
Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer

 ,
,  ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Significance of the Inflammatory Immune Infiltrate in PDAC
3. Importance of Murine Models in Studying the PDAC Tumor Microenvironment
4. Introduction to Macrophages in PDAC
5. Introduction to Neutrophils in PDAC
6. Spatial Arrangement of Macrophages and Neutrophils in the PDAC TME
7. Macrophage and Neutrophil Crosstalk in the PDAC TME
8. Sequelae of Macrophages and Neutrophils in PDAC
8.1. Generation of Hypoxia
8.2. Vascular Remodeling
8.3. Fibrosis
8.4. Immunosuppression
8.5. Tumor Progression and Metastasis
8.6. Treatment Resistance
9. Treatments Targeting Macrophages and Neutrophils

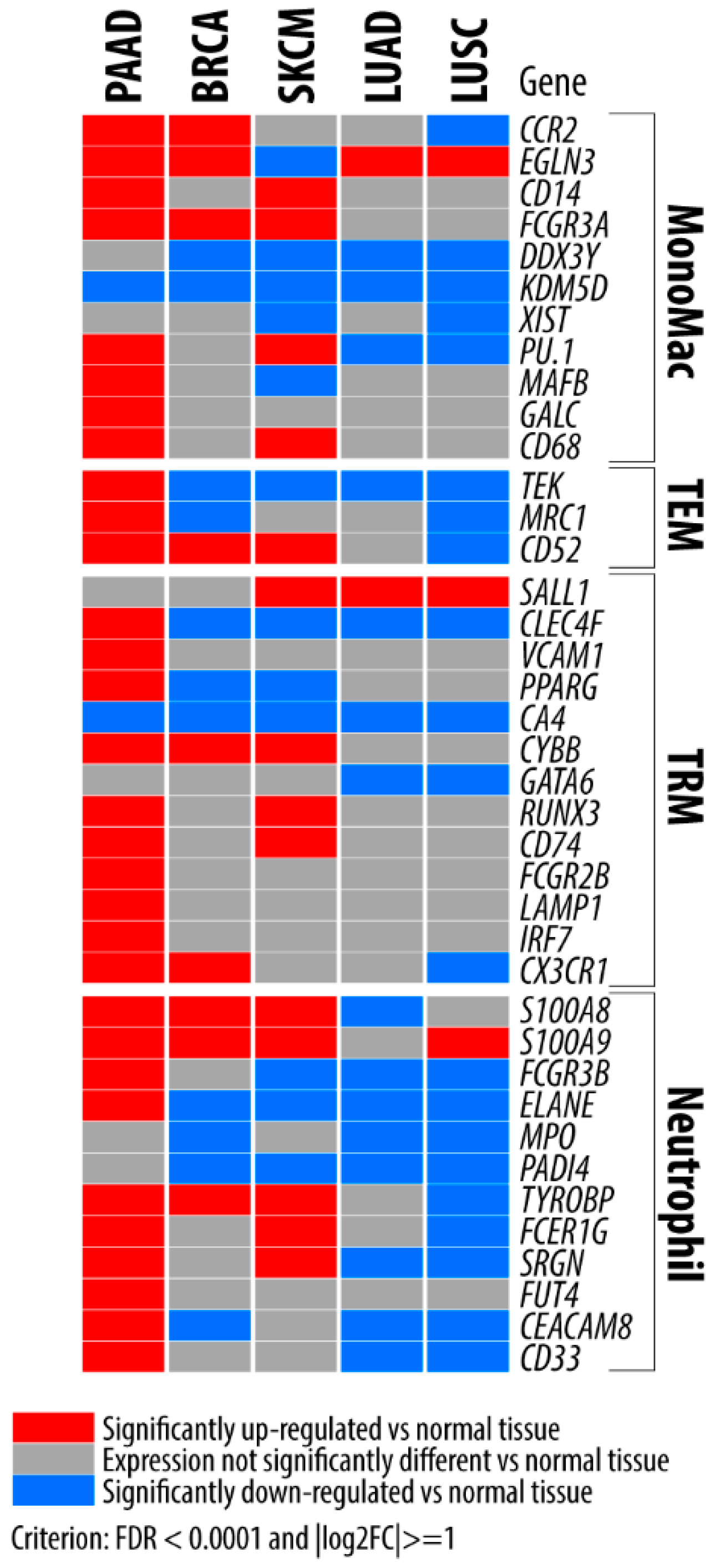{kind=link}
{kind=link}
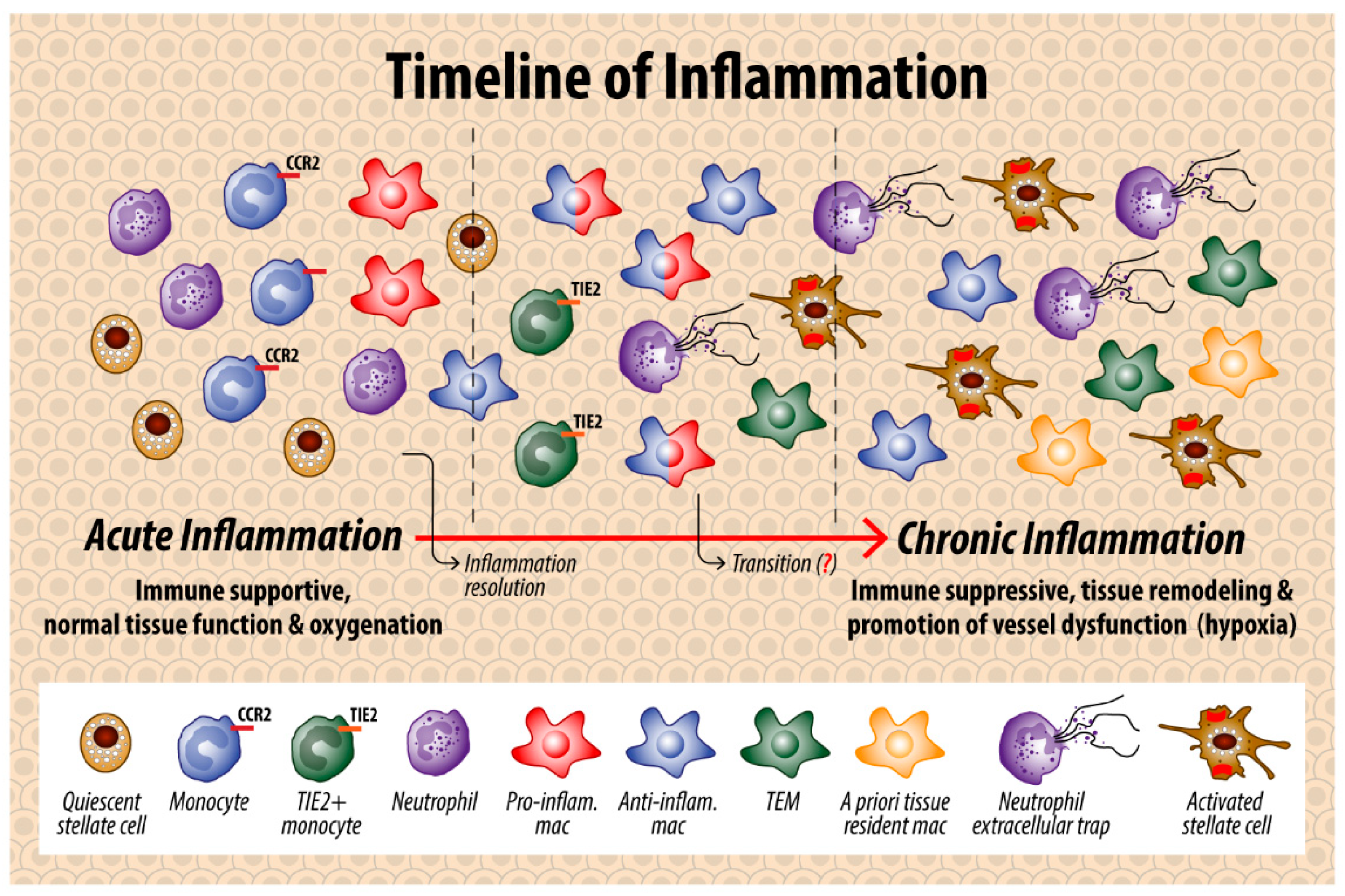{kind=link}
{kind=link}
| Mechanism | Treatment | References |
|---|---|---|
| Macrophage Reprogramming | IFNγ | [218] |
| MMP-9 Inhibitor | [219] | |
| CD40 agonistic antibody | [220,221,222,223] | |
| Nanomicelle PI3k-γ and CSF1-R Inhibition | [224] | |
| Pomalidomide | [225] | |
| Trabectedin | [235] | |
| Macrophage Depletion | Lurbectedin | [216] |
| CSF1 Inhibition | [226] | |
| CCR2 Antagonism | [70,186,226] | |
| CCL2 Antibody | [227] | |
| Liposomal Clodronate | [152,228] | |
| Neutrophil Reprogramming | IFNβ | [230] |
| TGFβ | [84] | |
| Neutrophil Depletion | CXCR2 Inhibition | [70] |
| Lorlatinib | [71] | |
| NETosis Inhibition | Dnase | [87] |
| (Hydroxy)Chloroquine | [103,231,232,233,234] | |
| IL-17/IL-17R Blockade | [4] | |
| PAD4 Inhibition | [89] |
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-Mediated MDSC Tumor Trafficking Enhances Anti-PD1 Efficacy. Sci. Transl. Med. 2014, 6, 237ra67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chandra, V.; Sanchez, E.R.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ Tumor-Infiltrating Lymphocytes Together with CD4+ Tumor-Infiltrating Lymphocytes and Dendritic Cells Improve the Prognosis of Patients with Pancreatic Adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases During the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ademmer, K.; Ebert, M.; Ostermeyer, M.-; Friess, H.; Büchler, M.W.; Schubert, W.; Malfertheiner, P. Effector T lymphocyte subsets in human pancreatic cancer: Detection of CD8+ CD18+ cells and CD8+ CD103+ cells by multi-epitope imaging. Clin. Exp. Immunol. 1998, 112, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Porembka, M.; Mitchem, J.; Belt, B.A.; Hsieh, C.-S.; Lee, H.-M.; Herndon, J.; Gillanders, W.E.; Linehan, D.C.; Goedegebuure, P. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 2012, 61, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Farren, M.; Mace, T.A.; Geyer, S.; Mikhail, S.; Wu, C.; Ciombor, K.K.; Tahiri, S.; Ahn, D.; Noonan, A.; Villalonacalero, M.A.; et al. Systemic Immune Activity Predicts Overall Survival in Treatment-Naïve Patients with Metastatic Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 2565–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Castino, G.F.; Cortese, N.; Capretti, G.; Serio, S.; Di Caro, G.; Mineri, R.; Magrini, E.; Grizzi, F.; Cappello, P.; Novelli, F.; et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. OncoImmunology 2016, 5, e1085147. [Google Scholar] [CrossRef] [Green Version]
- Hirooka, S.; Yanagimoto, H.; Satoi, S.; Yamamoto, T.; Toyokawa, H.; Yamaki, S.; Yui, R.; Inoue, K.; Michiura, T.; Kwon, A.-H. The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer. Res. 2011, 31, 3827–3834. [Google Scholar] [PubMed]
- Yanagimoto, H.; Takai, S.; Satoi, S.; Toyokawa, H.; Takahashi, K.; Terakawa, N.; Kwon, A.-H.; Kamiyama, Y. Impaired function of circulating dendritic cells in patients with pancreatic cancer. Clin. Immunol. 2005, 114, 52–60. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2019, 58, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Strouch, M.J.; Cheon, E.C.; Salabat, M.R.; Krantz, S.B.; Gounaris, E.; Melstrom, L.G.; Dangi-Garimella, S.; Wang, E.; Munshi, H.G.; Khazaie, K.; et al. Crosstalk between Mast Cells and Pancreatic Cancer Cells Contributes to Pancreatic Tumor Progression. Clin. Cancer Res. 2010, 16, 2257–2265. [Google Scholar] [CrossRef] [Green Version]
- Esposito, I.; Menicagli, M.; Funel, N.; Bergmann, F.; Boggi, U.; Mosca, F.; Bevilacqua, G.; Campani, D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J. Clin. Pathol. 2004, 57, 630–636. [Google Scholar] [CrossRef] [Green Version]
- Trovato, R.; Fiore, A.; Sartori, S.; Canè, S.; Giugno, R.; Cascione, L.; Paiella, S.; Salvia, R.; De Sanctis, F.; Poffe, O.; et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J. Immunother. Cancer 2019, 7, 255. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.; Liu, C.L.; Bratman, S.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Available online: https://xenabrowser.net/datapages/?dataset=TcgaTargetGtex_gene_expected_count&host=https%3A%2F%2Ftoil.xenahubs.net&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443 (accessed on 8 April 2021).
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://storage.googleapis.com/gtex_analysis_v8/annotations/GTEx_Analysis_v8_Annotations_SampleAttributesDS.txt (accessed on 11 April 2021).
- Available online: https://gdc.cancer.gov/resources-tcga-users/tcga-code-tables/sample-type-codes (accessed on 11 April 2021).
- Xie, D.; Xie, K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015, 2, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spear, S.; Candido, J.B.; McDermott, J.R.; Ghirelli, C.; Maniati, E.; Beers, S.A.; Balkwill, F.R.; Kocher, H.; Capasso, M. Discrepancies in the Tumor Microenvironment of Spontaneous and Orthotopic Murine Models of Pancreatic Cancer Uncover a New Immunostimulatory Phenotype for B Cells. Front. Immunol. 2019, 10, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westphalen, C.B.; Olive, K.P. Genetically Engineered Mouse Models of Pancreatic Cancer. Cancer J. 2012, 18, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.L.; Miller, A.L.; Yoon, K.J. Patient-Derived Xenograft Models of Pancreatic Cancer: Overview and Comparison with Other Types of Models. Cancers 2020, 12, 1327. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Anver, M.R.; Biankin, A.; Boivin, G.P.; Furth, E.E.; Furukawa, T.; Klein, A.; Klimstra, D.S.; et al. Pathology of Genetically Engineered Mouse Models of Pancreatic Exocrine Cancer: Consensus Report and Recommendations. Cancer Res. 2006, 66, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Fu, S.; Mastio, J.; Dominguez, G.A.; Purohit, A.; Kossenkov, A.; Lin, C.; Alicea-Torres, K.; Sehgal, M.; Nefedova, Y.; et al. Unique pattern of neutrophil migration and function during tumor progression. Nat. Immunol. 2018, 19, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Walters, D.M.; Stokes, J.B.; Adair, S.J.; Stelow, E.B.; Borgman, C.A.; Lowrey, B.T.; Xin, W.; Blais, E.M.; Lee, J.K.; Papin, J.A.; et al. Clinical, Molecular and Genetic Validation of a Murine Orthotopic Xenograft Model of Pancreatic Adenocarcinoma Using Fresh Human Specimens. PLoS ONE 2013, 8, e77065. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herdon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Chong, Z.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef] [PubMed]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Hailman, E.; Lichenstein, H.S.; Wurfel, M.M.; Miller, D.S.; Johnson, D.A.; Kelley, M.; Busse, L.A.; Zukowski, M.M.; Wright, S.D. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J. Exp. Med. 1994, 179, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Kapellos, T.S.; Bonaguro, L.; Gemünd, I.; Reusch, N.; Saglam, A.; Hinkley, E.R.; Schultze, J.L. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 2019, 10, 2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical Patrolling Monocyte Function in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Calderon, B.; Carrero, J.A.; Ferris, S.; Sojka, D.K.; Moore, L.; Epelman, S.; Murphy, K.M.; Yokoyama, W.M.; Randolph, G.J.; Unanue, E.R. The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med. 2015, 212, 1497–1512. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; De Palma, M.; Naldini, L. Tie2-Expressing Monocytes and Tumor Angiogenesis: Regulation by Hypoxia and Angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberger, K.J.; Forget, M.A.; Bobko, A.A.; Mihalik, N.E.; Gencheva, M.; Roda, J.M.; Cole, S.L.; Mo, X.; Hoblitzell, E.H.; Evans, R.; et al. Hypoxia-Inducible Factor α Subunits Regulate Tie2-Expressing Macrophages That Influence Tumor Oxygen and Perfusion in Murine Breast Cancer. J. Immunol. 2020, 205, 2301–2311. [Google Scholar] [CrossRef] [PubMed]
- Sfiligoi, C.; de Luca, A.; Cascone, I.; Sorbello, V.; Fuso, L.; Ponzone, R.; Biglia, N.; Audero, E.; Arisio, R.; Bussolino, F.; et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int. J. Cancer 2003, 103, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Nouso, K.; Miyahara, K.; Oyama, A.; Wada, N.; Dohi, C.; Takeuchi, Y.; Yasunaka, T.; Onishi, H.; Ikeda, F.; et al. Monitoring serum proangiogenic cytokines from hepatocellular carcinoma patients treated with sorafenib. J. Gastroenterol. Hepatol. 2019, 34, 1081–1087. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; He, Q.; Luo, C.; Qian, L. Diagnostic and prognostic potential of serum angiopoietin-2 expression in human breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 660–664. [Google Scholar]
- Goede, V.; Coutelle, O.; Neuneier, J.; Reinacher-Schick, A.; Schnell, R.; Koslowsky, T.C.; Weihrauch, M.R.; Cremer, B.; Kashkar, H.; Odenthal, M.; et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br. J. Cancer 2010, 103, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Naumnik, W.; Chyczewska, E.; Ossolinska, M. Serum Levels of Angiopoietin-1, Angiopoietin-2, and Their Receptor Tie-2 in Patients with Nonsmall Cell Lung Cancer During Chemotherapy. Cancer Investig. 2009, 27, 741–746. [Google Scholar] [CrossRef]
- Atanasov, G.; Pötner, C.; Aust, G.; Schierle, K.; Dietel, C.; Benzing, C.; Krenzien, F.; Bartels, M.; Eichfeld, U.; Schmelzle, M.; et al. TIE2-expressing monocytes and M2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget 2018, 9, 29715–29726. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.S.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Venneri, M.A.; Roca, C.; Naldini, L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat. Med. 2003, 9, 789–795. [Google Scholar] [CrossRef]
- Kurahara, H.; Takao, S.T.; Kuwahata, T.; Nagai, T.; Ding, Q.; Maeda, K.; Shinchi, H.; Mataki, Y.; Maemura, K.; Matsuyama, T.; et al. Clinical significance of folate receptor beta-expressing tumor-associated macrophages in pancreatic cancer. Ann. Surg. Oncol. 2012, 19, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, H.; Ouyang, R.; Yang, Y.; Peng, J. Prognostic significance of the systemic immune-inflammation index in pancreatic carcinoma patients: A meta-analysis. Biosci. Rep. 2021, 41, BSR20204401. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Jung, E.-K.; Park, S.J.; Jeong, S.; Kim, I.-H.; Lee, M.-A. Neutrophil-to-lymphocyte ratio and carbohydrate antigen 19-9 as prognostic markers for advanced pancreatic cancer patients receiving first-line chemotherapy. World J. Gastrointest. Oncol. 2021, 13, 915–928. [Google Scholar] [CrossRef]
- Murthy, P.; Zenati, M.S.; Al Abbas, A.I.; Rieser, C.J.; Bahary, N.; Lotze, M.T.; ZehIII, H.J.; Zureikat, A.; Boone, B.A. Prognostic Value of the Systemic Immune-Inflammation Index (SII) After Neoadjuvant Therapy for Patients with Resected Pancreatic Cancer. Ann. Surg. Oncol. 2019, 27, 898–906. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, T.; Huang, L.; Wang, H.; Zhang, L.; Wang, Z.; Cui, Y. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher malignancy and worse prognosis. Biochem. Biophys. Res. Commun. 2018, 501, 313–319. [Google Scholar] [CrossRef]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef] [Green Version]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key mediators of tumour angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Teijeira, A.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e8. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.R.; Strøbech, J.E.; Horton, E.R.; Jackstadt, R.; Laitala, A.; Bravo, M.C.; Maltese, G.; Jensen, A.R.D.; Reuten, R.; Rafaeva, M.; et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat. Commun. 2021, 12, 3414. [Google Scholar] [CrossRef] [PubMed]
- Gaida, M.M.; Günther, F.; Wagner, C.; Friess, H.; Giese, N.A.; Schmidt, J.; Hänsch, G.M.; Wente, M.N. Expression of the CXCR6 on polymorphonuclear neutrophils in pancreatic carcinoma and in acute, localized bacterial infections. Clin. Exp. Immunol. 2008, 154, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Reibman, J.; Meixler, S.; Lee, T.C.; Gold, L.I.; Cronstein, B.N.; Haines, K.A.; Kolasinski, S.L.; Weissmann, G. Transforming growth factor beta 1, a potent chemoattractant for human neutrophils, bypasses classic signal-transduction pathways. Proc. Natl. Acad. Sci. USA 1991, 88, 6805–6809. [Google Scholar] [CrossRef] [Green Version]
- Brkovic, A.; Pelletier, M.; Girard, D.; Sirois, M.G. Angiopoietin chemotactic activities on neutrophils are regulated by PI-3K activation. J. Leukoc. Biol. 2007, 81, 1093–1101. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, C.; Maliba, R.; Favier, J.; Théoret, J.-F.; Merhi, Y.; Sirois, M.G. Angiopoietins can directly activate endothelial cells and neutrophils to promote proinflammatory responses. Blood 2005, 105, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Dumas, E.; Martel, C.; Neagoe, P.-E.; Bonnefoy, A.; Sirois, M.G. Angiopoietin-1 but not angiopoietin-2 promotes neutrophil viability: Role of interleukin-8 and platelet-activating factor. Biochim. et Biophys. Acta 2012, 1823, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Pekarek, L.A.; Starr, B.A.; Toledano, A.Y.; Schreiber, H. Inhibition of tumor growth by elimination of granulocytes. J. Exp. Med. 1995, 181, 435–440. [Google Scholar] [CrossRef]
- Shojaei, F.; Singh, M.; Thompson, J.D.; Ferrara, N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc. Natl. Acad. Sci. USA 2008, 105, 2640–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17--a new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Ohms, M.; Möller, S.; Laskay, T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes In Vitro. Front. Immunol. 2020, 11, 532. [Google Scholar] [CrossRef]
- Casbon, A.-J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegue, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–E575. [Google Scholar] [CrossRef] [Green Version]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J., III. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. OncoImmunology 2019, 8, e1605822. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, C.; McMaster, W.R.; Girard, D.; Descoteaux, A. Leishmania donovaniPromastigotes Evade the Antimicrobial Activity of Neutrophil Extracellular Traps. J. Immunol. 2010, 185, 4319–4327. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Xu, H.-X.; Zhang, S.-R.; Li, H.; Wang, W.-Q.; Gao, H.-L.; Wu, C.-T.; Xu, J.-Z.; Qi, Z.-H.; Li, S.; et al. Tumor-Infiltrating NETs Predict Postsurgical Survival in Patients with Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2019, 26, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, A.K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, S.S.; Dumas, E.; Vulesevic, B.; Neagoe, P.-E.; White, M.; Sirois, M.G. Synthesis of Human Neutrophil Extracellular Traps Contributes to Angiopoietin-Mediated In Vitro Proinflammatory and Proangiogenic Activities. J. Immunol. 2018, 200, 3801–3813. [Google Scholar] [CrossRef] [PubMed]
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- McInturff, A.M.; Cody, M.J.; Elliott, E.A.; Glenn, J.W.; Rowley, J.W.; Rondina, M.T.; Yost, C.C. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood 2012, 120, 3118–3125. [Google Scholar] [CrossRef] [Green Version]
- Khawaja, A.A.; Chong, D.L.; Sahota, J.; Mikolasch, T.A.; Pericleous, C.; Ripoll, V.M.; Booth, H.L.; Khan, S.; Rodriguez-Justo, M.; Giles, I.P.; et al. Identification of a Novel HIF-1alpha-alphaMbeta2 Integrin-NET Axis in Fibrotic Interstitial Lung Disease. Front. Immunol. 2020, 11, 2190. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, Y.; Tu, K.; Xu, Q.; Yang, Y.; Salman, S.; Le, N.; Lu, H.; Chen, C.; Zhu, Y.; et al. Histone citrullination by PADI4 is required for HIF-dependent transcriptional responses to hypoxia and tumor vascularization. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Branitzki-Heinemann, K.; Möllerherm, H.; Völlger, L.; Husein, D.M.; de Buhr, N.; Blodkamp, S.; Reuner, F.; Brogden, G.; Naim, H.Y.; von Köckritz-Blickwede, M. Formation of Neutrophil Extracellular Traps under Low Oxygen Level. Front. Immunol. 2016, 7, 518. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.; Brogden, G.; Jerjomiceva, N.; Brodesser, S.; Naim, H.Y.; von Köckritz-Blickwede, M. Lipid alterations in human blood-derived neutrophils lead to formation of neutrophil extracellular traps. Eur. J. Cell Biol. 2014, 93, 347–354. [Google Scholar] [CrossRef]
- Boone, B.A.; Orlichenko, L.; Schapiro, N.E.; Loughran, P.; Gianfrate, G.C.; Ellis, J.T.; Singhi, A.D.; Kang, R.; Tang, D.; Lotze, M.T.; et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015, 22, 326–334. [Google Scholar] [CrossRef] [Green Version]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-Polarized Tumor-Associated Macrophage in Pancreatic Cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef]
- Reid, M.D.; Basturk, O.; Thirabanjasak, D.; Hruban, R.H.; Klimstra, D.S.; Bagci, P.; Altinel, D.; Adsay, V. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod. Pathol. 2011, 24, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding Macrophage Entry into Hypoxic Tumor Areas by Sema3A/Nrp1 Signaling Blockade Inhibits Angiogenesis and Restores Antitumor Immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Li, Y.; Li, Z.; Huang, C.; Yang, Y.; Lang, M.; Cao, J.; Jiang, W.; Xu, Y.; Dong, J.; et al. Hypoxia Inducible Factor 1 (HIF-1) Recruits Macrophage to Activate Pancreatic Stellate Cells in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2016, 17, 799. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Landers, R.J.; Underwood, C.E.; Harris, A.L.; Lewis, C.E. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J. Pathol. 2000, 192, 150–158. [Google Scholar] [CrossRef]
- Roiniotis, J.; Dinh, H.; Masendycz, P.; Turner, A.; Elsegood, C.L.; Scholz, G.M.; Hamilton, J.A. Hypoxia Prolongs Monocyte/Macrophage Survival and Enhanced Glycolysis Is Associated with Their Maturation under Aerobic Conditions. J. Immunol. 2009, 182, 7974–7981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Marchesi, F.; Basso, G.; Rahal, D.; Delconte, G.; Catalano, M.; Cappello, P.; et al. Early expression of the fractalkine receptor CX3CR1 in pancreatic carcinogenesis. Br. J. Cancer 2013, 109, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta 2013, 1832, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Silverpil, E.; Glader, P.; Hansson, M.; Lindén, A. Impact of Interleukin-17 on Macrophage Phagocytosis of Apoptotic Neutrophils and Particles. Inflammation 2010, 34, 1–9. [Google Scholar] [CrossRef]
- Chen, K.; Murao, A.; Arif, A.; Takizawa, S.; Jin, H.; Jiang, J.; Aziz, M.; Wang, P. Inhibition of Efferocytosis by Extracellular CIRP–Induced Neutrophil Extracellular Traps. J. Immunol. 2021, 206, 797–806. [Google Scholar] [CrossRef]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lämmermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frossard, J.L.; Lenglet, S.; Montecucco, F.; Steffens, S.; Galan, K.; Pelli, G.; Spahr, L.; Mach, F.; Hadengue, A. Role of CCL-2, CCR-2 and CCR-4 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. J. Clin. Pathol. 2011, 64, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Li, Y.; Niu, Z.; Zong, Y.; Wang, M.; Yao, L.; Lu, Z.; Liao, Q.; Zhao, Y. Atorvastatin (Lipitor) attenuates the effects of aspirin on pancreatic cancerogenesis and the chemotherapeutic efficacy of gemcitabine on pancreatic cancer by promoting M2 polarized tumor associated macrophages. J. Exp. Clin. Cancer Res. 2016, 35, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefkowitz, D.L.; Lefkowitz, S.S. Macrophage–neutrophil interaction: A paradigm for chronic inflammation revisited. Immunol. Cell Biol. 2001, 79, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Bayuh, R.; Levy, L.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunother. 2013, 62, 1745–1756. [Google Scholar] [CrossRef]
- Qin, F.; Liu, X.; Chen, J.; Huang, S.; Wei, W.; Zou, Y.; Liu, X.; Deng, K.; Mo, S.; Chen, J.; et al. Anti-TGF-beta attenuates tumor growth via polarization of tumor associated neutrophils towards an anti-tumor phenotype in colorectal cancer. J. Cancer 2020, 11, 2580–2592. [Google Scholar] [CrossRef]
- Chen, S.-J.; Lian, G.-D.; Li, J.-J.; Zhang, Q.-B.; Zeng, L.-J.; Yang, K.-G.; Huang, C.-M.; Li, Y.-Q.; Chen, Y.-T.; Huang, K.-H. Tumor-driven like macrophages induced by conditioned media from pancreatic ductal adenocarcinoma promote tumor metastasis via secreting IL-8. Cancer Med. 2018, 7, 5679–5690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meher, A.K.; Spinosa, M.; Davis, J.P.; Pope, N.; Laubach, V.E.; Su, G.; Serbulea, V.; Leitinger, N.; Ailawadi, G.; Upchurch, G.R. Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Whyte, M.K.; Haslett, C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J. Leukoc. Biol. 1993, 54, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Kabala, P.A.; Malvar-Fernández, B.; Lopes, A.P.; Carvalheiro, T.; Hartgring, S.A.Y.; Tang, M.W.; Conde, C.; Baeten, D.L.; Sleeman, M.; Tak, P.P.; et al. Promotion of macrophage activation by Tie2 in the context of the inflamed synovia of rheumatoid arthritis and psoriatic arthritis patients. Rheumatology 2019, 59, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Connolly, D.T.; Heuvelman, D.M.; Nelson, R.; Olander, J.V.; Eppley, B.L.; Delfino, J.J.; Siegel, N.R.; Leimgruber, R.M.; Feder, J. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Investig. 1989, 84, 1470–1478. [Google Scholar] [CrossRef]
- Vaupel, P. The Role of Hypoxia-Induced Factors in Tumor Progression. Oncologist 2004, 9 (Suppl. 5), 10–17. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Huang, C.; Li, Z.; Li, N.; Li, Y.; Chang, A.; Zhao, T.; Wang, X.; Wang, H.; Gao, S.; Yang, S.; et al. Interleukin 35 Expression Correlates with Microvessel Density in Pancreatic Ductal Adenocarcinoma, Recruits Monocytes, and Promotes Growth and Angiogenesis of Xenograft Tumors in Mice. Gastroenterology 2018, 154, 675–688. [Google Scholar] [CrossRef]
- Bergers, G.; Javaherian, K.; Lo, K.-M.; Folkman, J.; Hanahan, D. Effects of Angiogenesis Inhibitors on Multistage Carcinogenesis in Mice. Science 1999, 284, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Karagiannis, G.S.; Pignatelli, J.; Smith, B.D.; Kadioglu, E.; Wise, S.C.; Hood, M.M.; Kaufman, M.D.; Leary, C.B.; Lu, W.-P.; et al. The Selective Tie2 Inhibitor Rebastinib Blocks Recruitment and Function of Tie2Hi Macrophages in Breast Cancer and Pancreatic Neuroendocrine Tumors. Mol. Cancer Ther. 2017, 16, 2486–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, G.; Baluk, P.; McDonald, D.M. Determinants of endothelial cell phenotype in venules. Microcirculation 2000, 7, 67–80. [Google Scholar] [CrossRef] [PubMed]
- McCourt, M.; Wang, J.H.; Sookhai, S.; Redmond, H.P. Proinflammatory Mediators Stimulate Neutrophil-Directed Angiogenesis. Arch. Surg. 1999, 134, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.D.; Lebovic, D.I.; Garrett, E.; Taylor, R.N. Neutrophils infiltrating the endometrium express vascular endothelial growth factor: Potential role in endometrial angiogenesis. Fertil. Steril. 2000, 74, 107–112. [Google Scholar] [CrossRef]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, J.; Wu, C.-F.; Leschner, A.S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-beta. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Benelli, R.; Morini, M.; Carrozzino, F.; Ferrari, N.; Minghelli, S.; Santi, L.; Cassatella, M.; Noonan, D.M.; Albini, A. Neutrophils as a key cellular target for angiostatin: Implications for regulation of angiogenesis and inflammation. FASEB J. 2002, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Raghuwanshi, S.K.; Smith, N.; Rivers, E.J.; Thomas, A.J.; Sutton, N.; Hu, Y.; Mukhopadhyay, S.; Chen, X.L.; Leung, T.; Richardson, R.M.; et al. G protein-coupled receptor kinase 6 deficiency promotes angiogenesis, tumor progression, and metastasis. J. Immunol. 2013, 190, 5329–5336. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [Green Version]
- Schomber, T.; Kopfstein, L.; Djonov, V.; Albrecht, I.; Baeriswyt, V.; Strittmatter, K.; Christofori, G. Placental growth factor-1 attenuates vascular endothelial growth factor-A-dependent tumor angiogenesis during beta cell carcinogenesis. Cancer Res. 2007, 67, 10840–10848. [Google Scholar] [CrossRef] [Green Version]
- Matsui, A.; Yokoo, H.; Negishi, Y.; Endo-Takahashi, Y.; Chun, N.A.L.; Kadouchi, I.; Suzuki, R.; Maruyama, K.; Aramaki, Y.; Semba, K.; et al. CXCL17 expression by tumor cells recruits CD11b+Gr1 high F4/80- cells and promotes tumor progression. PLoS ONE 2012, 7, e44080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Doerschuk, C.M. Neutrophil-Induced Changes in the Biomechanical Properties of Endothelial Cells: Roles of ICAM-1 and Reactive Oxygen Species. J. Immunol. 2000, 164, 6487–6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massena, S.; Christoffersson, G.; Vågesjö, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Hidalgo, C.H.; Giraud, A.; Lomei, J.; Weström, S.; et al. Identification and characterization of VEGF-A–responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Soloviev, D.A.; Hazen, S.L.; Szpak, D.; Bledzka, K.M.; Ballantyne, C.M.; Plow, E.F.; Pluskota, E. Dual role of the leukocyte integrin alphaMbeta2 in angiogenesis. J. Immunol. 2014, 193, 4712–4721. [Google Scholar] [CrossRef] [Green Version]
- Keklikoglou, I.; Kadioglu, E.; Bissinger, S.; Langlois, B.; Bellotti, A.; Orend, G.; Ries, C.H.; De Palma, M. Periostin Limits Tumor Response to VEGFA Inhibition. Cell Rep. 2018, 22, 2530–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.; Michl, P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2017, 66, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Qi, F.; Zhang, S.; Ma, X.; Wang, S.; Wang, C.; Fu, Y.; Luo, Y. Adrenomedullin promotes the growth of pancreatic ductal adenocarcinoma through recruitment of myelomonocytic cells. Oncotarget 2016, 7, 55043–55056. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See, P.; Luis, B.S.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. OncoImmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Nadella, V.; Singh, S.; Jain, A.; Jain, M.; Vasquez, K.M.; Sharma, A.; Tanwar, P.; Rath, G.K.; Prakash, H. Low dose radiation primed iNOS + M1macrophages modulate angiogenic programming of tumor derived endothelium. Mol. Carcinog. 2018, 57, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Roberts, R.; Galloway, M.; Wang, Y.; Cohn, D.E.; Marsh, C.B. GM-CSF Induces Expression of Soluble VEGF Receptor-1 from Human Monocytes and Inhibits Angiogenesis in Mice. Immun. 2004, 21, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Eubank, T.D.; Roberts, R.D.; Khan, M.; Curry, J.M.; Nuovo, G.J.; Kuppusamy, P.; Marsh, C.B. Granulocyte Macrophage Colony-Stimulating Factor Inhibits Breast Cancer Growth and Metastasis by Invoking an Anti-Angiogenic Program in Tumor-Educated Macrophages. Cancer Res. 2009, 69, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Eubank, T.D.; Roda, J.M.; Liu, H.; O’Neil, T.; Marsh, C.B. Opposing roles for HIF-1α and HIF-2α in the regulation of angiogenesis by mononuclear phagocytes. Blood 2011, 117, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Roda, J.M.; Sumner, L.A.; Evans, R.; Phillips, G.S.; Marsh, C.B.; Eubank, T.D. Hypoxia-Inducible Factor-2α Regulates GM-CSF–Derived Soluble Vascular Endothelial Growth Factor Receptor 1 Production from Macrophages and Inhibits Tumor Growth and Angiogenesis. J. Immunol. 2011, 187, 1970–1976. [Google Scholar] [CrossRef] [Green Version]
- Roda, J.; Wang, Y.; Sumner, L.A.; Phillips, G.S.; Marsh, C.B.; Eubank, T.D. Stabilization of HIF-2α Induces sVEGFR-1 Production from Tumor-Associated Macrophages and Decreases Tumor Growth in a Murine Melanoma Model. J. Immunol. 2012, 189, 3168–3177. [Google Scholar] [CrossRef] [Green Version]
- Mihalik, N.E.; Wen, S.; Driesschaert, B.; Eubank, T.D. Formulation and In Vitro Characterization of PLGA/PLGA-PEG Nanoparticles Loaded with Murine Granulocyte-Macrophage Colony-Stimulating Factor. AAPS PharmSciTech 2021, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.-H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; Van Bruggen, N.; et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nat. Cell Biol. 2007, 450, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Curtis, V.F.; Wang, H.; Yang, P.; McLendon, R.E.; Li, X.; Zhou, Q.-Y.; Wang, X.-F. A PK2/Bv8/PROK2 antagonist suppresses tumorigenic processes by inhibiting angiogenesis in glioma and blocking myeloid cell infiltration in pancreatic cancer. PLoS ONE 2013, 8, e54916. [Google Scholar]
- Christoffersson, G.; Vågesjö, E.; Vandooren, J.; Lidén, M.; Massena, S.; Reinert, R.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9–delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef] [PubMed]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.D.; Meng, X.; Fullerton, D.A.; Moore, E.E.; Lee, J.H.; Ao, L.; Silliman, C.C.; Barnett, C.C. Activation state of stromal inflammatory cells in murine metastatic pancreatic adenocarcinoma. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R1067–R1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Dufort, C.C.; DelGiorno, K.E.; Carlson, M.A.; Osgood, R.J.; Zhao, C.; Huang, Z.; Thompson, C.B.; Connor, R.J.; Thanos, C.D.; Brockenbrough, J.S.; et al. Interstitial Pressure in Pancreatic Ductal Adenocarcinoma Is Dominated by a Gel-Fluid Phase. Biophys. J. 2016, 110, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Zeiher, A.M. Netting Insights into Fibrosis. N. Engl. J. Med. 2017, 376, 1475–1477. [Google Scholar] [CrossRef]
- Zhang, S.; Jia, X.; Zhang, Q.; Zhang, L.; Yang, J.; Hu, C.; Shi, J.; Jiang, X.; Lu, J.; Shen, H. Neutrophil extracellular traps activate lung fibroblast to induce polymyositis-related interstitial lung diseases via TLR9-miR-7-Smad2 pathway. J. Cell Mol. Med. 2020, 24, 1658–1669. [Google Scholar] [CrossRef]
- Suzuki, M.; Ikari, J.; Anazawa, R.; Tanaka, N.; Katsumata, Y.; Shimada, A.; Suzuki, E.; Tatsumi, K. PAD4 Deficiency Improves Bleomycin-induced Neutrophil Extracellular Traps and Fibrosis in Mouse Lung. Am. J. Respir. Cell Mol. Biol. 2020, 63, 806–818. [Google Scholar] [CrossRef]
- Martinod, K.; Witsch, T.; Erpenbeck, L.; Savchenko, A.; Hayashi, H.; Cherpokova, D.; Gallant, M.; Mauler, M.; Cifuni, S.M.; Wagner, D.D. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 2017, 214, 439–458. [Google Scholar] [CrossRef]
- Kuyl, E.V.; Shu, F.; Sosa, B.R.; Lopez, J.D.; Qin, D.; Pannellini, T.; Ivashkiv, L.B.; Greenblatt, M.B.; Bostrom, M.P.G.; Yang, X. Inhibition of PAD4 mediated neutrophil extracellular traps prevents fibrotic osseointegration failure in a tibial implant murine model: An animal study. Bone Jt. J. 2021, 103 (Suppl. 7), 135–144. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Eubank, T.; Lukomski, S.; Boone, B. Immune Cell Modulation of the Extracellular Matrix Contributes to the Pathogenesis of Pancreatic Cancer. Biomolecules 2021, 11, 901. [Google Scholar] [CrossRef]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.; Grahovac, J.; Kao, S.; et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016, 6, 852–869. [Google Scholar] [CrossRef] [Green Version]
- Mayer, P.; Dinkic, C.; Jesenofsky, R.; Klauss, M.; Schirmacher, P.; Dapunt, U.; Hackert, T.; Uhle, F.; Hänsch, G.M.; Gaida, M.M. Changes in the microarchitecture of the pancreatic cancer stroma are linked to neutrophil-dependent reprogramming of stellate cells and reflected by diffusion-weighted magnetic resonance imaging. Theranostics 2018, 8, 13–30. [Google Scholar] [CrossRef]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int. J. Oncol. 2019, 56, 596–605. [Google Scholar] [CrossRef]
- Detlefsen, S.; Sipos, B.; Feyerabend, B.; Klöppel, G. Fibrogenesis in alcoholic chronic pancreatitis: The role of tissue necrosis, macrophages, myofibroblasts and cytokines. Mod. Pathol. 2006, 19, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhang, B.; Chen, L.; Xia, W.; Liu, G.; Zhu, X.; Ren, C.; Liu, W.; Lu, P. Essential Contribution of Macrophage Tie2 Signalling in a Murine Model of Laser-Induced Choroidal Neovascularization. Sci. Rep. 2020, 10, 9613. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Zhu, Q.; Zheng, S.; Li, G.; Li, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Ochi, A.; Heindel, D.W.; Lee, K.B.; Zambirinis, C.P.; Pandian, G.S.B.; Savadkar, S.; et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat. Med. 2017, 23, 556–567. [Google Scholar] [CrossRef]
- Sica, A.; Saccani, A.; Bottazzi, B.; Polentarutti, N.; Vecchi, A.; Damme, J.V.; Mantovani, A. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J. Immunol. 2000, 164, 762–767. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.B.; Morton, J.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Rice, C.M.; Davies, L.C.; Subleski, J.J.; Maio, N.; Gonzalez-Cotto, M.; Andrews, C.; Patel, N.L.; Palmieri, E.M.; Weiss, J.M.; Lee, J.-M.; et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun. 2018, 9, 5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hu, L.P.; Qin, W.T.; Yang, Q.; Chen, D.Y.; Li, Q.; Zhou, K.X.; Huang, P.Q.; Xu, C.J.; Li, J.; et al. Identification of a subset of immunosuppressive P2RX1-negative neutrophils in pancreatic cancer liver metastasis. Nat. Commun. 2021, 12, 174. [Google Scholar] [CrossRef]
- Sippel, T.R.; White, J.; Nag, K.; Tsvankin, V.; Klaassen, M.; Kleinschmidt-DeMasters, B.; Waziri, A. Neutrophil Degranulation and Immunosuppression in Patients with GBM: Restoration of Cellular Immune Function by Targeting Arginase I. Clin. Cancer Res. 2011, 17, 6992–7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xu, X.; Shi, M.; Chen, Y.; Yu, D.; Zhao, C.; Gu, Y.; Yang, B.; Guo, S.; Ding, G.; et al. CD13hi Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. OncoImmunology 2017, 6, e1258504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Cowburn, A.; Crosby, A.; Macias, D.; Branco, C.B.M.; Colaço, R.D.; Southwood, M.; Toshner, M.; Alexander, L.E.C.; Morrell, N.; Chilvers, E.; et al. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2018, 113, 8801–8806. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.M.; Reed, L.F.; Krasnick, B.A.; Miranda-Carboni, G.; Fields, R.C.; Bi, Y.; Elahi, A.; Ajidahun, A.; Dickson, P.V.; DeNeve, J.L.; et al. IL23 and TGF-ß diminish macrophage associated metastasis in pancreatic carcinoma. Sci. Rep. 2018, 8, 5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, A.; Wang, S.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshiruma, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Krüger, U.; Becker, T.; Ebsen, M.; Röcken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.-Y.; Shi, X.-Y.; Huang, W.; Ruan, T.-Y.; Xie, P.; Ding, J.-L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef]
- Yin, Z.; Ma, T.; Huang, B.; Lin, L.; Zhou, Y.; Yan, J.; Zou, Y.; Chen, S. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-beta signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 310. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer -Active Players in Tumor Progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Gaida, M.M.; Steffen, T.G.; Günther, F.; Tschaharganeh, D.F.; Felix, K.; Bergmann, F.; Schirmacher, P.; Hänsch, G.M. Polymorphonuclear neutrophils promote dyshesion of tumor cells and elastase-mediated degradation of E-cadherin in pancreatic tumors. Eur. J. Immunol. 2012, 42, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Steffen, T.; Giese, T.; Giese, N.; Longerich, T.; Schirmacher, P.; Hänsch, G.M.; Gaida, M.M. Epithelial-to-Mesenchymal Transition in Pancreatic Ductal Adenocarcinoma and Pancreatic Tumor Cell Lines: The Role of Neutrophils and Neutrophil-Derived Elastase. Clin. Dev. Immunol. 2012, 2012, 720768. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Ferri, L.E. Neutrophils Promote Liver Metastasis via Mac-1–Mediated Interactions with Circulating Tumor Cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.; Berden, J.H.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arter. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Kajioka, H.; Kagawa, S.; Ito, A.; Yoshimoto, M.; Sakamoto, S.; Kikuchi, S.; Kuroda, S.; Yoshida, R.; Umeda, Y.; Noma, K.; et al. Targeting neutrophil extracellular traps with thrombomodulin prevents pancreatic cancer metastasis. Cancer Lett. 2021, 497, 1–13. [Google Scholar] [CrossRef]
- Bulle, A.; Dekervel, J.; Deschuttere, L.; Nittner, D.; Libbrecht, L.; Janky, R.; Plaisance, S.; Topal, B.; Coosemans, A.; Lambrechts, D.; et al. Gemcitabine Recruits M2-Type Tumor-Associated Macrophages into the Stroma of Pancreatic Cancer. Transl. Oncol. 2020, 13, 100743. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.-J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Xian, G.; Zhao, J.; Qin, C.; Zhang, Z.; Lin, Y.; Su, Z. Simvastatin attenuates macrophage-mediated gemcitabine resistance of pancreatic ductal adenocarcinoma by regulating the TGF-beta1/Gfi-1 axis. Cancer Lett. 2017, 385, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Q.; Peng, J.; Wang, M.; Gao, X.; Liao, Q.; Zhao, Y. Pancreatic cancer-educated macrophages protect cancer cells from complement-dependent cytotoxicity by up-regulation of CD59. Cell Death Dis. 2019, 10, 836. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, M.V.; Guillén, M.J.; Lopez-Casas, P.P.; Sarno, F.; Gallardo, A.; Álamo, P.; Cuevas, C.; Hidalgo, M.; Galmarini, C.M.; Allavena, P.; et al. Lurbinectedin induces depletion of tumor-associated macrophages (TAM), an essential component of its in vivo synergism with gemcitabine. Dis. Model. Mech. 2016, 9, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahler, J.C.; Tazzyman, S.; Erez, N.; Chen, Y.-Y.; Murdoch, C.; Nozawa, H.; Lewis, C.E.; Hanahan, D. Plasticity in Tumor-Promoting Inflammation: Impairment of Macrophage Recruitment Evokes a Compensatory Neutrophil Response. Neoplasia 2008, 10, 329-IN2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duluc, D.; Corvaisier, M.; Blanchard, S.; Catala, L.; Descamps, P.; Gamelin, E.; Ponsoda, S.; Delneste, Y.; Hebbar, M.; Jeannin, P. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int. J. Cancer 2009, 125, 367–373. [Google Scholar] [CrossRef]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9–expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.S.; Poudel, B.; Torres, E.R.; Sidhom, J.-W.; Robinson, T.M.; Christmas, B.; Scott, B.; Cruz, K.; Woolman, S.; Wall, V.Z.; et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell–Mediated Anticancer Activity. Cancer Immunol. Res. 2019, 7, 428–442. [Google Scholar] [CrossRef]
- Rech, A.J.; Dada, H.; Kotzin, J.J.; Henao-Mejia, J.; Minn, A.J.; Twyman-Saint Victor, C.; Vonderheide, R.H. Radiotherapy and CD40 Activation Separately Augment Immunity to Checkpoint Blockade in Cancer. Cancer Res. 2018, 78, 4282–4291. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control Release 2020, 321, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Bastea, L.I.; Liou, G.-Y.; Pandey, V.; Fleming, A.K.; Von Roemeling, C.A.; Doeppler, H.; Li, Z.; Qiu, Y.; Edenfield, B.; Copland, J.A.; et al. Pomalidomide Alters Pancreatic Macrophage Populations to Generate an Immune-Responsive Environment at Precancerous and Cancerous Lesions. Cancer Res. 2019, 79, 1535–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Zeisberger, S.M.; Odermatt, B.; Marty, C.; Zehnder-Fjallman, A.H.M.; Ballmer-Hofer, K.; Schwendener, R.A. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 2006, 95, 272–281. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef]
- Murthy, P.; Singhi, A.D.; Ross, M.A.; Loughran, P.; Paragomi, P.; Papachristou, G.I.; Whitcomb, D.C.; Zureikat, A.; Lotze, M.T.; Iii, H.J.Z.; et al. Enhanced Neutrophil Extracellular Trap Formation in Acute Pancreatitis Contributes to Disease Severity and Is Reduced by Chloroquine. Front. Immunol. 2019, 10, 28. [Google Scholar] [CrossRef]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.; Doerfler, W.R.; Ellis, J.T.; Liang, X.; Ross, M.A.; Wallace, C.T.; Sperry, J.L.; Lotze, M.T.; et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel with or Without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]




| Macrophage Subtype | Function | Murine Markers | Human Markers |
|---|---|---|---|
| M1-like | Immunosupportive | CD80+ CD86+ MHCII+ iNOS+ | CD80+ CD86+ HLA-DR+ iNOS+ |
| M2-like/TAM | Anti-inflammatory, Immunosuppressive | CD206+ ARG1+ FRβ+ | CD206+ CD204+ CD163+ ARG1+ FRβ+ |
| TEM | Anti-inflammatory, Immunosuppressive, Angiogenesis | TIE2+ CD31− | TIE2+ CD31− |
| MonoMac | Pro-inflammatory, Immunosupportive | CCR2+ Ly6Chi CX3CR1lo | CCR2+ CD14++ CD16− |
| TRM | Cloak microlesions, Tissue remodeling | CX3CR1hi Ly6Clo CCR2− | CX3CR1+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pratt, H.G.; Steinberger, K.J.; Mihalik, N.E.; Ott, S.; Whalley, T.; Szomolay, B.; Boone, B.A.; Eubank, T.D. Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer. Cancers 2022, 14, 194. https://doi.org/10.3390/cancers14010194
Pratt HG, Steinberger KJ, Mihalik NE, Ott S, Whalley T, Szomolay B, Boone BA, Eubank TD. Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer. Cancers. 2022; 14(1):194. https://doi.org/10.3390/cancers14010194
Chicago/Turabian StylePratt, Hillary G., Kayla J. Steinberger, Nicole E. Mihalik, Sascha Ott, Thomas Whalley, Barbara Szomolay, Brian A. Boone, and Timothy D. Eubank. 2022. "Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer" Cancers 14, no. 1: 194. https://doi.org/10.3390/cancers14010194