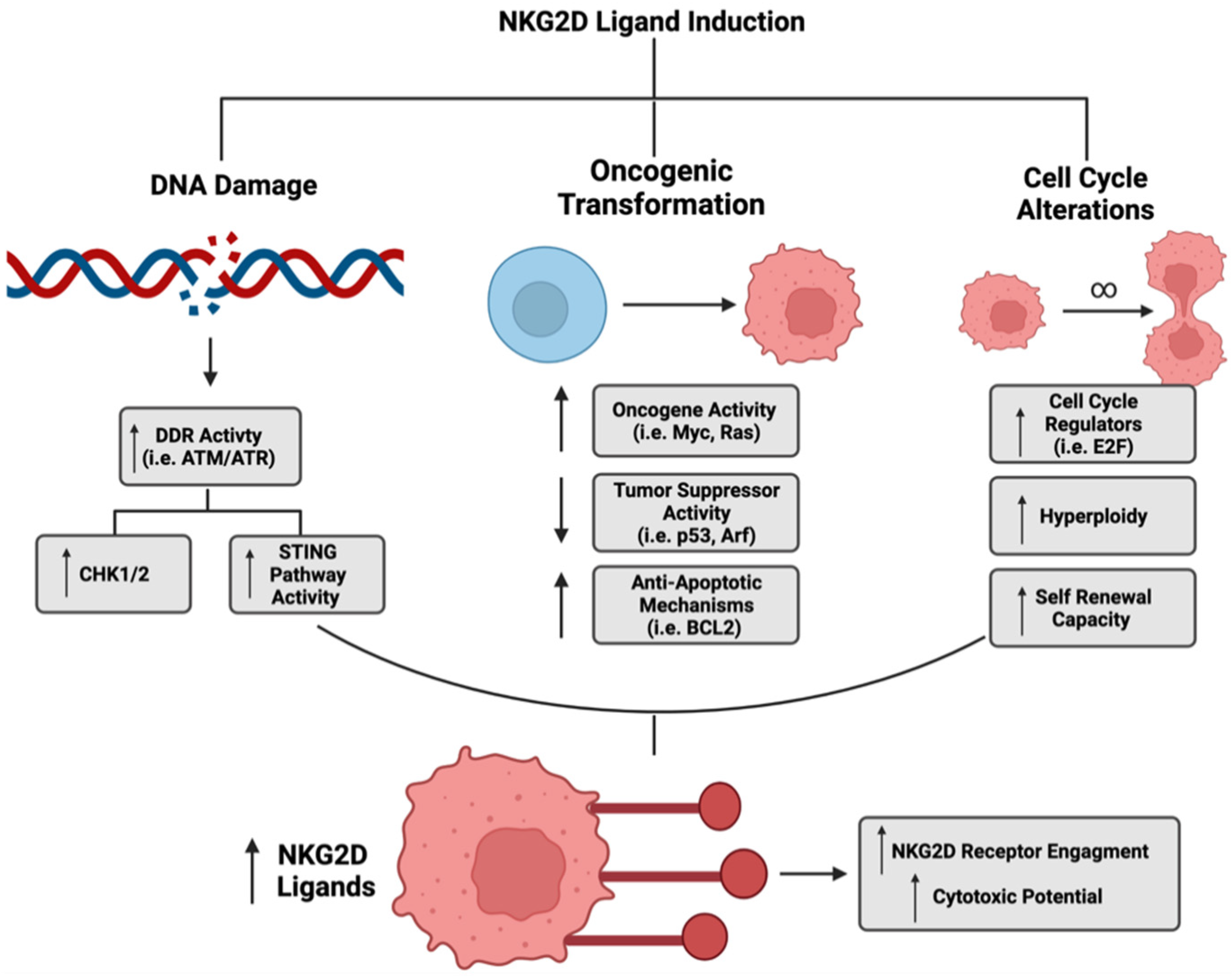{kind=link}
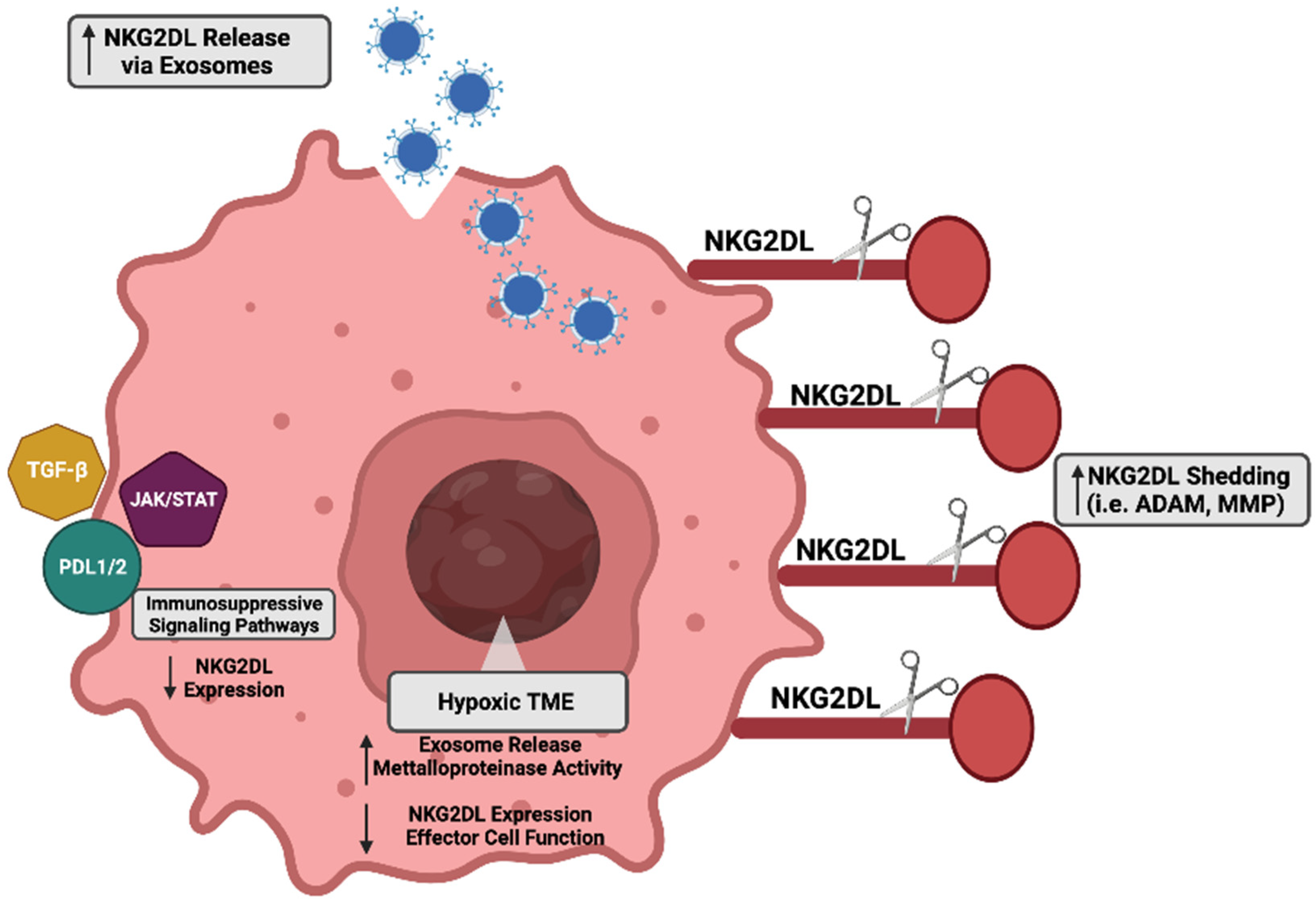{kind=link}
Table 1.
Current clinical trials utilizing the NKG2D stress response pathway.
| Study Title | Disease Targeted | Therapeutic Approach | Clinical Trials Identifier |
|---|---|---|---|
| NKG2D-based CAR T-cells Immunotherapy for Patient with r/r NKG2DL+ Solid Tumors | Hepatocellular Carcinoma, Glioblastoma, Medulloblastoma, Colon Cancer | Autologous genetically modified anti-NKG2DLs CAR transduced T cells | NCT05131763 |
| NKG2D CAR-T Cell Therapy for Patients With Relapsed and/or Refractory Acute Myeloid Leukemia | Acute Myeloid Leukemia | NKG2D CAR T cells | NCT04658004 |
| Pilot Study of NKG2D CAR-T in Treating Patients with Recurrent Glioblastoma | Recurrent Glioblastoma | NKG2D CAR T cells | NCT04717999 |
| alloSHRINK—Standard cHemotherapy Regimen and Immunotherapy with Allogeneic NKG2D-based CYAD-101 Chimeric Antigen Receptor T-cells | Unresectable Metastatic Colorectal Carcinoma | Allogeneic NKG2D-based CYAD-101 Chimeric antigen Receptor T-cells, 5-FU, leucovorin, oxaliplatin, irinotecan | NCT03692429 |
| Safety Study of Chimeric Antigen Receptor Modified T-cells Targeting NKG2D-Ligands | Acute Myeloid Leukemia, Multiple Myeloma Myelodysplastic Syndrome | NKG2D CAR T cells | NCT02203825 |
| Pilot Study of NKG2D-Ligand Targeted CAR-NK Cells in Patients With Metastatic Solid Tumours | Solid Tumors | CAR-NK Cells followed by IL-2 injection | NCT03415100 |
| Immunotherapy of CD8+ NKG2D+ AKT Cell With Chemotherapy to Pancreatic Cancer | Pancreatic Ductal Adenocarcinoma | CD8+, NKG2D+, AKT cells, Gemcitabine | NCT02929797 |
| NKG2D CAR-T(KD-025) in the Treatment of Relapsed or Refractory NKG2DL+ Tumors | Solid Tumor, Hepatocellular Carcinoma, Colorectal Carcinoma, Glioma | Autologous genetically modified anti-NKG2DLs CAR transduced T cells | NCT04550663 |
| Adoptive Cellular Immunotherapy Following Autologous Peripheral Blood Stem Cell Transplantation for Multiple Myeloma | Myeloma, Transplant Eligible Patients | Cytotoxic T-cells, IL-2, GM-CSF | NCT00439465 |
| Novel Gamma-Delta (γδ) T Cell Therapy for Treatment of Patients with Newly Diagnosed Glioblastoma (DRI) | Glioblastoma | Gene modified drug resistant immunotherapy (γδT Cell) administered | NCT04165941 |
Table 2.
Chemotherapies investigated in cancer for NKG2D ligand induction.
| Chemotherapy | Cancer Type | Ligands Induced | Reference(s) |
|---|---|---|---|
| Cisplatin | Lung | MICA/B; ULB2/5/6 | [185,221] |
| Bortezomib | Multiple Myeloma AML, ALL | MICA/B; ULBP1/2/3/5/6 | [222,223,224] |
| Gemcitabine | Lung, Hepatocellular, Colorectal | MICA/B; ULBP1/2/3/5/6 | [225,226,227] |
| 5-Fluorouracil | Pancreatic Cancer, Lung | MICA/B; ULBP1/2/4/5/6 | [228,229] |
| Pemetrexed | Lung | MICA/B; ULBP2/5/6 | [230] |
| Vemurafenib | Melanoma | MICA; ULBP2 | [231] |
| Decitabine | Osteosarcoma, IDH Mutant Glioma | MICB; ULBP1/3 | [105,232,233] |
| Temozolomide | Glioblastoma | MICA/MICB; ULBP1/2/3/4 | [93,191,192] |
| Metformin | Leukemia | ULBP1 | [234] |
| Gefitinib | Lung | MICA; ULBP1/2 | [226,235] |
| Erlotinib | Lung | MICB; ULBP1 | [236] |
| Dacarbazine | Melanoma | Rae-1; Mult-1 | [237,238] |
| Sunitinib | Nasopharyngeal | MICA/B; ULBP1/2/3 | [239,240] |
| Trabectedin | Multiple Myeloma | MICA/B; ULBP1 | [241] |
| Sulforaphane | Breast, Adenocarcinoma, Lymphoma | MICA/B | [242] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jones, A.B.; Rocco, A.; Lamb, L.S.; Friedman, G.K.; Hjelmeland, A.B. Regulation of NKG2D Stress Ligands and Its Relevance in Cancer Progression. Cancers 2022, 14, 2339. https://doi.org/10.3390/cancers14092339
AMA Style
Jones AB, Rocco A, Lamb LS, Friedman GK, Hjelmeland AB. Regulation of NKG2D Stress Ligands and Its Relevance in Cancer Progression. Cancers. 2022; 14(9):2339. https://doi.org/10.3390/cancers14092339
Chicago/Turabian StyleJones, Amber B., Abbey Rocco, Lawrence S. Lamb, Gregory K. Friedman, and Anita B. Hjelmeland. 2022. "Regulation of NKG2D Stress Ligands and Its Relevance in Cancer Progression" Cancers 14, no. 9: 2339. https://doi.org/10.3390/cancers14092339
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.