
Blocking Gi/o-Coupled Signaling Eradicates Cancer Stem Cells and Sensitizes Breast Tumors to HER2-Targeted Therapies to Inhibit Tumor Relapse



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mouse Studies
2.3. Fluorescence-Activated Cell Sorting
2.4. Flow Cytometry
2.5. Cell Lines
2.6. Isolation of Mammary Epithelial Cells
2.7. Lentiviral Constructs
2.8. Establishment of Stable Cell Lines
2.9. Tumorsphere Culture
2.10. Western Blotting Analysis
2.11. Statistics
3. Results
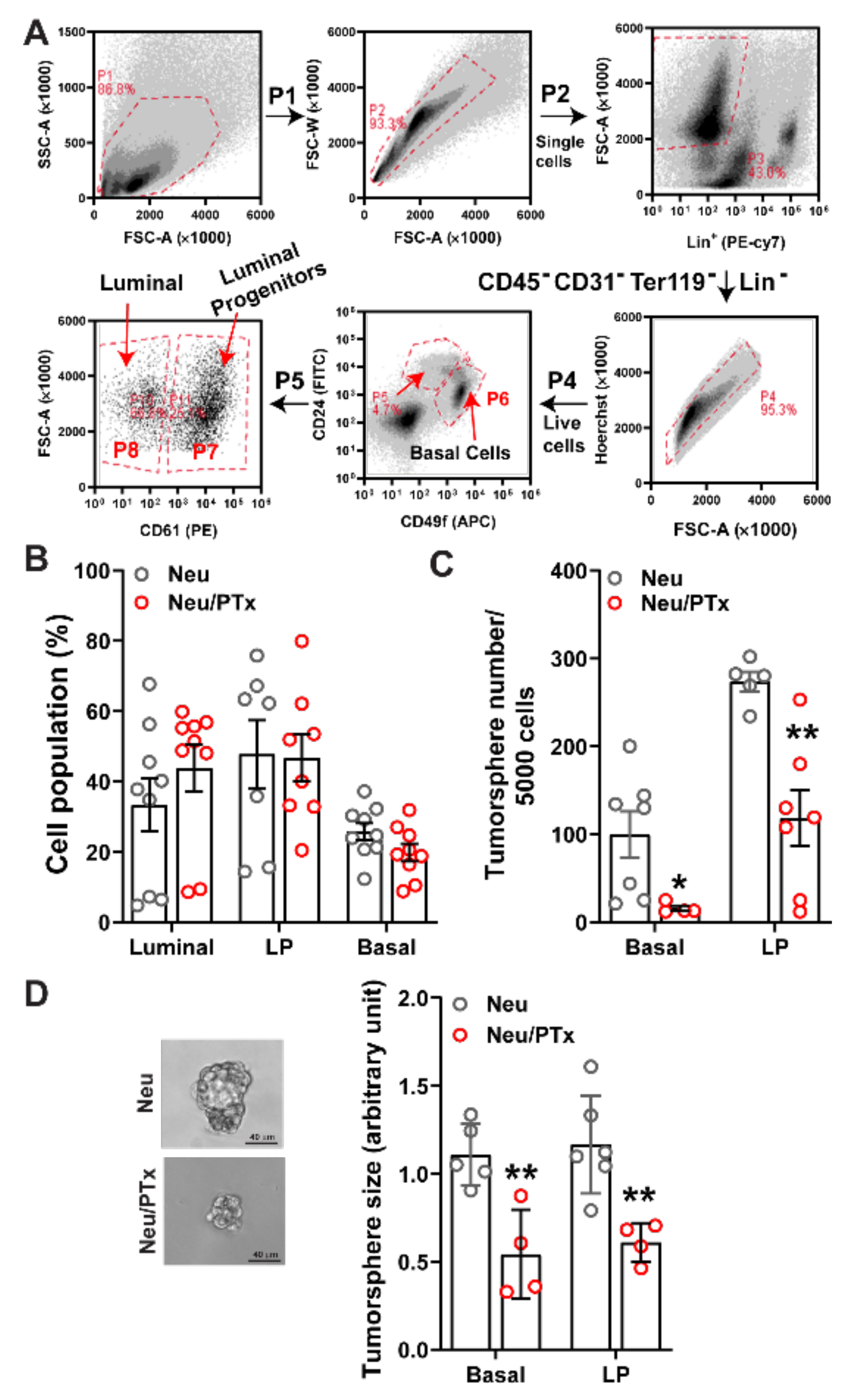
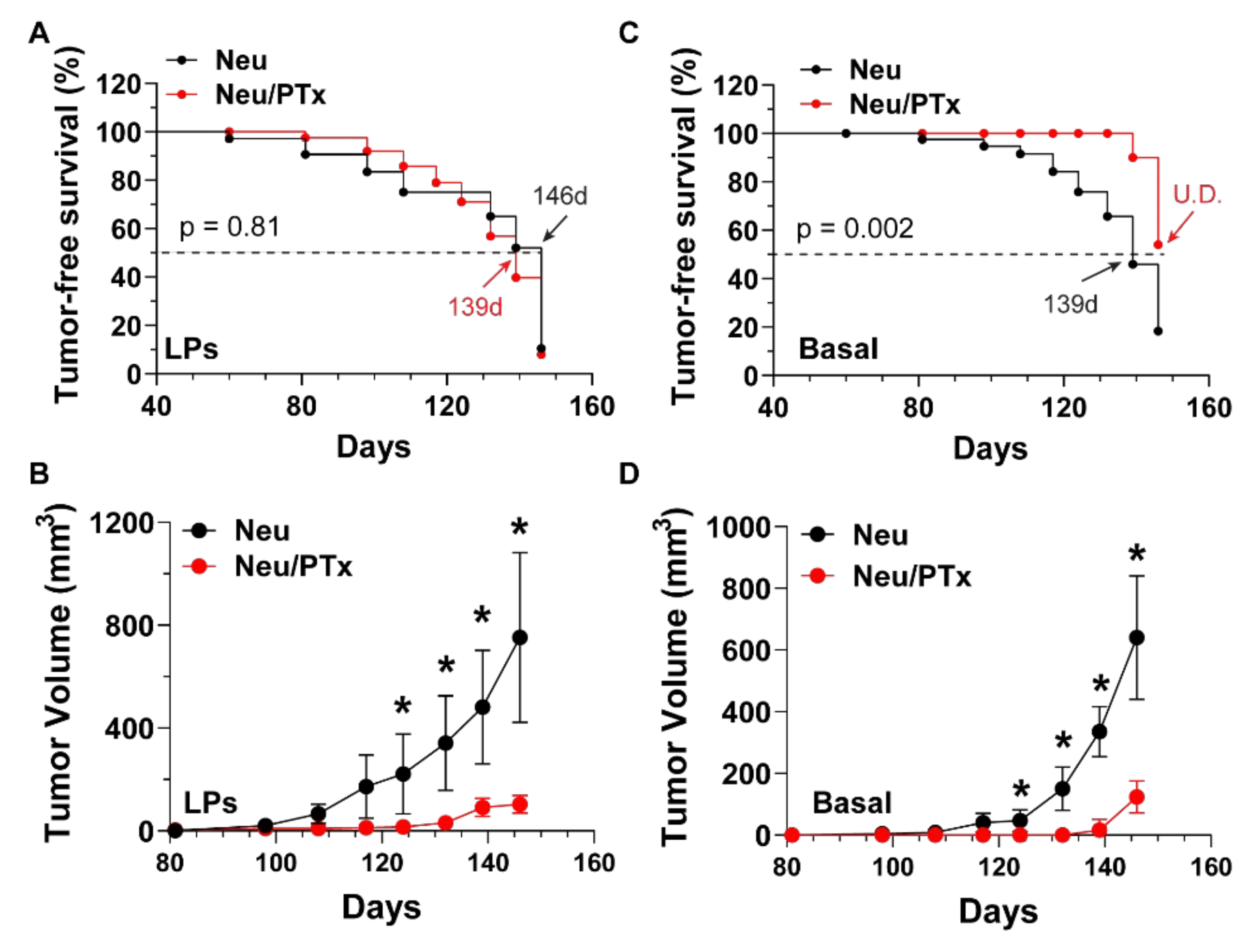
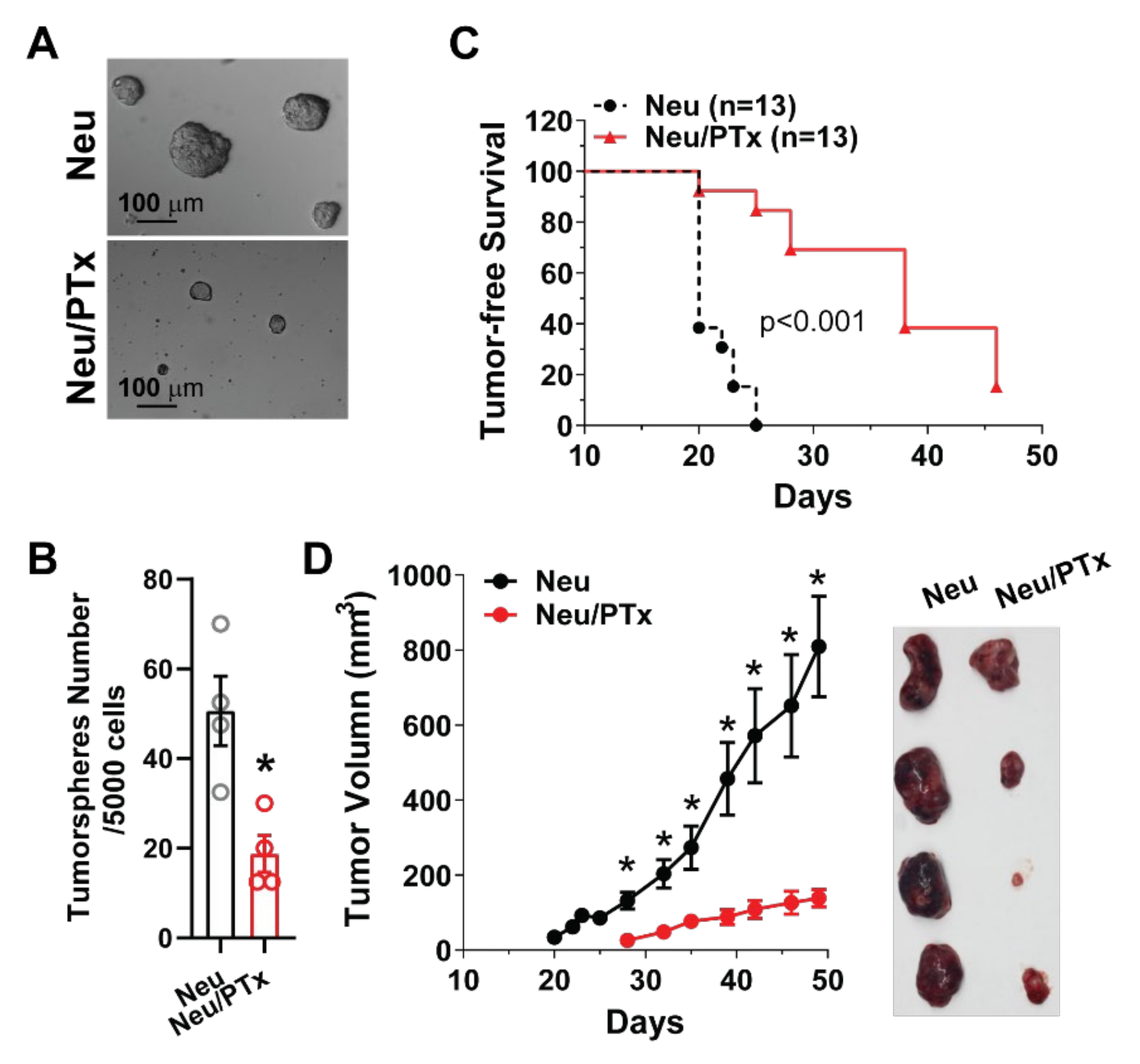
3.1. Blocking Gi/o-GPCR Signaling Suppresses the Tumorigenicity of HER2-Induced CSCs
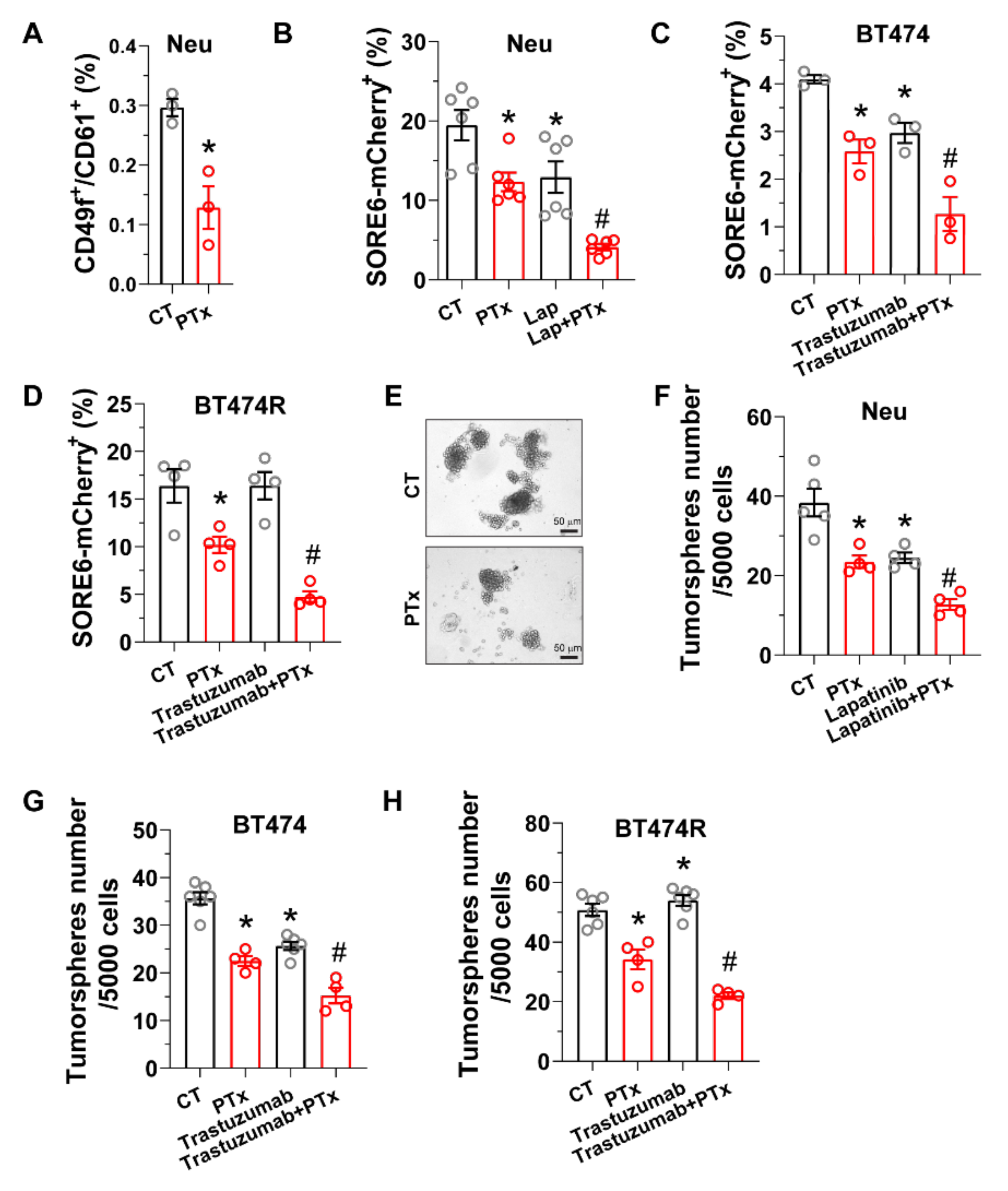
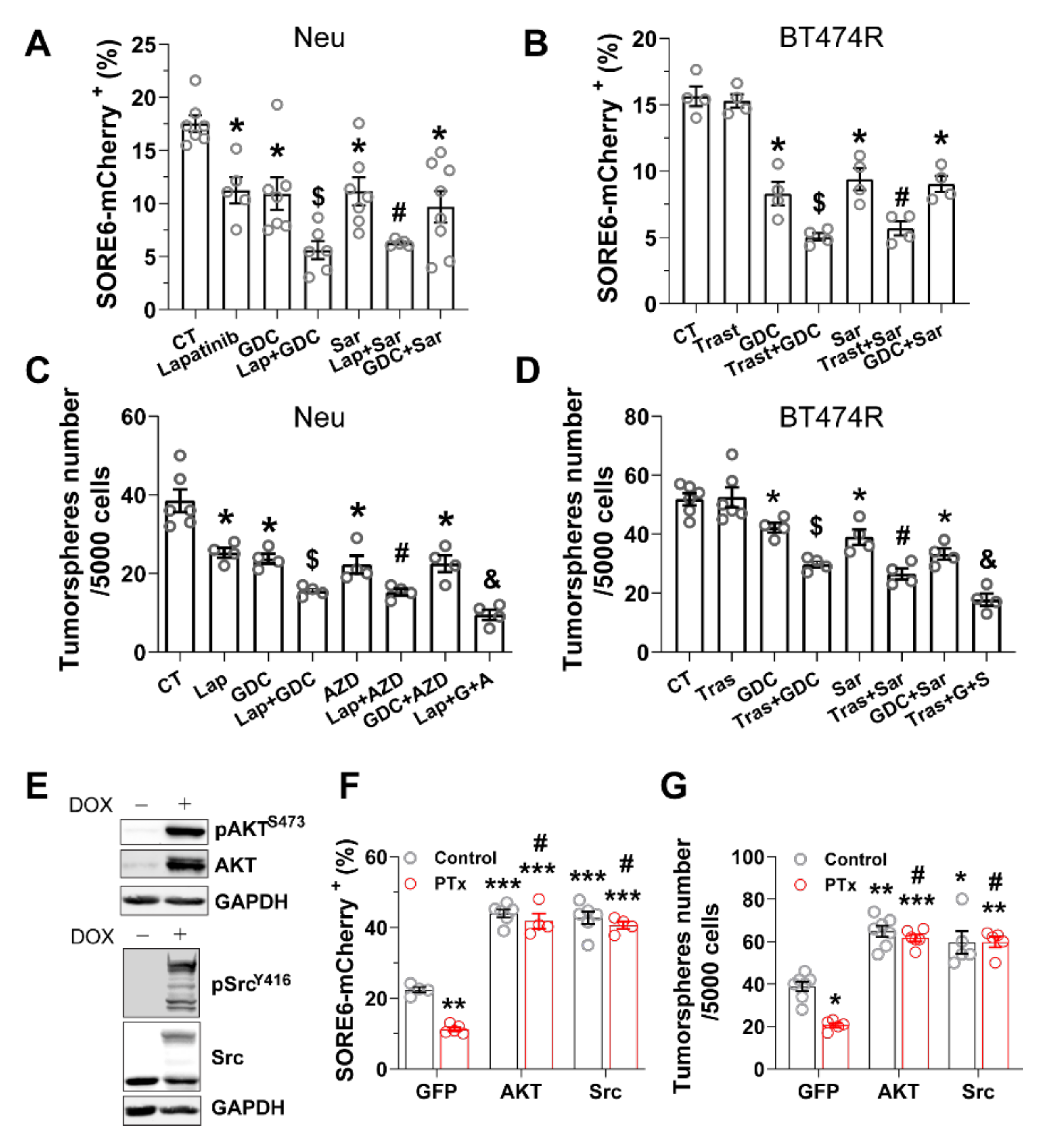
3.2. Blocking Gi/o-GPCR Signaling Enhances CSC Sensitivity to HER2-Targeted Therapy
3.3. PI3K/AKT and Src Pathways Regulate CSC Sensitivity to HER2-Targeted Therapy
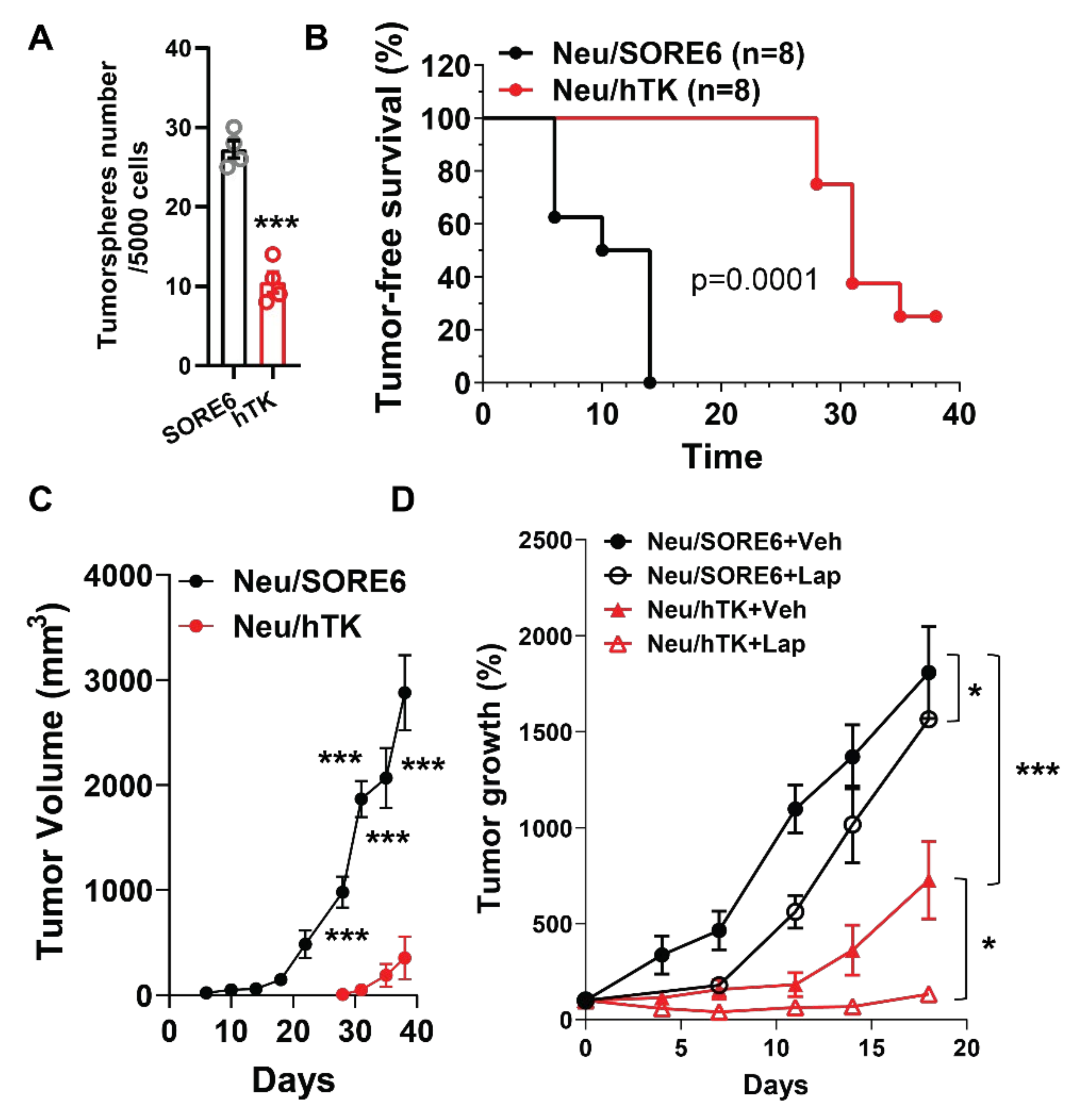
3.4. CSCs Elimination Suppresses Initiation and Progression by HER2+ Tumors and Enhances Lapatinib Therapeutic Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Han, X.; Liu, S.; Wang, L. CSCs in Breast Cancer-One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers 2019, 11, 1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef] [Green Version]
- Korkaya, H.; Wicha, M.S. HER2 and breast cancer stem cells: More than meets the eye. Cancer Res. 2013, 73, 3489–3493. [Google Scholar] [CrossRef] [Green Version]
- Pupa, S.M.; Ligorio, F.; Cancila, V.; Franceschini, A.; Tripodo, C.; Vernieri, C.; Castagnoli, L. HER2 Signaling and Breast Cancer Stem Cells: The Bridge behind HER2-Positive Breast Cancer Aggressiveness and Therapy Refractoriness. Cancers 2021, 13, 4778. [Google Scholar] [CrossRef]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013, 73, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef] [Green Version]
- Martin-Castillo, B.; Lopez-Bonet, E.; Cuyas, E.; Vinas, G.; Pernas, S.; Dorca, J.; Menendez, J.A. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): Towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget 2015, 6, 32317–32338. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Yang, L.; Liu, H.; Luo, X. Cancer stem cell-targeted therapeutic approaches for overcoming trastuzumab resistance in HER2-positive breast cancer. Stem Cells 2021, 39, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, A.; Bhola, N.E.; Sutton, C.; Ghosh, R.; Kuba, M.G.; Dave, B.; Chang, J.C.; Arteaga, C.L. Trastuzumab-resistant cells rely on a HER2-PI3K-FoxO-survivin axis and are sensitive to PI3K inhibitors. Cancer Res. 2013, 73, 1190–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daverey, A.; Drain, A.P.; Kidambi, S. Physical Intimacy of Breast Cancer Cells with Mesenchymal Stem Cells Elicits Trastuzumab Resistance through Src Activation. Sci. Rep. 2015, 5, 13744. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef]
- Choi, H.Y.; Saha, S.K.; Kim, K.; Kim, S.; Yang, G.M.; Kim, B.; Kim, J.H.; Cho, S.G. G protein-coupled receptors in stem cell maintenance and somatic reprogramming to pluripotent or cancer stem cells. BMB Rep. 2015, 48, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Lynch, J.R.; Wang, J.Y. G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. Int. J. Mol. Sci. 2016, 17, 707. [Google Scholar] [CrossRef] [Green Version]
- Insel, P.A.; Sriram, K.; Gorr, M.W.; Wiley, S.Z.; Michkov, A.; Salmeron, C.; Chinn, A.M. GPCRomics: An Approach to Discover GPCR Drug Targets. Trends Pharmacol. Sci. 2019, 40, 378–387. [Google Scholar] [CrossRef]
- Campbell, A.P.; Smrcka, A.V. Targeting G protein-coupled receptor signalling by blocking G proteins. Nat. Rev. Drug Discov. 2018, 17, 789–803. [Google Scholar] [CrossRef]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Farnie, G.; Bundred, N.J.; Simoes, B.M.; Shergill, A.; Landberg, G.; Howell, S.J.; Clarke, R.B. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin. Cancer Res. 2013, 19, 643–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, R.R.; Yadav, P.; Sahay, D.; Bhargava, D.K.; Creighton, C.J.; Yazdanfard, S.; Al-Rawi, A.; Yadav, V.; Qin, L.; Nanda, S.; et al. GPCRs profiling and identification of GPR110 as a potential new target in HER2+ breast cancer. Breast Cancer Res. Treat. 2018, 170, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Lyu, C.; Ye, Y.; Lensing, M.M.; Wagner, K.U.; Weigel, R.J.; Chen, S. Targeting Gi/o protein-coupled receptor signaling blocks HER2-induced breast cancer development and enhances HER2-targeted therapy. JCI Insight 2021, 6, e150532. [Google Scholar] [CrossRef] [PubMed]
- Smrcka, A.V. Molecular targeting of Galpha and Gbetagamma subunits: A potential approach for cancer therapeutics. Trends Pharmacol. Sci. 2013, 34, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Sun, Z.; Runne, C.; Madsen, J.; Domann, F.; Henry, M.; Lin, F.; Chen, S. A critical role of Gbetagamma in tumorigenesis and metastasis of breast cancer. J. Biol. Chem. 2011, 286, 13244–13254. [Google Scholar] [CrossRef] [Green Version]
- Ram, P.T.; Iyengar, R. G protein coupled receptor signaling through the Src and Stat3 pathway: Role in proliferation and transformation. Oncogene 2001, 20, 1601–1606. [Google Scholar] [CrossRef] [Green Version]
- Arora, P.; Cuevas, B.D.; Russo, A.; Johnson, G.L.; Trejo, J. Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 2008, 27, 4434–4445. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Adelstein, R.S. LPA(1) -induced migration requires nonmuscle myosin II light chain phosphorylation in breast cancer cells. J. Cell Physiol. 2011, 226, 2881–2893. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wolff, D.W.; Wei, T.; Wang, B.; Deng, C.; Kirui, J.K.; Jiang, H.; Qin, J.; Abel, P.W.; Tu, Y. Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 2009, 69, 5743–5751. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Tan, W.; Dillenburg-Pilla, P.; Armando, S.; Amornphimoltham, P.; Simaan, M.; Weigert, R.; Molinolo, A.A.; Bouvier, M.; Gutkind, J.S. A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci. Signal. 2011, 4, ra60. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef]
- Vaddi, P.K.; Stamnes, M.A.; Cao, H.; Chen, S. Elimination of SOX2/OCT4-Associated Prostate Cancer Stem Cells Blocks Tumor Development and Enhances Therapeutic Response. Cancers 2019, 11, 1331. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Tang, X.; Sun, Z.; Chen, S. Upregulated WDR26 serves as a scaffold to coordinate PI3K/ AKT pathway-driven breast cancer cell growth, migration, and invasion. Oncotarget 2016, 7, 17854–17869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavo, G.; van der Goot, F.G. The bacterial toxin toolkit. Nat. Rev. Mol. Cell Biol. 2001, 2, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Asselin-Labat, M.L.; Sutherland, K.D.; Barker, H.; Thomas, R.; Shackleton, M.; Forrest, N.C.; Hartley, L.; Robb, L.; Grosveld, F.G.; van der Wees, J.; et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 2007, 9, 201–209. [Google Scholar] [CrossRef]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef]
- Zhang, W.; Tan, W.; Wu, X.; Poustovoitov, M.; Strasner, A.; Li, W.; Borcherding, N.; Ghassemian, M.; Karin, M. A NIK-IKKalpha module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013, 23, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Lo, P.K.; Kanojia, D.; Liu, X.; Singh, U.P.; Berger, F.G.; Wang, Q.; Chen, H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFbeta signaling. Oncogene 2012, 31, 2614–2626. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Raviv, A.; Esposito, D.; Flanders, K.C.; Daniel, C.; Nghiem, B.T.; Garfield, S.; Lim, L.; Mannan, P.; Robles, A.I.; et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Rep. 2015, 4, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Zotano, A.; Mayer, I.A.; Arteaga, C.L. PI3K/AKT/mTOR: Role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016, 35, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthuswamy, S.K. Trastuzumab resistance: All roads lead to SRC. Nat. Med. 2011, 17, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Rexer, B.N.; Arteaga, C.L. Optimal targeting of HER2-PI3K signaling in breast cancer: Mechanistic insights and clinical implications. Cancer Res. 2013, 73, 3817–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Thust, R.; Kaina, B. Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: Critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene 2002, 21, 2141–2153. [Google Scholar] [CrossRef] [Green Version]
- Mangmool, S.; Kurose, H. G(i/o) protein-dependent and -independent actions of Pertussis Toxin (PTX). Toxins 2011, 3, 884–899. [Google Scholar] [CrossRef] [Green Version]
- Andreev, J.; Galisteo, M.L.; Kranenburg, O.; Logan, S.K.; Chiu, E.S.; Okigaki, M.; Cary, L.A.; Moolenaar, W.H.; Schlessinger, J. Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J. Biol. Chem. 2001, 276, 20130–20135. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Eckhart, A.D.; Eguchi, S.; Koch, W.J. Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J. Biol. Chem. 2002, 277, 32116–32123. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef] [Green Version]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity—A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.K.; Simoes, B.M.; Howell, S.J.; Farnie, G.; Clarke, R.B. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. BCR 2013, 15, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, S.; Khawer, M.; Rafique, S.; Naz, Z.; Saleem, K. The current status of anti-GPCR drugs against different cancers. J. Pharm. Anal. 2020, 10, 517–521. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, C.; Ye, Y.; Weigel, R.J.; Chen, S. Blocking Gi/o-Coupled Signaling Eradicates Cancer Stem Cells and Sensitizes Breast Tumors to HER2-Targeted Therapies to Inhibit Tumor Relapse. Cancers 2022, 14, 1719. https://doi.org/10.3390/cancers14071719
Lyu C, Ye Y, Weigel RJ, Chen S. Blocking Gi/o-Coupled Signaling Eradicates Cancer Stem Cells and Sensitizes Breast Tumors to HER2-Targeted Therapies to Inhibit Tumor Relapse. Cancers. 2022; 14(7):1719. https://doi.org/10.3390/cancers14071719
Chicago/Turabian StyleLyu, Cancan, Yuanchao Ye, Ronald J. Weigel, and Songhai Chen. 2022. "Blocking Gi/o-Coupled Signaling Eradicates Cancer Stem Cells and Sensitizes Breast Tumors to HER2-Targeted Therapies to Inhibit Tumor Relapse" Cancers 14, no. 7: 1719. https://doi.org/10.3390/cancers14071719