
Performance of 16S Metagenomic Profiling in Formalin-Fixed Paraffin-Embedded versus Fresh-Frozen Colorectal Cancer Tissues


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Library Preparation for Illumina MiSeq Sequencing
2.4. 16S rRNA Sequence Analysis
2.5. Statistical Analysis
2.6. Species-Level Taxonomy Inference and Phylogenetic Tree Reconstruction
2.7. Microbial In Situ Hybridization (ISH) and Image Analysis
3. Results
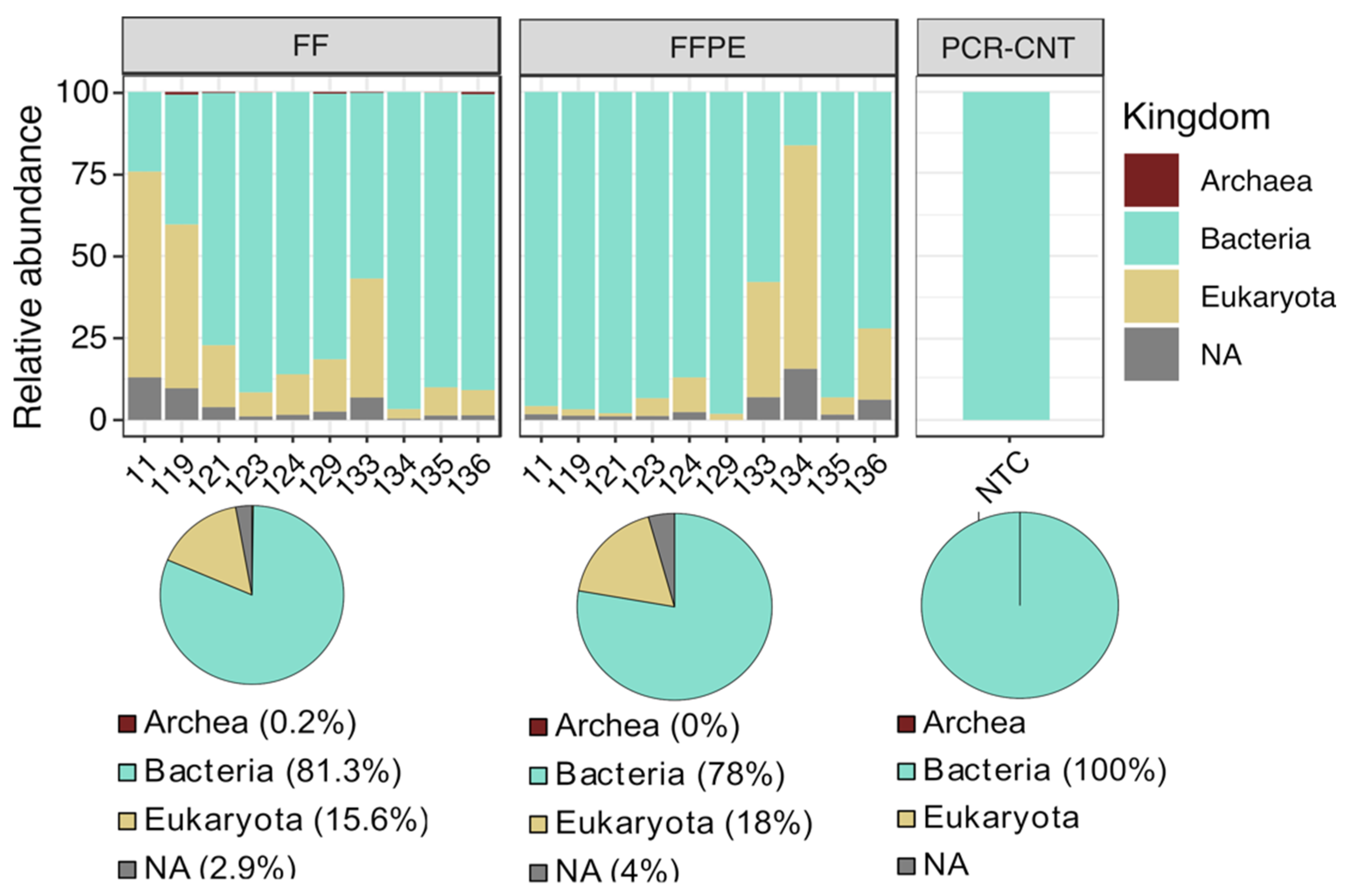
3.1. Data Processing and Quality Control
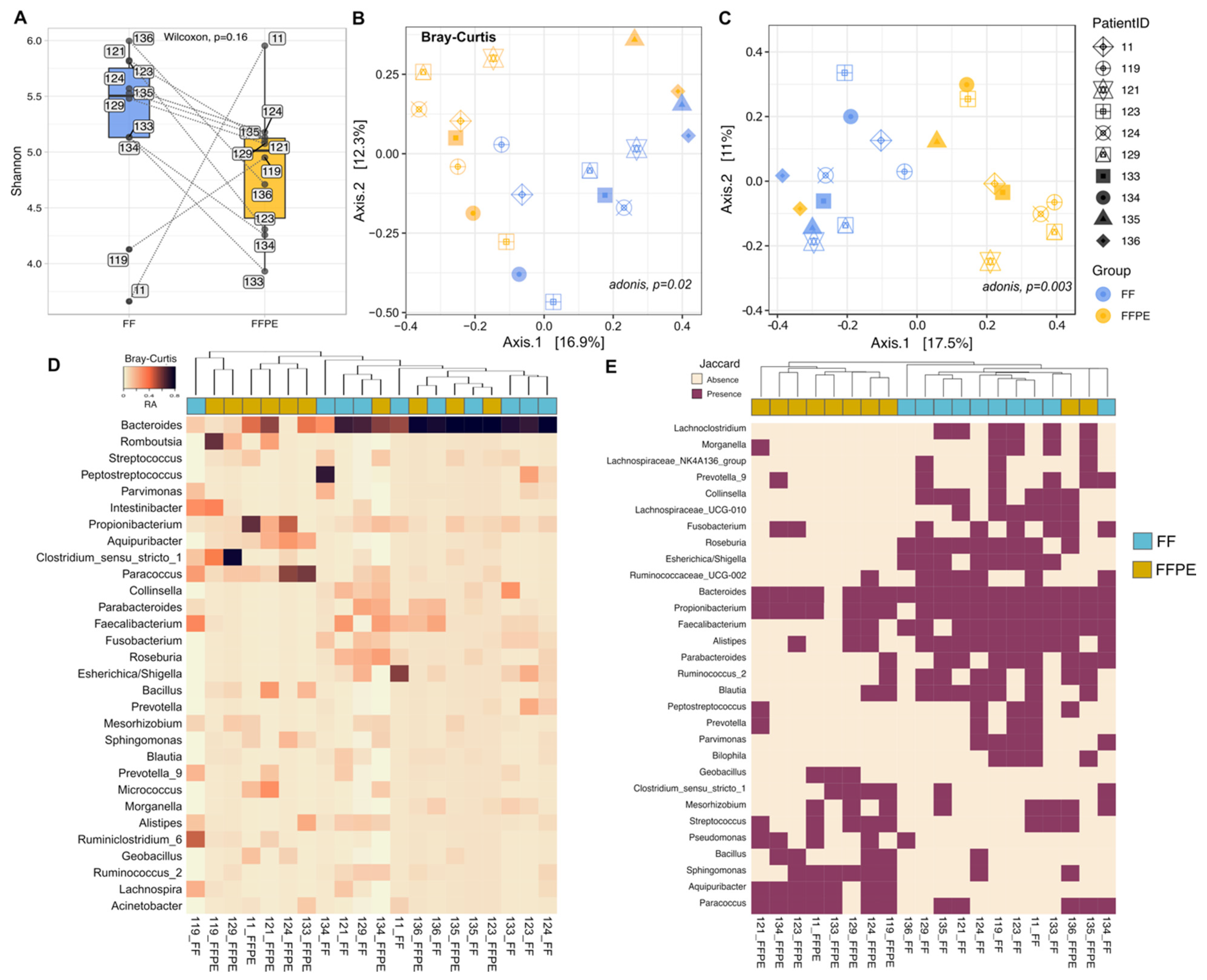
3.2. Comparison of the Microbial Diversity between FF and FFPE Tissues
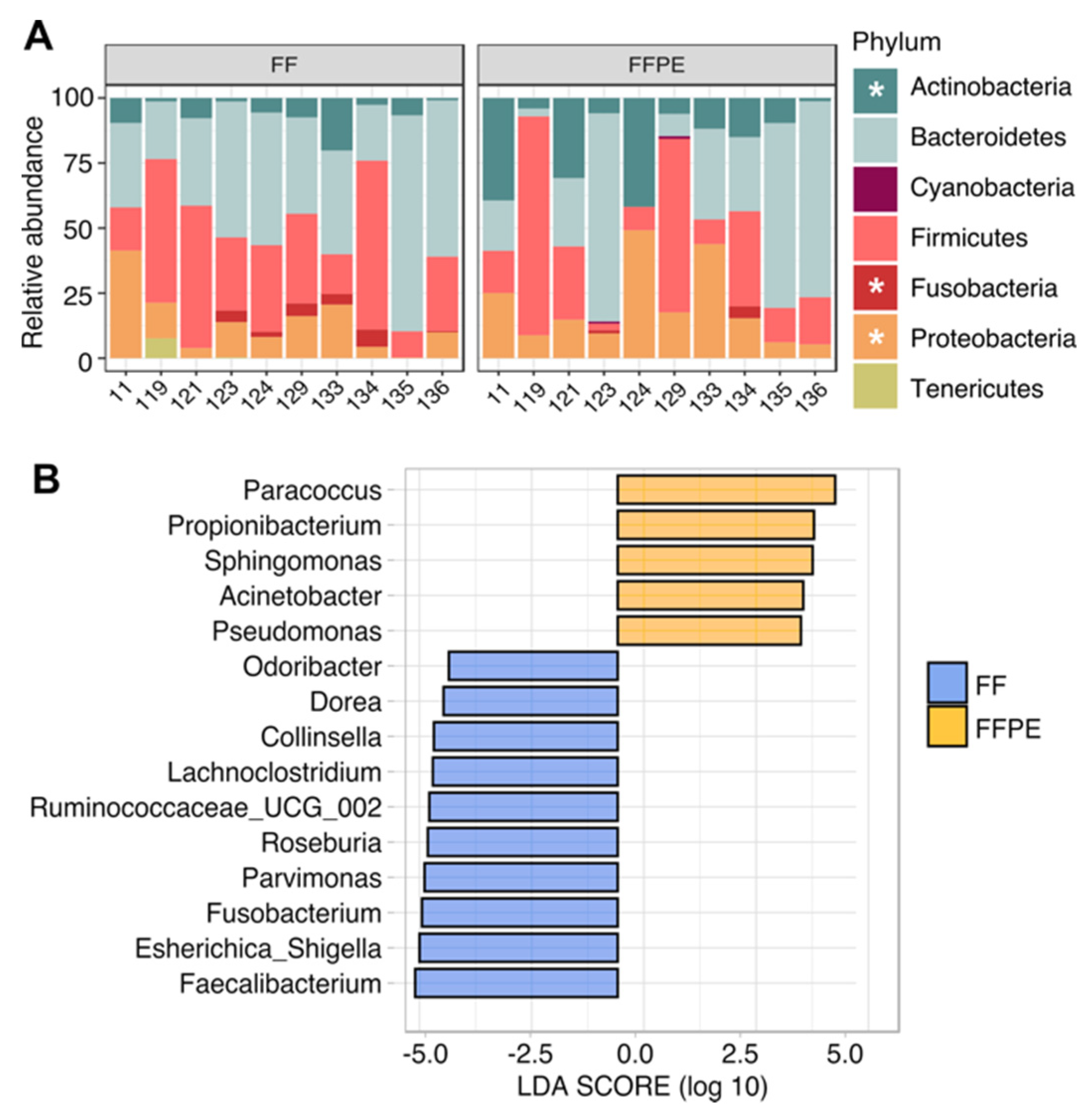
3.3. Taxonomic Profiling and Discriminant Taxa between FF and FFPE Tissue Samples
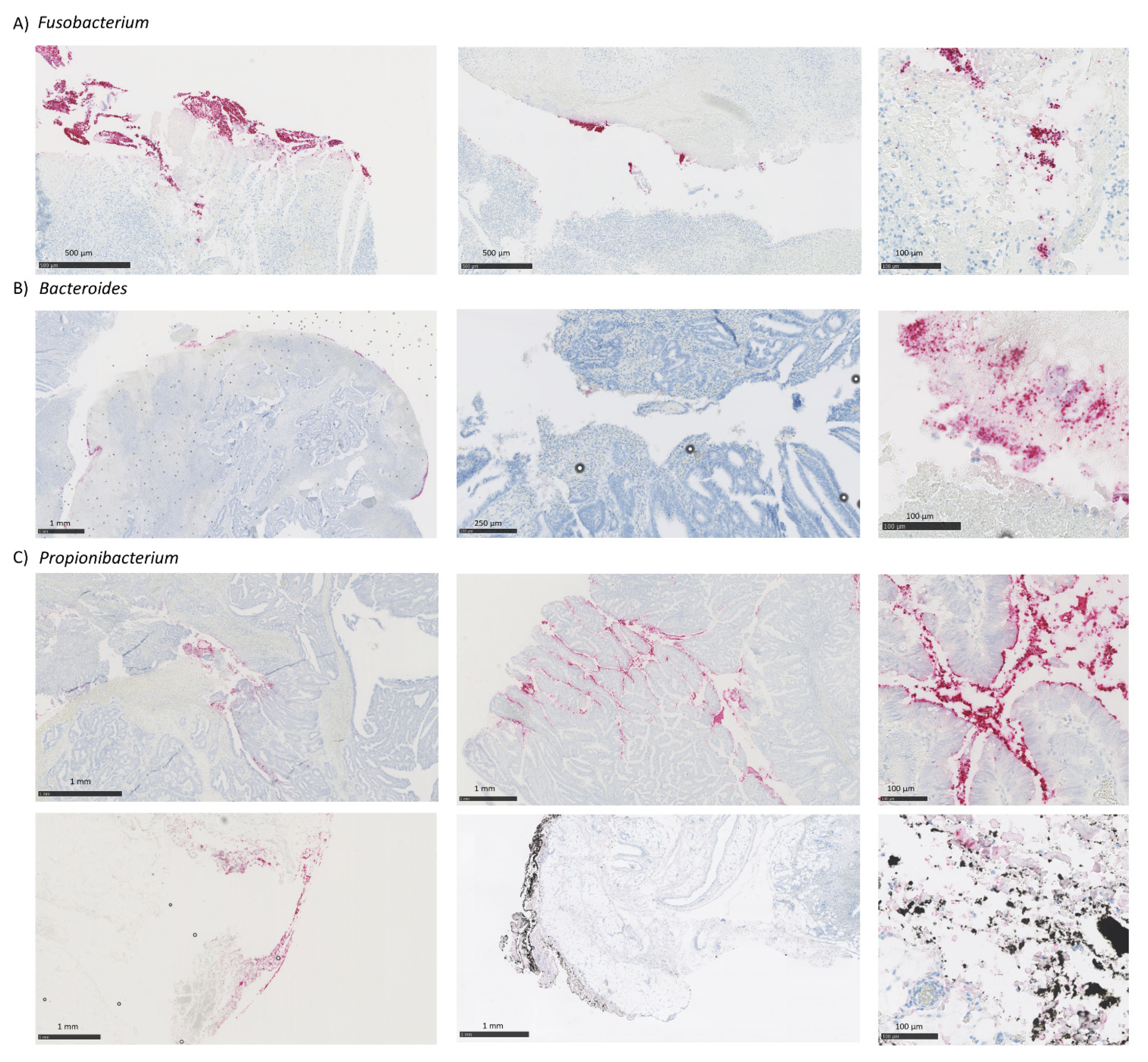
3.4. Putative Fusobacterium, Bacteroides and Propionibacterium Species Classification
3.5. Comparison of CRC-Associated Bacteria Characterized by RNA-ISH and 16S rRNA Sequencing
3.6. Quality Assessment of FFPE DNA Based on 16S rRNA Amplicon Profiling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 16S rRNA | 16S ribosomal RNA |
| ASV | Amplicon sequence variant |
| CRC | Colorectal cancer |
| DNA | Deoxyribonucleic acid |
| FF | Fresh frozen |
| FFPE | Formalin-fixed paraffin-embedded |
| HiPR-FISH | High Phylogenetic Resolution microbiome mapping by Fluorescence In-Situ Hybridization |
| RNA-ISH | RNA in situ hybridization |
| NCBI | National Center for Biotechnology Information |
| NGS | Next-generation sequencing |
| NTC | Negative template control |
| PCoA | Principal Coordinates Analysis |
| PCR | Polymerase chain reaction |
References
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, I.R. The role of the gastrointestinal microbiota in colorectal cancer. Curr. Pharm. Des. 2009, 15, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes 2017, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatumin colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.-O.; Sugai, T.; An, B.; et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhu, X.; Huang, X.; Murff, H.J.; Ness, R.M.; Seidner, D.L.; Sorgen, A.A.; Blakley, I.C.; Yu, C.; Dai, Q.; et al. On the robustness of inference of association with the gut microbiota in stool, rectal swab and mucosal tissue samples. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Debesa-Tur, G.; Pérez-Brocal, V.; Ruiz-Ruiz, S.; Castillejo, A.; Latorre, A.; Soto, J.L.; Moya, A. Metagenomic analysis of formalin-fixed paraffin-embedded tumor and normal mucosa reveals differences in the microbiome of colorectal cancer patients. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Racsa, L.D.; DeLeon-Carnes, M.; Hiskey, M.; Guarner, J. Identification of bacterial pathogens from formalin-fixed, paraffin-embedded tissues by using 16S sequencing: Retrospective correlation of results to clinicians’ responses. Hum. Pathol. 2017, 59, 132–138. [Google Scholar] [CrossRef]
- Hart, J.D.; Street, T.; Wrightson, J.M.; Moore, D.P.; Scott, A.G.; Crook, D.W.; Turner, G.D. 16S rRNA sequencing in molecular microbiological diagnosis of bacterial infections in the autopsy setting. Pathology 2014, 46, S113. [Google Scholar] [CrossRef]
- Betge, J.; Kerr, G.; Miersch, T.; Leible, S.; Erdmann, G.; Galata, C.L.; Zhan, T.; Gaiser, T.; Post, S.; Ebert, M.P.; et al. Amplicon sequencing of colorectal cancer: Variant calling in frozen and formalin-fixed samples. PLoS ONE 2015, 10, e0127146. [Google Scholar] [CrossRef] [Green Version]
- Robbe, P.; Popitsch, N.; Knight, S.J.L.; Antoniou, P.; Becq, J.; He, M.; Kanapin, A.; Samsonova, A.; Vavoulis, D.V.; Ross, M.T.; et al. Clinical whole-genome sequencing from routine formalin-fixed, paraffin-embedded specimens: Pilot study for the 100,000 genomes project. Genet. Med. 2018, 20, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.J.; Fatemizadeh, R.; Parsons, P.; Lamb, C.A.; Shady, D.A.; Petrosino, J.F.; Hair, A.B. Using formalin fixed paraffin embedded tissue to characterize the preterm gut microbiota in necrotising enterocolitis and spontaneous isolated perforation using marginal and diseased tissue. BMC Microbiol. 2019, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wagle, N.; Berger, M.F.; Davis, M.J.; Blumenstiel, B.; DeFelice, M.; Pochanard, P.; Ducar, M.; van Hummelen, P.; MacConaill, L.E.; Hahn, W.C.; et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2011, 2, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Pinto-Ribeiro, I.; Ferreira, R.M.; Pereira-Marques, J.; Pinto, V.; Macedo, G.; Carneiro, F.; Figueiredo, C. Evaluation of the use of formalin-fixed and paraffin-embedded archive gastric tissues for microbiota characterization using next-generation sequencing. Int. J. Mol. Sci. 2020, 21, 1096. [Google Scholar] [CrossRef] [Green Version]
- Emery, D.C.; Shoemark, D.; Batstone, T.E.; Waterfall, C.M.; Coghill, J.A.; Cerajewska, T.L.; Davies, M.; West, N.X.; Allen-Birt, S. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front. Aging Neurosci. 2017, 9, 195. [Google Scholar] [CrossRef] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC. A Quality Control Tool for High Throughput Se-Quence Data. Babraham Bioinformatics. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 October 2020).
- R Core Team. R: A Language and Environment for Statistical Computing (R Version 3.5.2); R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 16 September 2021).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-2. 2019. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 16 September 2021).
- Dray, S.; Dufour, A.-B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Bodenhofer, U.; Kothmeier, A.; Hochreiter, S. APCluster: An R package for affinity propagation clustering. Bioinformatics 2011, 27, 2463–2464. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Shao, L.; Liu, X.; Ji, F.; Mei, Y.; Cheng, Y.; Liu, F.; Yan, C.; Li, L.; Ling, Z. Alterations of gastric mucosal microbiota across different stomach microhabitats in a cohort of 276 patients with gastric cancer. EBioMedicine 2019, 40, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Citron, D.M. Update on the taxonomy and clinical aspects of the genus Fusobacterium. Clin. Infect. Dis. 2002, 35, S22–S27. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Jiang, B. Analysis of mucosa-associated microbiota in colorectal cancer. Med. Sci. Monit. 2017, 23, 4422–4430. [Google Scholar] [CrossRef] [Green Version]
- Coker, O.O.; Wu, W.K.K.; Wong, S.H.; Sung, J.J.; Yu, J. Altered gut archaea composition and interaction with bacteria are associated with colorectal cancer. Gastroenterology 2020, 159, 1459–1470. [Google Scholar] [CrossRef]
- Gethings-Behncke, C.; Coleman, H.G.; Jordao, H.W.T.; Longley, D.B.; Crawford, N.; Murray, L.J.; Kunzmann, A.T. Fusobac-terium nucleatum in the colorectum and its association with cancer risk and survival: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2020, 29, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [Green Version]
- Davidsson, S.; Mölling, P.; Rider, J.; Unemo, M.; Karlsson, M.G.; Carlsson, J.; Andersson, S.-O.; Elgh, F.; Söderquist, B.; Andrén, O. Frequency and typing of Propionibacterium acnes in prostate tissue obtained from men with and without prostate cancer. Infect. Agents Cancer 2016, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mollerup, S.; Friis-Nielsen, J.; Vinner, L.; Hansen, T.A.; Richter, S.R.; Fridholm, H.; Herrera, J.A.R.; Lund, O.; Brunak, S.; Izarzugaza, J.M.G.; et al. Propionibacterium acnes: Disease-causing agent or common contaminant? Detection in diverse patient samples by next-generation sequencing. J. Clin. Microbiol. 2016, 54, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Asp. Med. 2019, 69, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Hykin, S.M.; Bi, K.; McGuire, J.A. Fixing formalin: A method to recover genomic-scale DNA sequence data from formalin-fixed museum specimens using high-throughput sequencing. PLoS ONE 2015, 10, e0141579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, S.P.; Tangney, M.; Claesson, M. Sequence-based characterization of intratumoral bacteria—A guide to best practice. Front. Oncol. 2020, 10, 179. [Google Scholar] [CrossRef] [Green Version]
- Jervis-Bardy, J.; Leong, L.E.X.; Marri, S.; Smith, R.J.; Choo, J.M.; Smith-Vaughan, H.C.; Nosworthy, E.; Morris, P.S.; O’Leary, S.; Rogers, G.B.; et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 2015, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.T.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Wirth, U.; Garzetti, D.; Jochum, L.M.; Spriewald, S.; Kühn, F.; Ilmer, M.; Lee, S.M.L.; Niess, H.; Bazhin, A.V.; Andrassy, J.; et al. Microbiome analysis from paired mucosal and fecal samples of a colorectal cancer biobank. Cancers 2020, 12, 3702. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Sehn, J.K.; Abel, H.J.; Watson, M.A.; Pfeifer, J.D.; Duncavage, E.J. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J. Mol. Diagn. 2013, 15, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Fuks, G.; Elgart, M.; Amir, A.; Zeisel, A.; Turnbaugh, P.J.; Soen, Y.; Shental, N. Combining 16S rRNA gene variable regions enables high-resolution microbial community profiling. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Shi, Q.; Grodner, B.; Lenz, J.S.; Zipfel, W.; Brito, I.L.; de Vlaminck, I. Highly multiplexed spatial mapping of microbial communities. Nature 2020, 588, 676–681. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Data | FFPE | FF | NTC |
|---|---|---|---|
| # Raw reads | 916,957 | 417,435 | 67,621 |
| Mean raw reads | 83,359 | 46,381 | |
| # Filtered reads | 492,972 | 359,237 | 57,201 |
| Mean filtered reads | 44,815 | 39,915 | |
| # Final high-quality reads | 181,046 | 142,325 | 24,537 |
| Mean final high-quality reads | 16,458 | 15,814 | |
| % Chimeric/Raw reads | 79% | 67% | 64% |
| # Number ASVs | 389 | 493 |
| Bacteroides | Fusobacterium | Propionibacterium | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | RNA-ISH (FFPE) | 16S (FFPE) | 16S (FF) | RNA-ISH (FFPE) | 16S (FFPE) | 16S (FF) | RNA-ISH (FFPE) | 16S (FFPE) | 16S (FF) |
| 119 | − | + | + | + | − | − | + | + | + |
| 121 | − | + | + | − | − | − | + | + | + |
| 123 | + | + | + | + | + | + | + | + | + |
| 124 | − | − | + | + | − | + | + | + | + |
| 129 | + | + | + | + | − | + | + | + | + |
| 135 | − | + | + | − | − | − | + | + | + |
| 136 | + | + | + | + | − | + | + | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgognone, A.; Serna, G.; Noguera-Julian, M.; Alonso, L.; Parera, M.; Català-Moll, F.; Sanchez, L.; Fasani, R.; Paredes, R.; Nuciforo, P. Performance of 16S Metagenomic Profiling in Formalin-Fixed Paraffin-Embedded versus Fresh-Frozen Colorectal Cancer Tissues. Cancers 2021, 13, 5421. https://doi.org/10.3390/cancers13215421
Borgognone A, Serna G, Noguera-Julian M, Alonso L, Parera M, Català-Moll F, Sanchez L, Fasani R, Paredes R, Nuciforo P. Performance of 16S Metagenomic Profiling in Formalin-Fixed Paraffin-Embedded versus Fresh-Frozen Colorectal Cancer Tissues. Cancers. 2021; 13(21):5421. https://doi.org/10.3390/cancers13215421
Chicago/Turabian StyleBorgognone, Alessandra, Garazi Serna, Marc Noguera-Julian, Lidia Alonso, Mariona Parera, Francesc Català-Moll, Lidia Sanchez, Roberta Fasani, Roger Paredes, and Paolo Nuciforo. 2021. "Performance of 16S Metagenomic Profiling in Formalin-Fixed Paraffin-Embedded versus Fresh-Frozen Colorectal Cancer Tissues" Cancers 13, no. 21: 5421. https://doi.org/10.3390/cancers13215421