
Preliminary Study on the Sequencing of Whole Genomic Methylation and Transcriptome-Related Genes in Thyroid Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
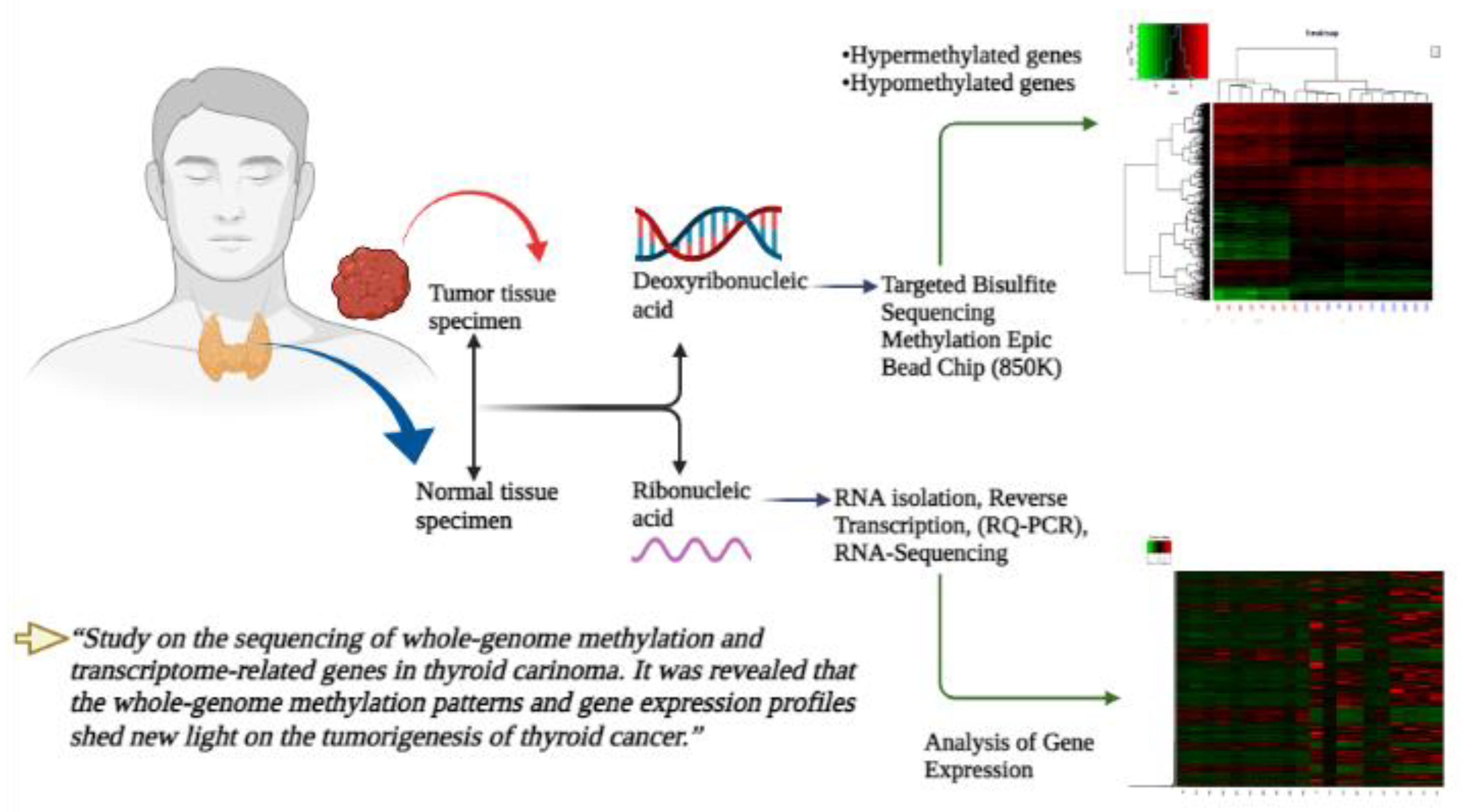
2. Material and Methods
2.1. Tissue Samples of Patients
2.2. MethylationEPIC BeadChip (850K)
2.3. RNA Sequencing (RNA-Seq)
2.4. Targeted Bisulfite Sequencing Assay
2.5. RNA Isolation, Reverse Transcription and Real-Time Quantitative PCR (RQ-PCR)
2.6. Analytical Statistics
3. Results
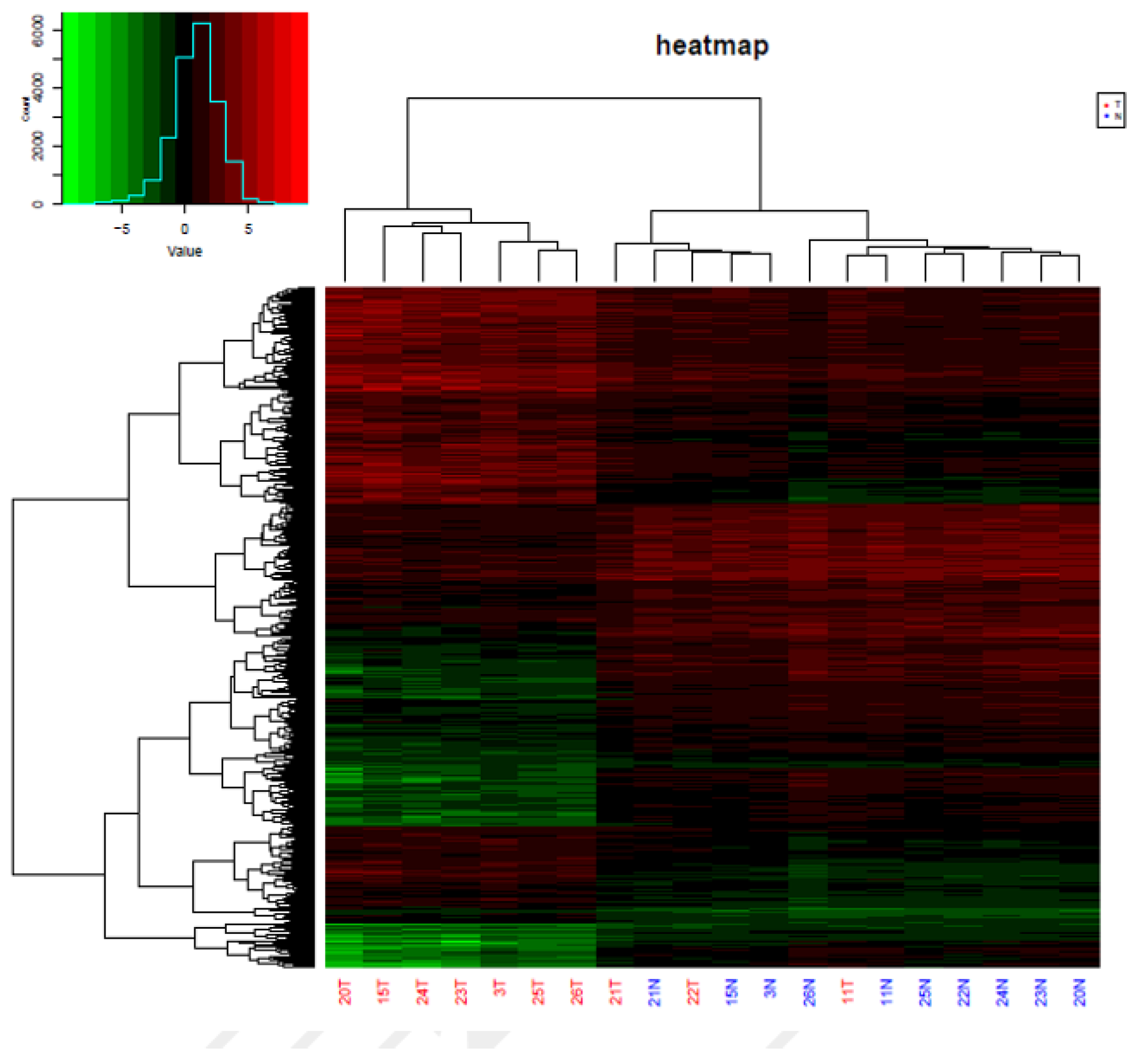
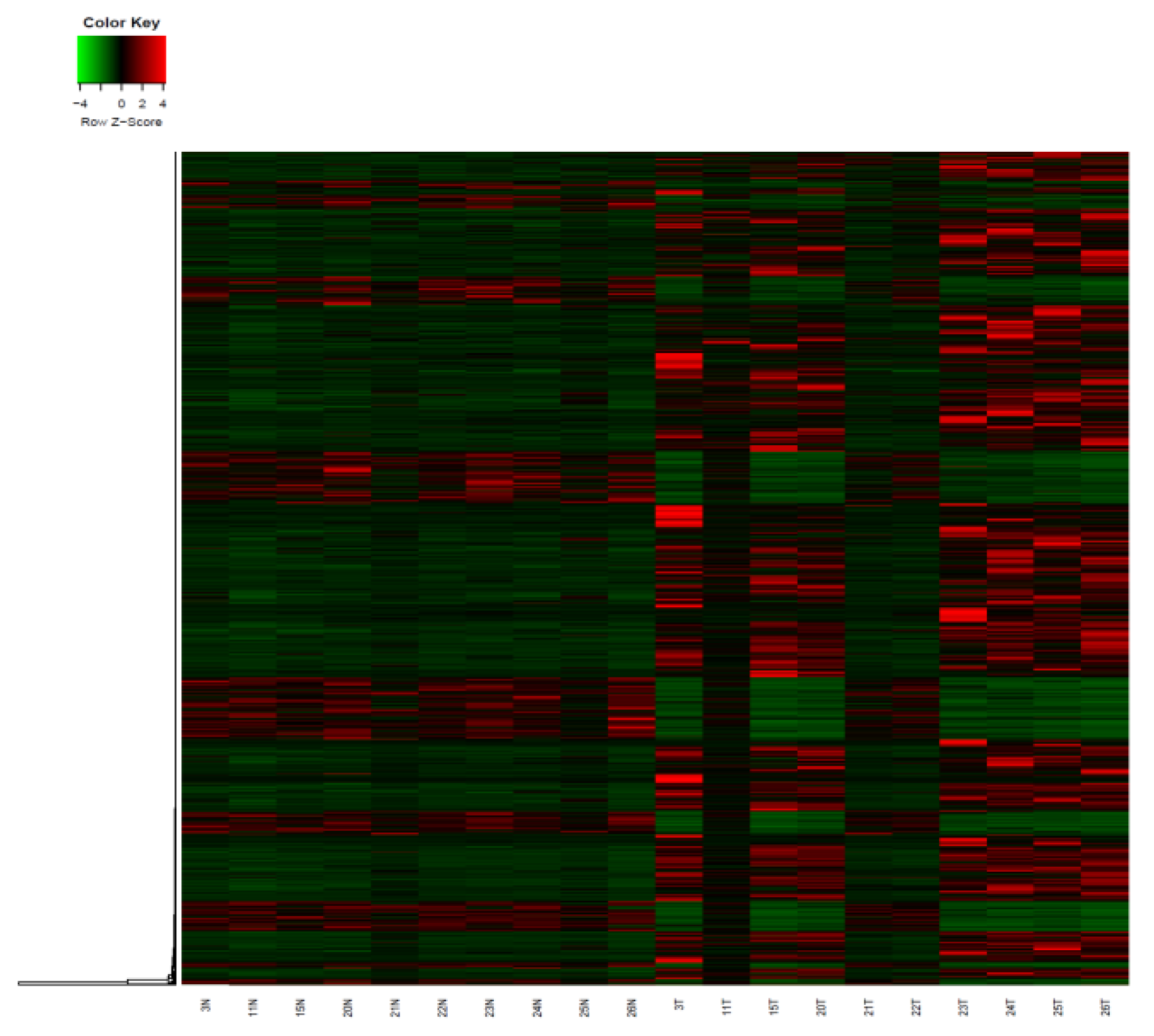
3.1. Analysis of Genome-Wide Methylation in the Thyroid Carcinoma Patients
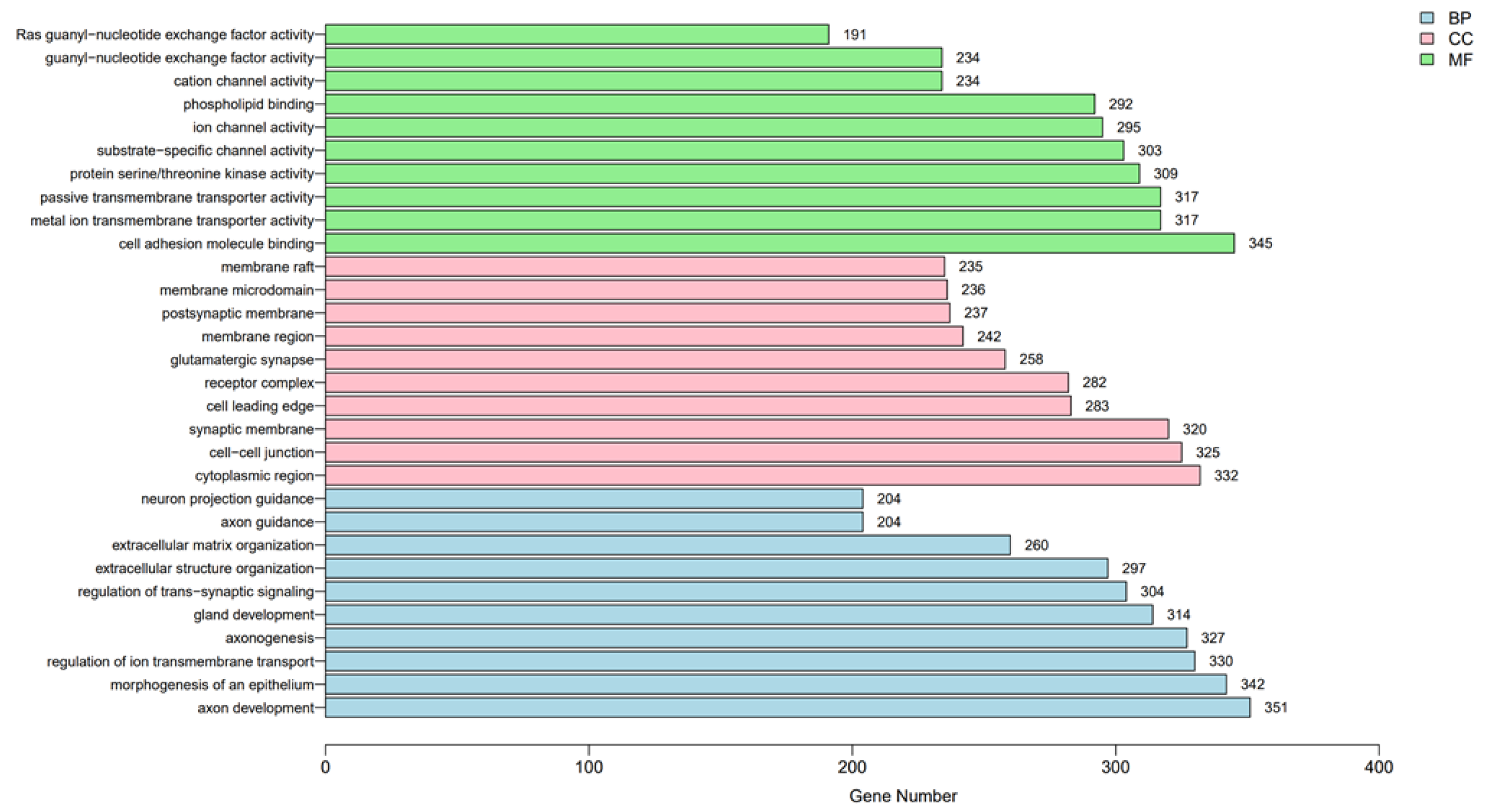
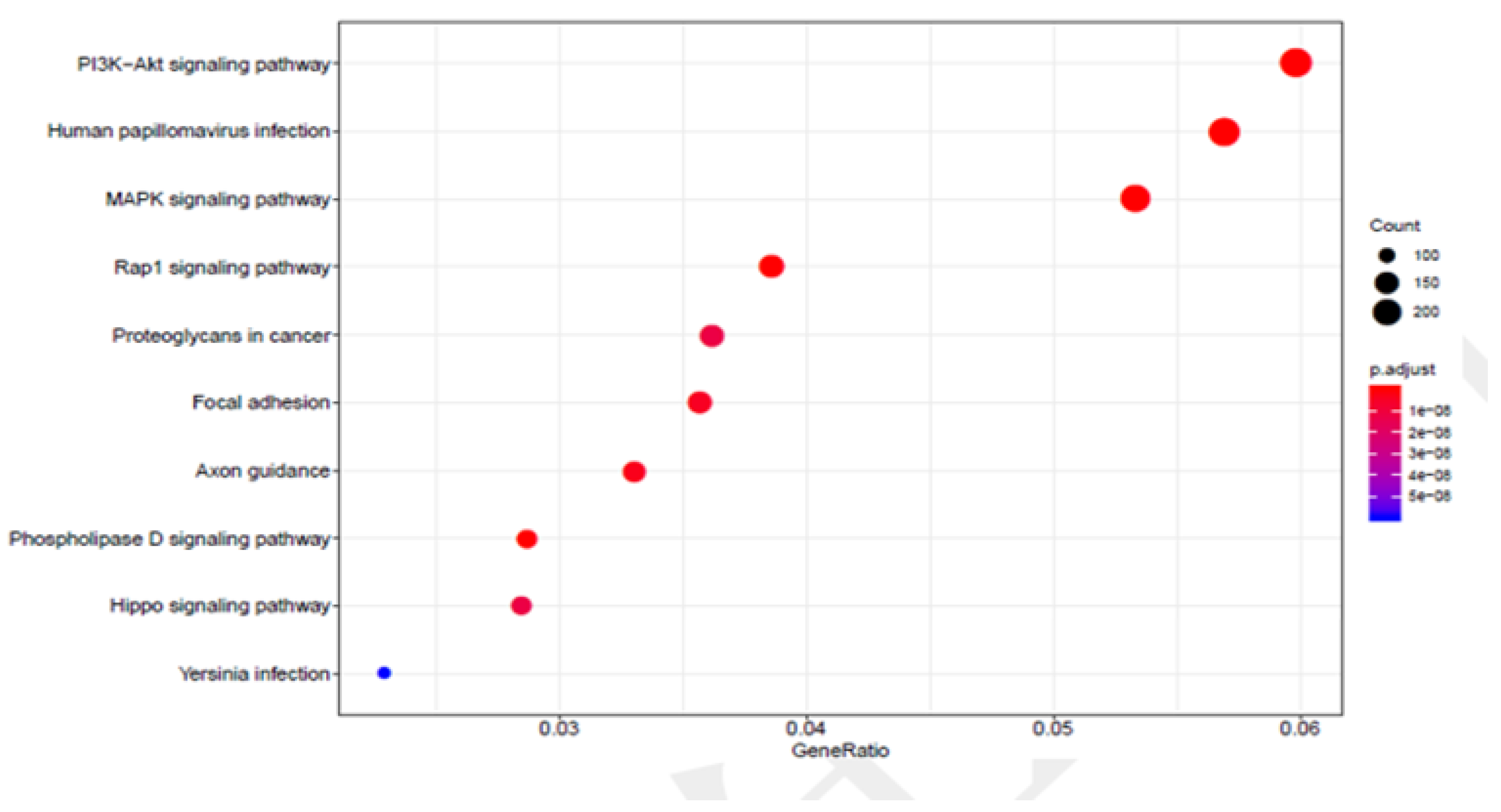
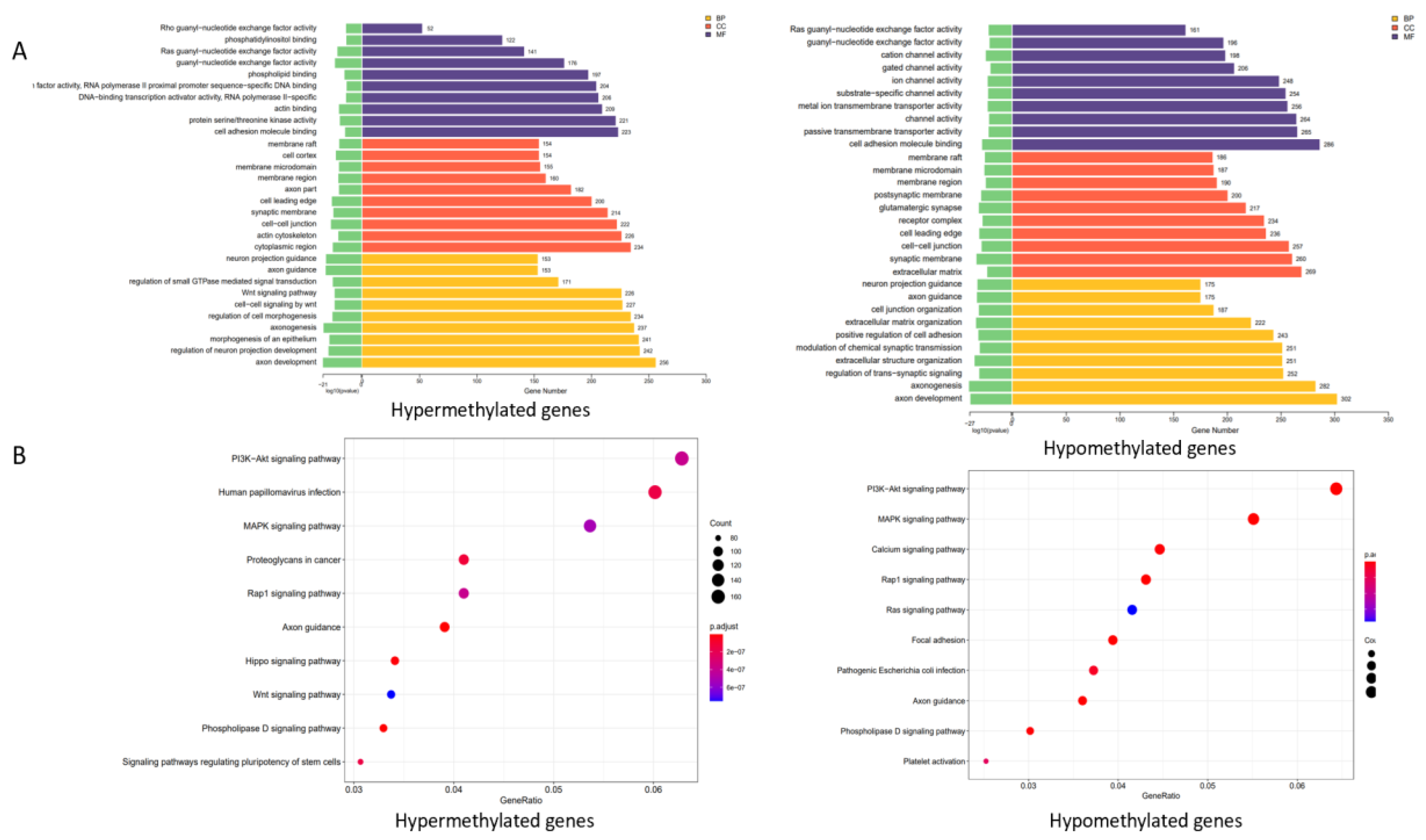
3.2. Functional Analysis of Genes Associated with DNA Methylation in Thyroid Cancer Patients
3.3. The Alterations in Gene Expression Related to Thyroid Carcinoma
3.4. Screening of Candidate Differentially Methylated Genes Related to Thyroid Cancer
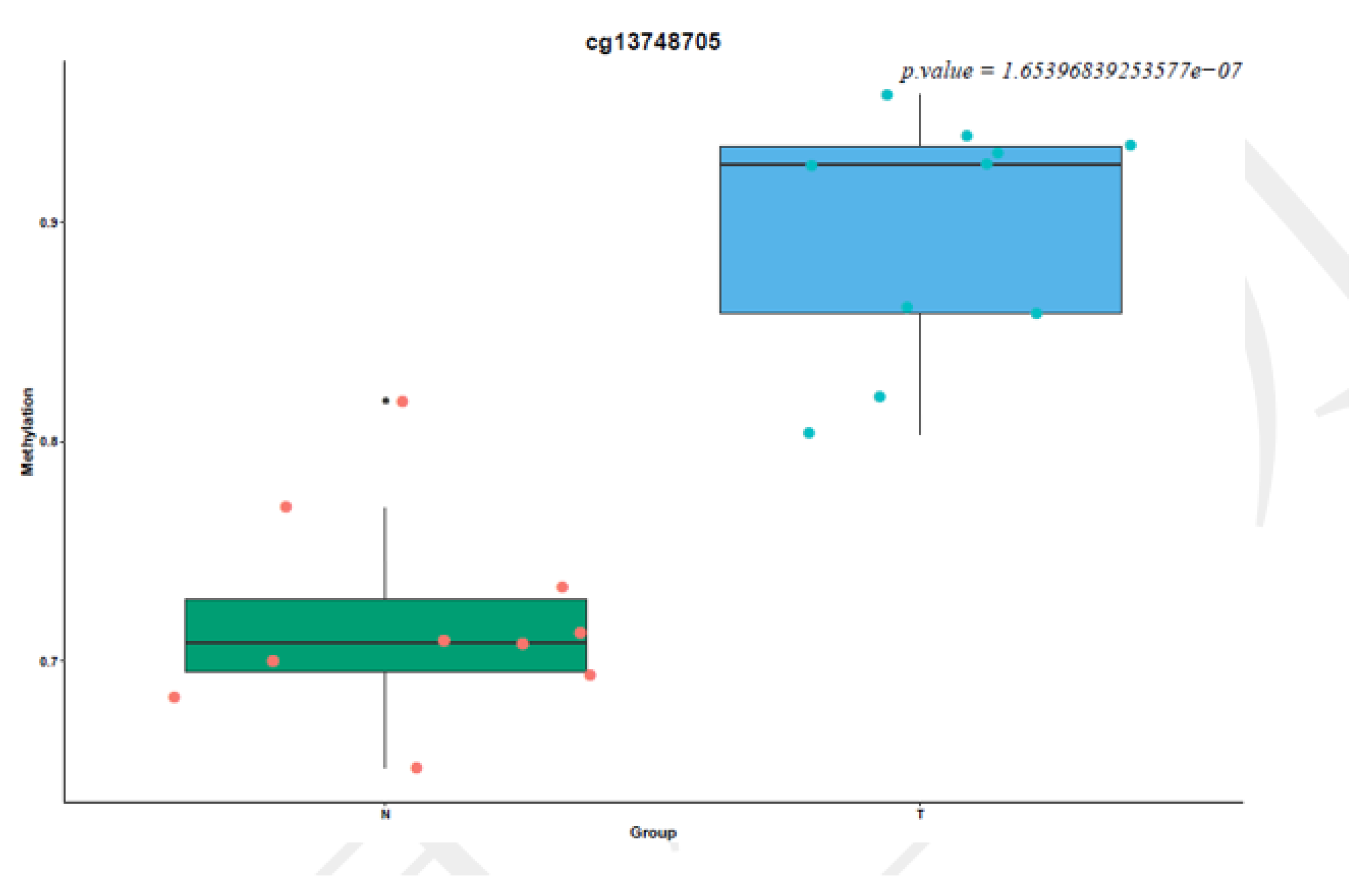
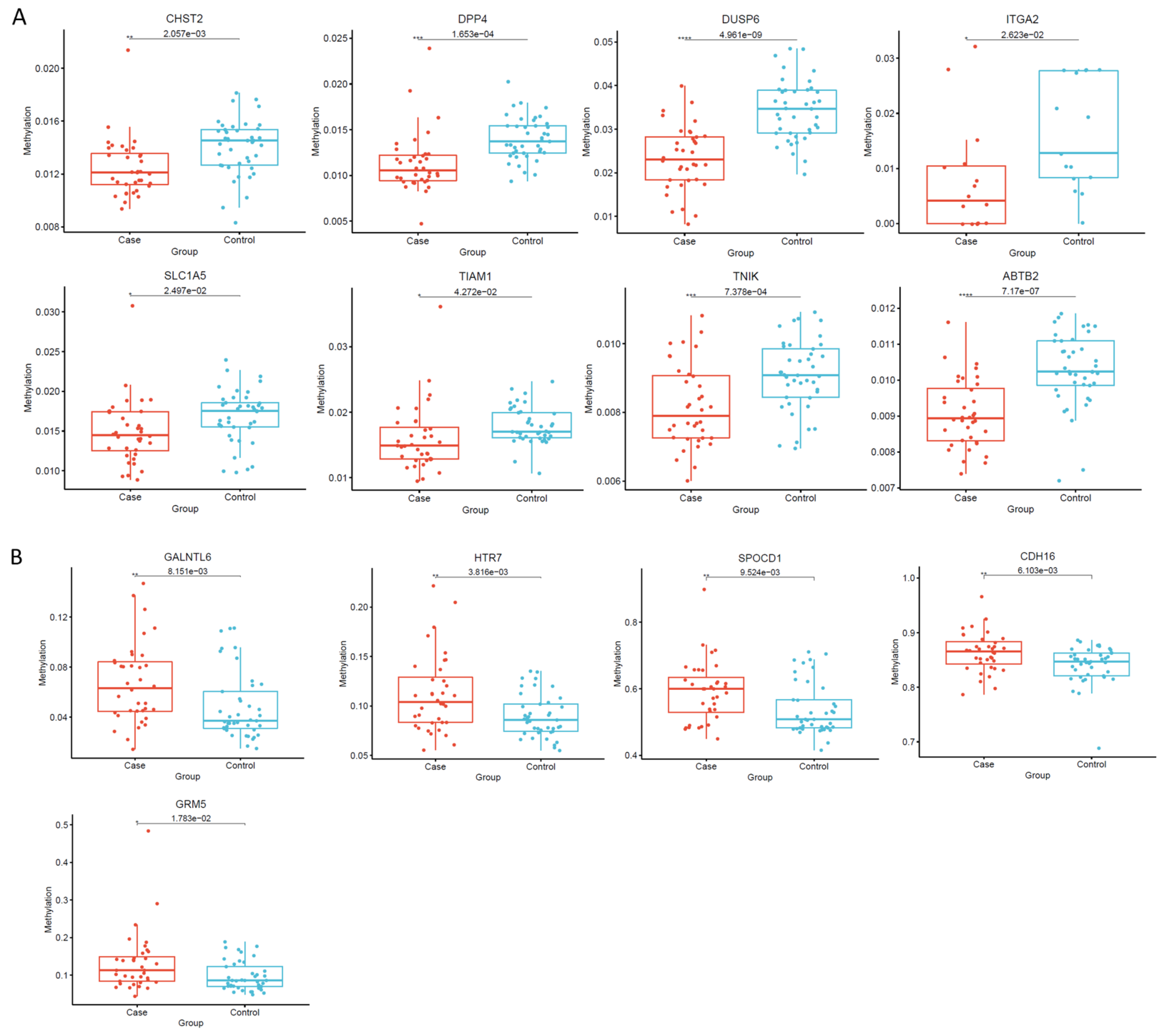
3.5. Identification and Validation of Thyroid Tumor-Related Genes by Targeted Bisulfite Sequencing and RQ-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in Thyroid Cancer Incidence and Mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- The American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer; Cooper, D.S.; Doherty, G.M.; Haugen, B.R.; Kloos, R.T.; Lee, S.L.; Mandel, S.J.; Mazzaferri, E.L.; McIver, B.; Pacini, F.; et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [Green Version]
- Riesco-Eizaguirre, G.; Santisteban, P. ENDOCRINE TUMOURS: Advances in the molecular pathogenesis of thyroid cancer: Lessons from the cancer genome. Eur. J. Endocrinol. 2016, 175, R203–R217. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Langst, G.; Manelyte, L. Chromatin Remodelers: From Function to Dysfunction. Genes 2015, 6, 299–324. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J.; et al. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Xu, X. DNA methylation and cognitive aging. Oncotarget 2015, 6, 13922–13932. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A. Cancer. Death and methylation. Nature 2001, 409, 141–143. [Google Scholar] [CrossRef]
- Aran, D.; Hellman, A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell 2013, 154, 11–13. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.H.; Lee, T.L.; Rennert, O.M.; Chan, W.Y. DNA methylation of cancer genome. Birth Defects Res. C Embryo Today 2009, 87, 335–350. [Google Scholar] [CrossRef] [Green Version]
- Schagdarsurengin, U.; Gimm, O.; Dralle, H.; Hoang-Vu, C.; Dammann, R. CpG island methylation of tumor-related promoters occurs preferentially in undifferentiated carcinoma. Thyroid 2006, 16, 633–642. [Google Scholar] [CrossRef]
- Guan, H.; Ji, M.; Hou, P.; Liu, Z.; Wang, C.; Shan, Z.; Teng, W.; Xing, M. Hypermethylation of the DNA mismatch repair gene hMLH1 and its association with lymph node metastasis and T1799A BRAF mutation in patients with papillary thyroid cancer. Cancer 2008, 113, 247–255. [Google Scholar] [CrossRef]
- Neumann, S.; Schuchardt, K.; Reske, A.; Reske, A.; Emmrich, P.; Paschke, R. Lack of correlation for sodium iodide symporter mRNA and protein expression and analysis of sodium iodide symporter promoter methylation in benign cold thyroid nodules. Thyroid 2004, 14, 99–111. [Google Scholar] [CrossRef]
- Galrao, A.L.; Camargo, R.Y.; Friguglietti, C.U.; Moraes, L.; Cerutti, J.M.; Serrano-Nascimento, C.; Suzuki, M.F.; Medeiros-Neto, G.; Rubio, I.G. Hypermethylation of a New Distal Sodium/Iodide Symporter (NIS) enhancer (NDE) is associated with reduced NIS expression in thyroid tumors. J. Clin. Endocrinol. Metab. 2014, 99, E944–E952. [Google Scholar] [CrossRef] [Green Version]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Gao, L.; Zhang, S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief. Bioinform. 2017, 18, 761–773. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.L.; Zhang, K.; Zhang, K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Walter, K.; Holcomb, T.; Januario, T.; Yauch, R.L.; Du, P.; Bourgon, R.; Seshagiri, S.; Amler, L.C.; Hampton, G.M.; David, S.S. Discovery and development of DNA methylation-based biomarkers for lung cancer. Epigenomics 2014, 6, 59–72. [Google Scholar] [CrossRef]
- Coppede, F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Lett. 2014, 342, 238–247. [Google Scholar] [CrossRef]
- Ashour, N.; Angulo, J.C.; Andres, G.; Alelu, R.; Gonzalez-Corpas, A.; Toledo, M.V.; Rodriguez-Barbero, J.M.; Lopez, J.I.; Sanchez-Chapado, M.; Ropero, S. A DNA hypermethylation profile reveals new potential biomarkers for prostate cancer diagnosis and prognosis. Prostate 2014, 74, 1171–1182. [Google Scholar] [CrossRef]
- Tahara, T.; Arisawa, T. DNA methylation as a molecular biomarker in gastric cancer. Epigenomics 2015, 7, 475–486. [Google Scholar] [CrossRef]
- Hou, P.; Liu, D.; Xing, M. Genome-wide alterations in gene methylation by the BRAF V600E mutation in papillary thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Guo, S.; Sun, J.; Huang, Z.; Zhu, T.; Zhang, H.; Gu, J.; He, Y.; Wang, W.; Ma, K.; et al. Methylcap-seq reveals novel DNA methylation markers for the diagnosis and recurrence prediction of bladder cancer in a Chinese population. PLoS ONE 2012, 7, e35175. [Google Scholar] [CrossRef]
- Guo, S.; Yan, F.; Xu, J.; Bao, Y.; Zhu, J.; Wang, X.; Wu, J.; Li, Y.; Pu, W.; Liu, Y.; et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin. Epigenetics 2015, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.Y.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.J.; Zhou, J.D.; Zhang, W.; Lin, J.; Ma, J.C.; Wen, X.M.; Yuan, Q.; Li, X.X.; Xu, Z.J.; Qian, J. H19 overexpression promotes leukemogenesis and predicts unfavorable prognosis in acute myeloid leukemia. Clin. Epigenetics 2018, 10, 47. [Google Scholar] [CrossRef]
- Zhou, J.D.; Wang, Y.X.; Zhang, T.J.; Li, X.X.; Gu, Y.; Zhang, W.; Ma, J.C.; Lin, J.; Qian, J. Identification and validation of SRY-box containing gene family member SOX30 methylation as a prognostic and predictive biomarker in myeloid malignancies. Clin. Epigenetics 2018, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.J.; Wen, X.M.; Zhou, J.D.; Gu, Y.; Xu, Z.J.; Guo, H.; Ma, J.C.; Yuan, Q.; Chen, Q.; Lin, J.; et al. SOX30 methylation correlates with disease progression in patients with chronic myeloid leukemia. OncoTargets Ther. 2019, 12, 4789–4794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.L.; Jeon, S.; Seo, E.H.; Bae, D.H.; Jeong, Y.M.; Kim, Y.; Bae, J.S.; Kim, S.K.; Jung, C.K.; Kim, Y.S. Comprehensive DNA Methylation Profiling Identifies Novel Diagnostic Biomarkers for Thyroid Cancer. Thyroid 2020, 30, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Gil, J.; Pérez-González, B.; Jordà, M. DNA methylation in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef]
- Zhao, L.; Jia, Y.; Liu, Y.; Han, B.; Wang, J.; Jiang, X. Integrated Bioinformatics Analysis of DNA Methylation Biomarkers in Thyroid Cancer Based on TCGA Database. Biochem. Genet. 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Canberk, S.; Lima, A.R.; Pinto, M.; Máximo, V. Translational Potential of Epigenetic-Based Markers on Fine-Needle Aspiration Thyroid Specimens. Front. Med. 2021, 8, 177. [Google Scholar] [CrossRef]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Lee, S.A.; Han, I.H.; Yoo, B.C.; Lee, S.H.; Park, J.Y.; Cha, I.H.; Kim, J.; Choi, S.W. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol. Rep. 2007, 17, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Wang, N.N.; Gu, L.; Yang, H.M.; Xia, N.; Zhang, H. The suppressive effect of metabotropic glutamate receptor 5 (mGlu5) inhibition on hepatocarcinogenesis. Biochimie 2012, 94, 2366–2375. [Google Scholar] [CrossRef]
- Sheng, X.; Liu, W.; Lu, Z.; Xu, M.; Li, R.; Zhong, R.; Li, Y.; Liu, T.; Zhang, S. HTR7 promotes laryngeal cancer growth through PI3K/AKT pathway activation. Ann. Transl. Med. 2021, 9, 840. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, H.; Hu, J.; Liu, Y.; Li, Z. SPOCD1 promotes cell proliferation and inhibits cell apoptosis in human osteosarcoma. Mol. Med. Rep. 2018, 17, 3218–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Yang, Y.; Yan, A.; Yang, Y. SPOCD1 accelerates ovarian cancer progression and inhibits cell apoptosis via the PI3K/AKT pathway. OncoTargets Ther. 2020, 13, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, F.; Umeda, S.; Ichimiya, T.; Kamiyama, S.; Hazawa, M.; Yasuda, T.; Nishihara, S.; Imai, T. Sulfation of keratan sulfate proteoglycan reduces radiation-induced apoptosis in human Burkitt’s lymphoma cell lines. FEBS Lett. 2013, 587, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, B.A.; Nardo, B.; Chieco, P.; Mazziotti, A.; Bolondi, L.; Cavallari, A. Aberrant dipeptidyl peptidase IV (DPP IV/CD26) expression in human hepatocellular carcinoma. J. Hepatol. 1997, 27, 337–345. [Google Scholar] [CrossRef]
- Lee, E.K.; Chung, K.W.; Yang, S.K.; Park, M.J.; Min, H.S.; Kim, S.W.; Kang, H.S. DNA methylation of MAPK signal-inhibiting genes in papillary thyroid carcinoma. Anticancer Res. 2013, 33, 4833–4839. [Google Scholar]
- Messina, S.; Frati, L.; Leonetti, C.; Zuchegna, C.; Di Zazzo, E.; Calogero, A.; Porcellini, A. Dual-specificity phosphatase DUSP6 has tumor-promoting properties in human glioblastomas. Oncogene 2011, 30, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Zhao, J.; Sun, Y.; Li, D.; Meng, Z.; Wang, B.; Fan, P.; Liu, Z.; Jin, X.; Wu, H. Overexpressed ITGA2 promotes malignant tumor aggression by up-regulating PD-L1 expression through the activation of the STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 485. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhao, Y.; Zhao, J.; Wu, S.; Jiang, Y.; Ma, H.; Zhang, T. Upregulated SLC1A5 promotes cell growth and survival in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 6006–6014. [Google Scholar]
- Wang, S.; Li, S.; Tang, Q.; Yang, S.; Wang, S.; Liu, J.; Yang, M.; Yang, X. Overexpression of Tiam1 promotes the progression of laryngeal squamous cell carcinoma. Oncol. Rep. 2015, 33, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.H.; Zhang, X.; Wang, H.; Zhang, L.; Chen, H.; Hu, M.; Dong, Z.; Zhu, G.; Qian, Z.; Fan, J.; et al. The essential role of TNIK gene amplification in gastric cancer growth. Oncogenesis 2014, 3, e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Hu, N.; Ma, L.; Li, W.; Cheng, X.; Zhang, Y.; Zhu, Y.; Yang, Y.; Peng, X.; Zou, D.; et al. ABTB2 Regulatory Variant as Predictor of Epirubicin-Based Neoadjuvant Chemotherapy in Luminal A Breast Cancer. Front. Oncol. 2020, 10, 1950. [Google Scholar] [CrossRef] [PubMed]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arechederra, M.; Daian, F.; Yim, A.; Bazai, S.K.; Richelme, S.; Dono, R.; Saurin, A.J.; Habermann, B.H.; Maina, F. Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 2018, 9, 3164. [Google Scholar] [CrossRef]
- Yang, Y.F.; Yu, B.; Zhang, X.X.; Zhu, Y.H. Identification of TNIK as a novel potential drug target in thyroid cancer based on protein druggability prediction. Medicine 2021, 100, e25541. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, M.A.; Li, M.; Lin, J.; Zhang, G.; Chen, M.; Moazzam, N.F.; Qian, W. Preliminary Study on the Sequencing of Whole Genomic Methylation and Transcriptome-Related Genes in Thyroid Carcinoma. Cancers 2022, 14, 1163. https://doi.org/10.3390/cancers14051163
Iqbal MA, Li M, Lin J, Zhang G, Chen M, Moazzam NF, Qian W. Preliminary Study on the Sequencing of Whole Genomic Methylation and Transcriptome-Related Genes in Thyroid Carcinoma. Cancers. 2022; 14(5):1163. https://doi.org/10.3390/cancers14051163
Chicago/Turabian StyleIqbal, Muhammad Asad, Mingyang Li, Jiang Lin, Guoliang Zhang, Miao Chen, Nida Fatima Moazzam, and Wei Qian. 2022. "Preliminary Study on the Sequencing of Whole Genomic Methylation and Transcriptome-Related Genes in Thyroid Carcinoma" Cancers 14, no. 5: 1163. https://doi.org/10.3390/cancers14051163