
Molecular Signature Expands the Landscape of Driver Negative Thyroid Cancers

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
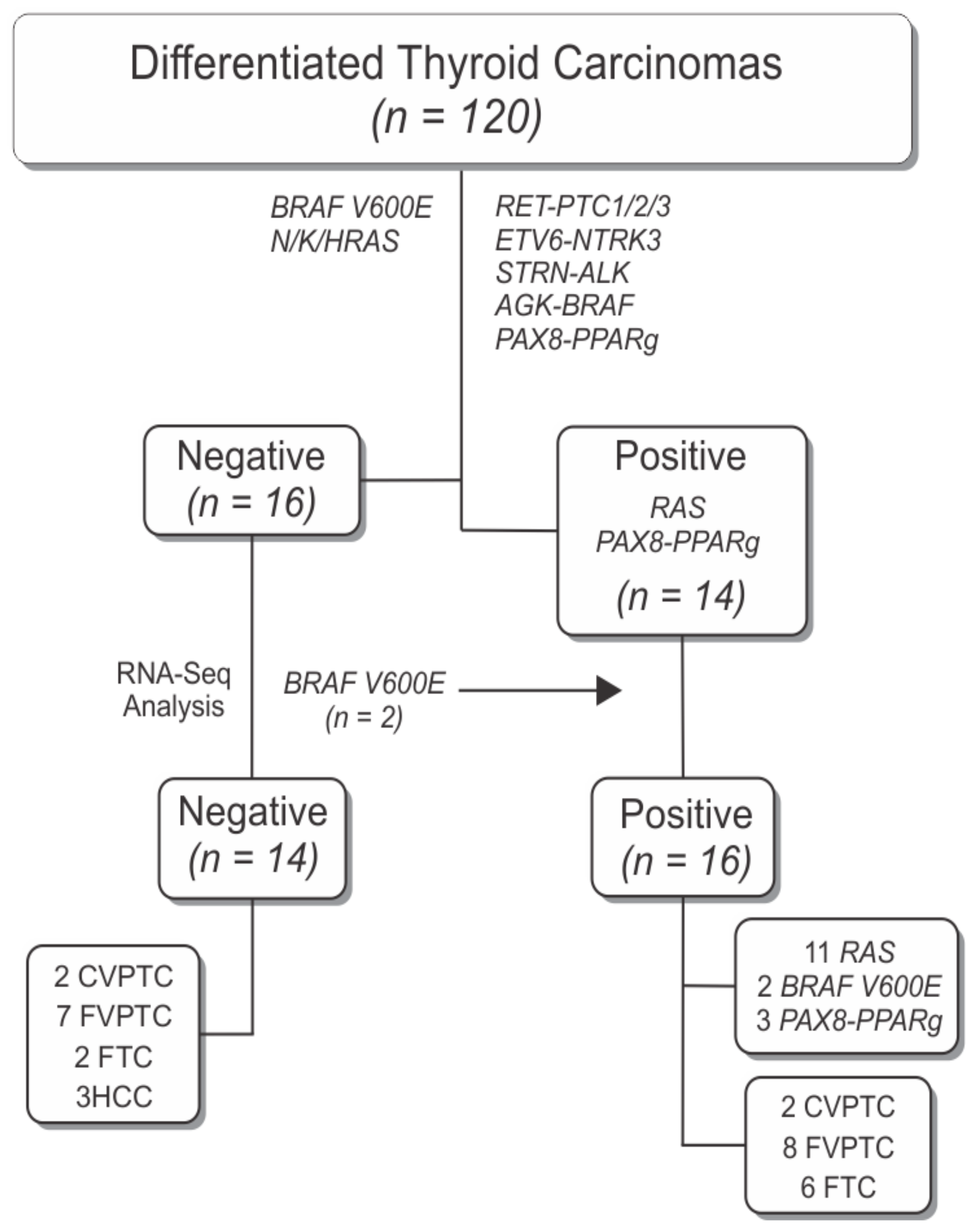
2.1. Patient Cohort
2.2. RNA Isolation and Library Preparation
2.3. Variant Calling and Gene Expression Analysis
2.4. Fusion Gene Analysis
2.5. Confirmation of Selected Variants by Sanger Sequencing
2.6. Sample Cluster Analysis
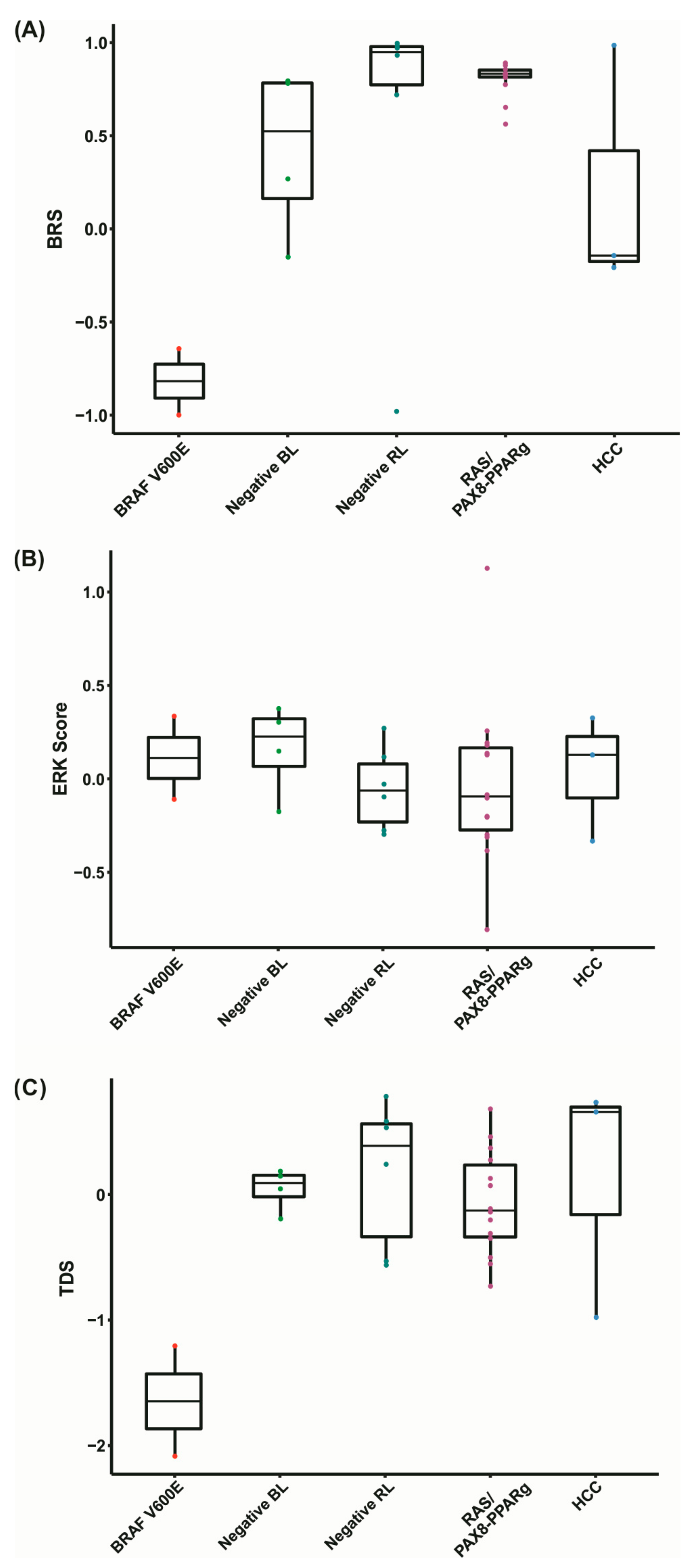
2.7. BRAF V600E-RAS Score (BRS)
2.8. Thyroid Differentiation Score (TDS) and ERK Score
2.9. TMB Calculation
2.10. Differential Gene Expression Analysis
2.11. Differential Expression Analysis on PTC from the TCGA Cohort
2.12. Pathway Enrichment Analysis
2.13. Evaluation of Tumor-Infiltrating Immune Cells
2.14. Statistics
3. Results
3.1. RNA-Sequencing Framework
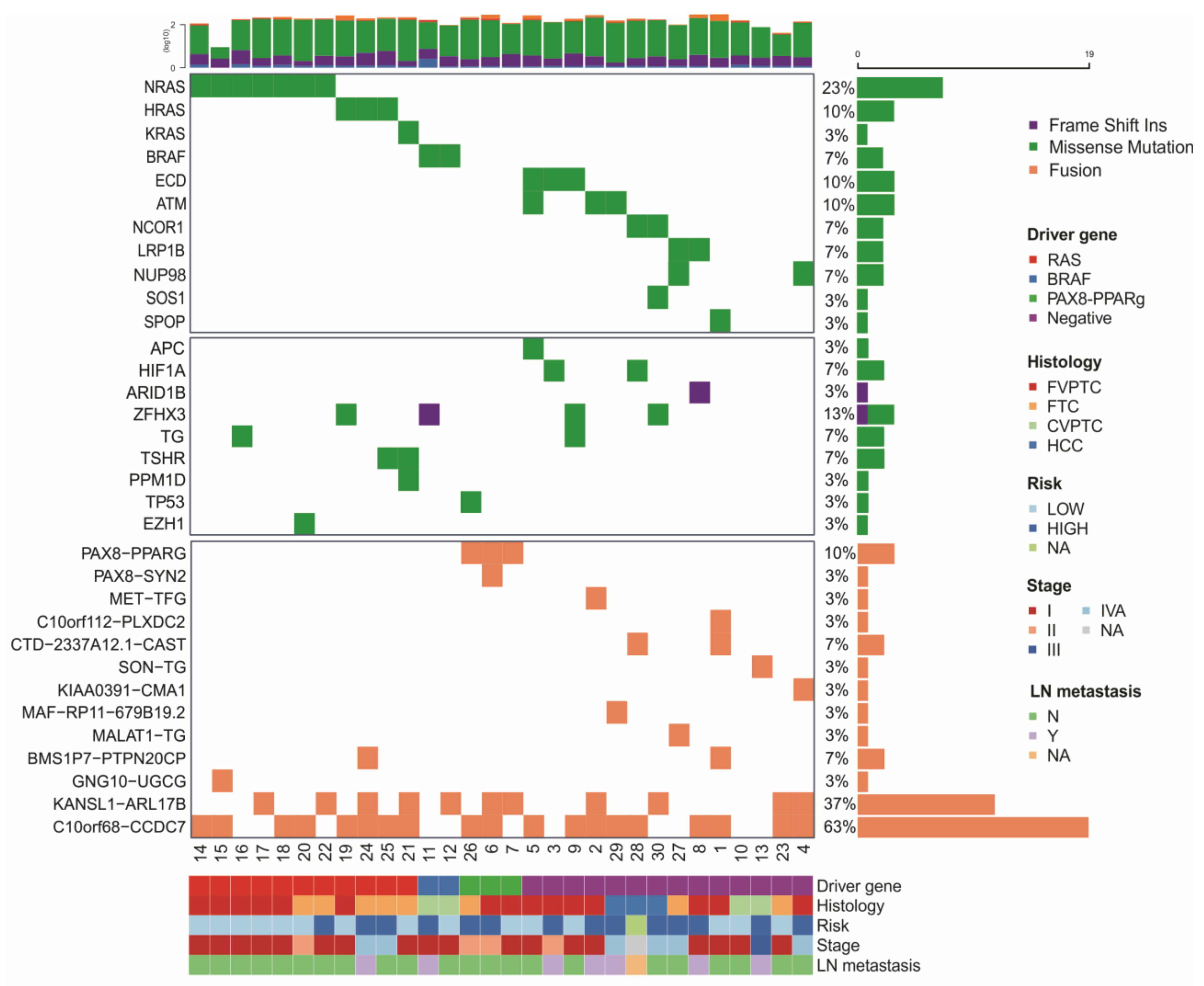
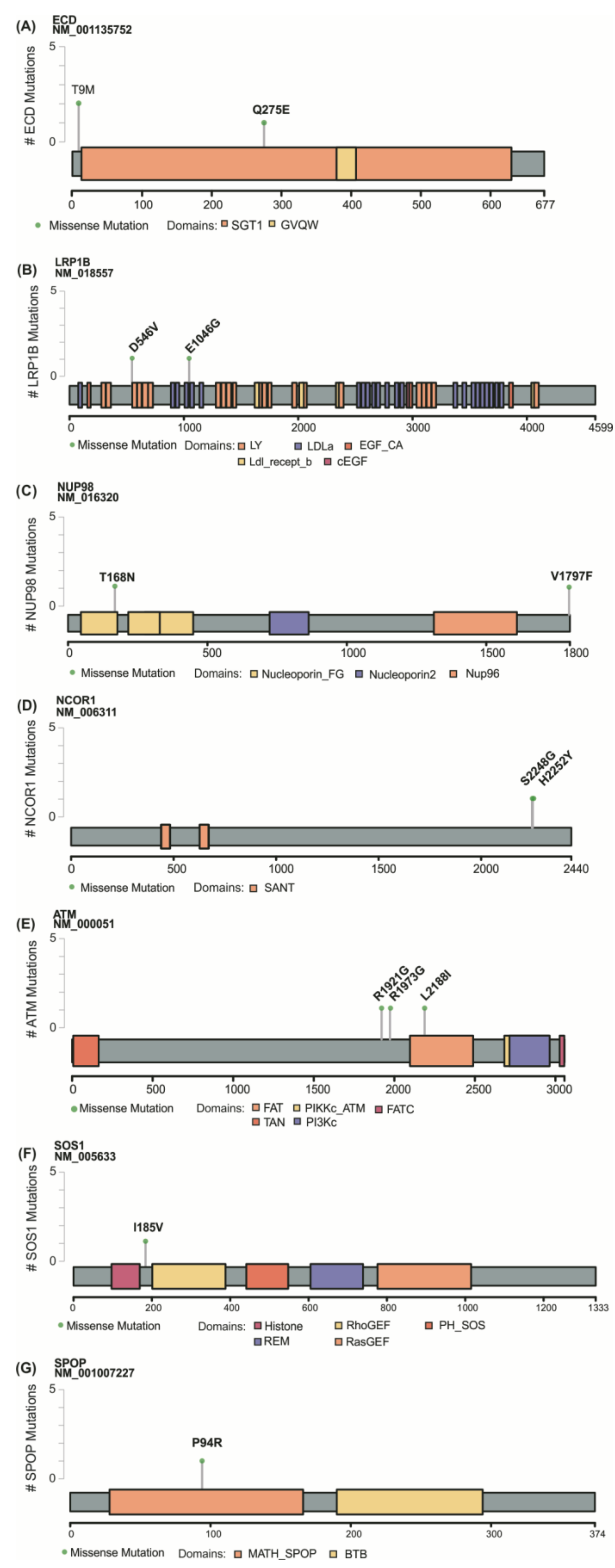
3.2. Identification of Novel Mutations in Thyroid Cancer
3.3. Gene Fusions in Thyroid Cancer
3.4. Technical and Experimental Validation by Sanger
3.5. In Silico Analysis
3.6. Negative Tumors Have Distinctive Expression Profiles
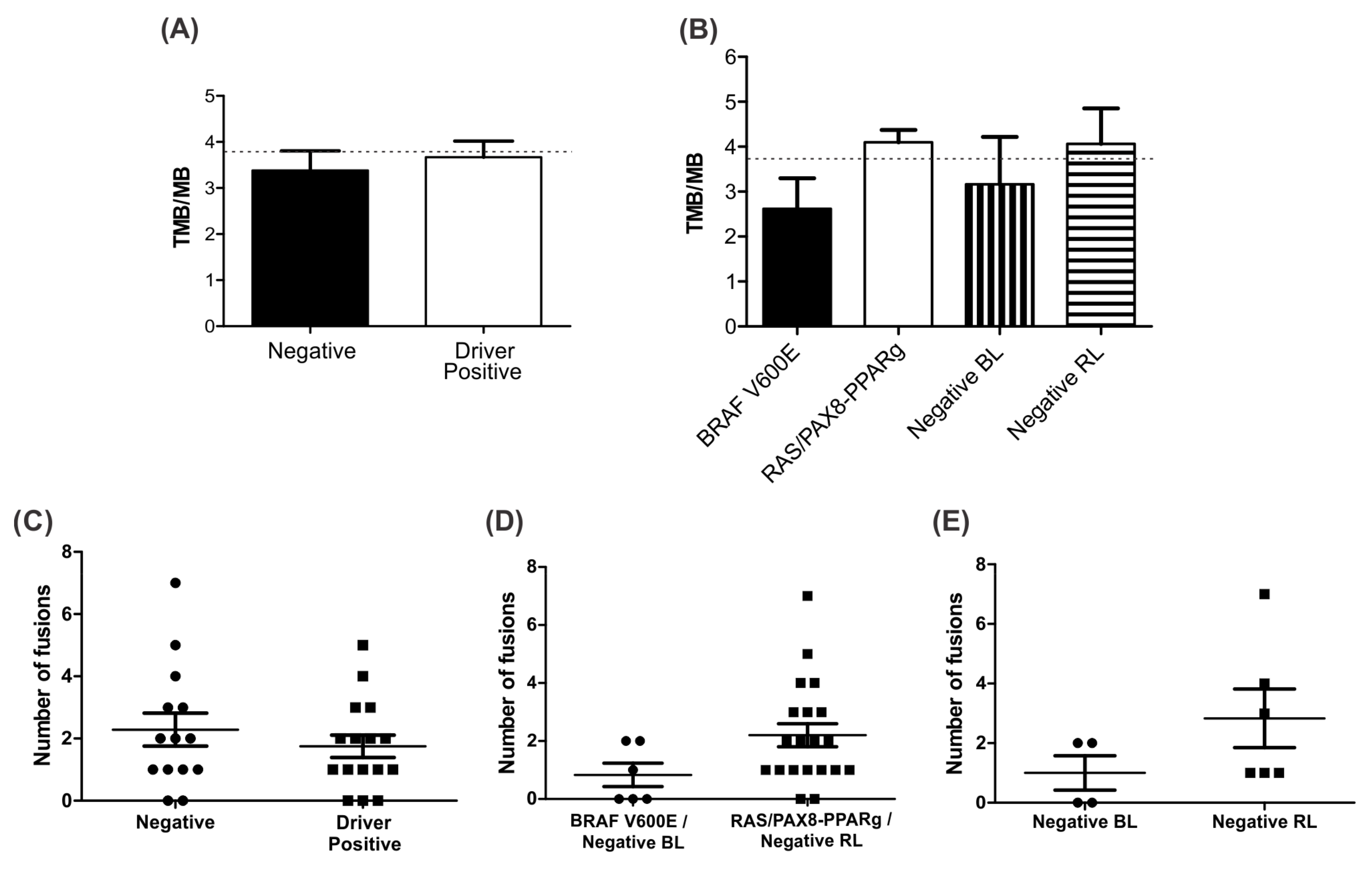
3.7. Negative BL Tumors Have Lower TMB Compared to Negative RL
3.8. Identification of Differentially Expressed Genes
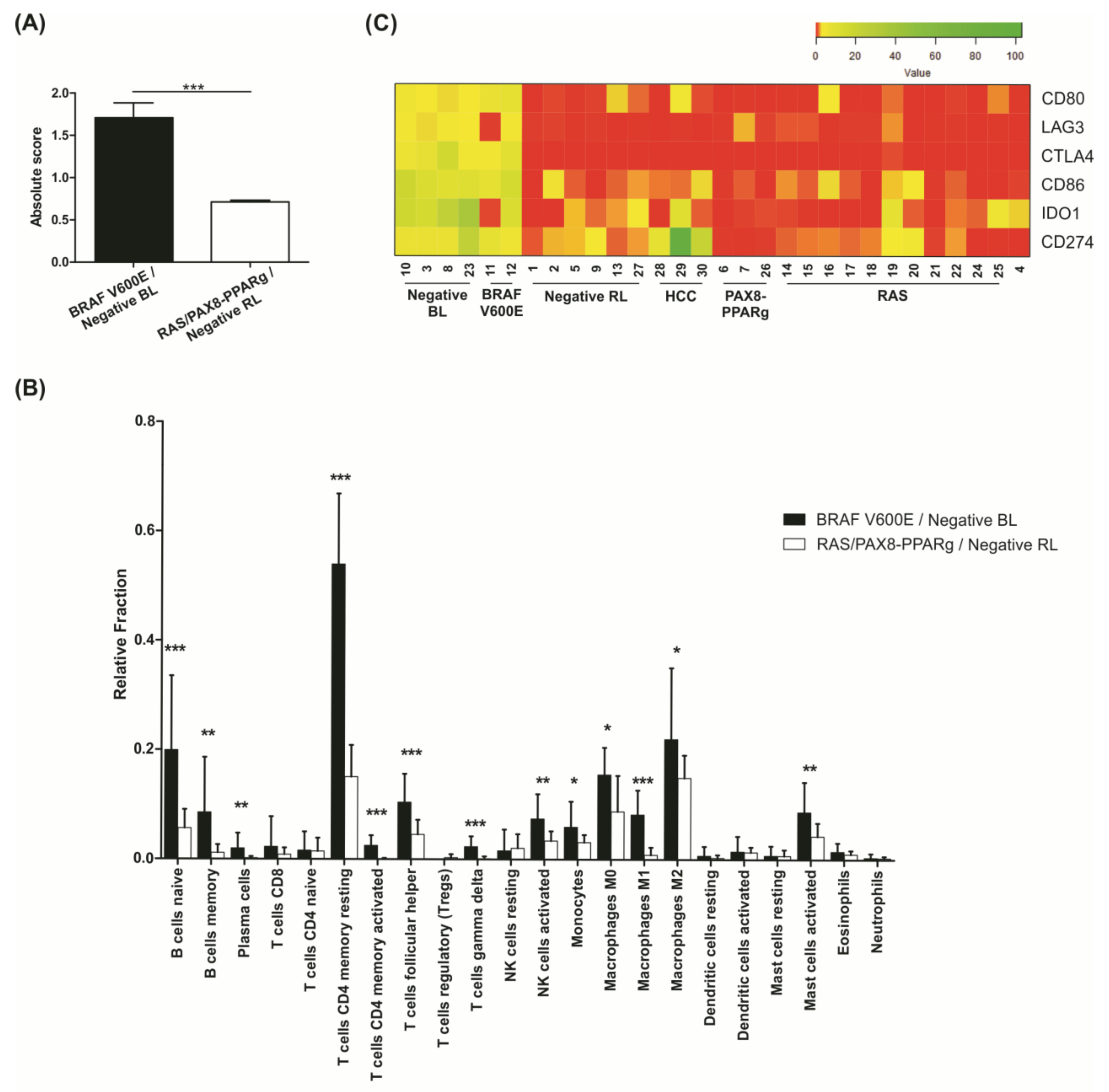
3.9. Negative BL Tumors Have a High Expression of Immune System Components
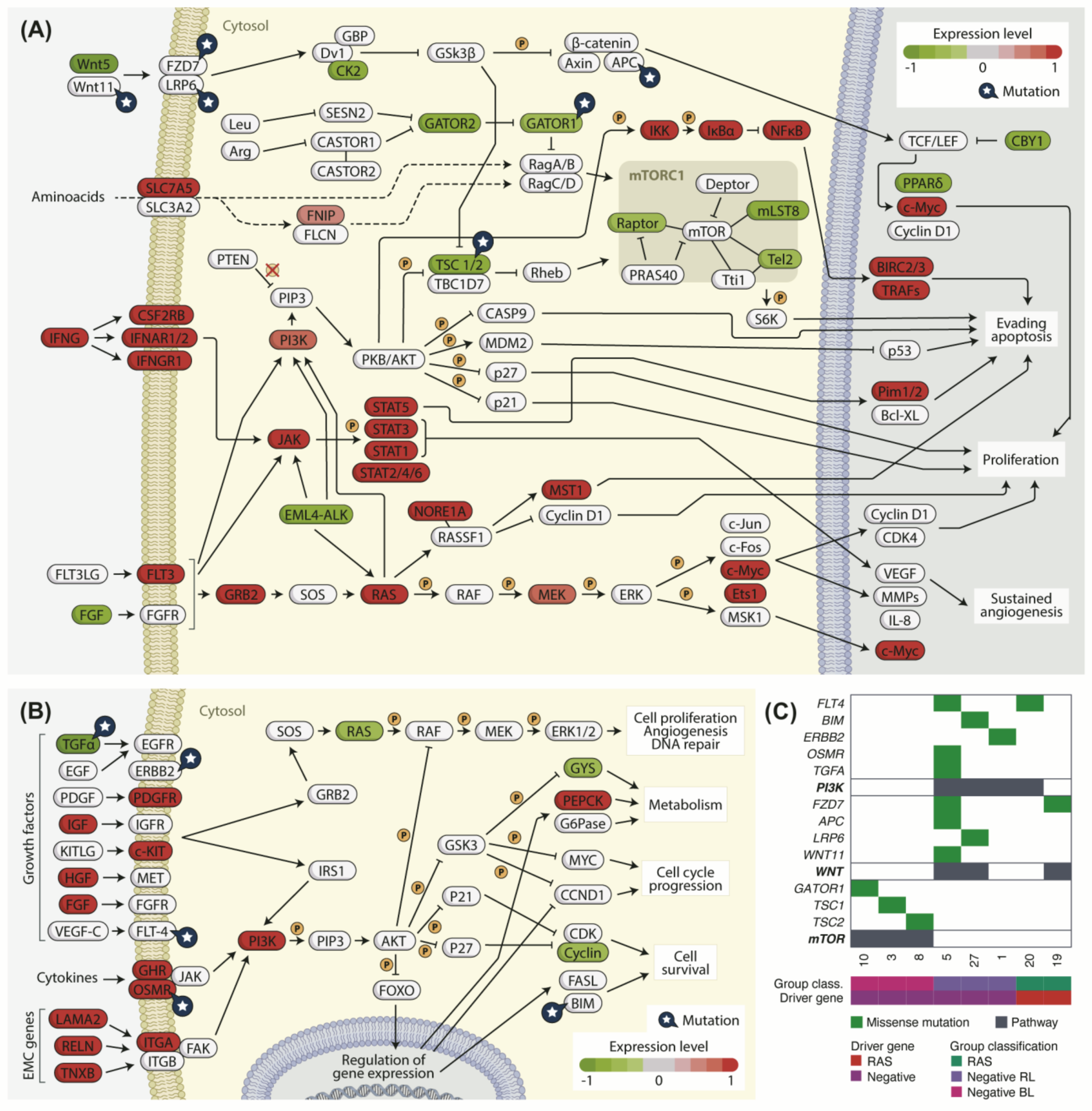
3.10. Cancer-Related Pathways Are Differently Regulated in Negative BL and RL Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kitahara, C.M.; Sosa, J.A. Understanding the ever-changing incidence of thyroid cancer. Nat. Rev. Endocrinol. 2020, 16, 617–618. [Google Scholar] [CrossRef]
- Cancer in Brazil—Data from the Population-Based Registries. Available online: https://www.inca.gov.br/en/publications/booklets/cancer-brazil-data-population-based-registries-vol4 (accessed on 18 August 2021).
- Asa, S.L. The Current Histologic Classification of Thyroid Cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 1–22. [Google Scholar] [CrossRef]
- Schmidbauer, B.; Menhart, K.; Hellwig, D.; Grosse, J. Differentiated Thyroid Cancer—Treatment: State of the Art. Int. J. Mol. Sci. 2017, 18, 1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macerola, E.; Poma, A.M.; Vignali, P.; Basolo, A.; Ugolini, C.; Torregrossa, L.; Santini, F.; Basolo, F. Molecular Genetics of Follicular-Derived Thyroid Cancer. Cancers 2021, 13, 1139. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Singh, B.; Tallini, G.; Carlson, D.L.; Katabi, N.; Shaha, A.; Tuttle, R.M.; Ghossein, R.A. Follicular variant of papillary thyroid carcinoma. Cancer 2006, 107, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Amendoeira, I.; Maia, T.; Sobrinho-Simões, M. Non-invasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP): Impact on the reclassification of thyroid nodules. Endocr. Relat. Cancer 2018, 25, R247–R258. [Google Scholar] [CrossRef] [Green Version]
- Basolo, F.; Macerola, E.; Ugolini, C.; Poller, D.N.; Baloch, Z. The Molecular Landscape of Noninvasive Follicular Thyroid Neoplasm With Papillary-like Nuclear Features (NIFTP): A Literature Review. Adv. Anat. Pathol. 2017, 24, 252–258. [Google Scholar] [CrossRef]
- Yoo, S.-K.; Lee, S.; Kim, S.; Jee, H.-G.; Kim, B.-A.; Cho, H.; Song, Y.S.; Cho, S.W.; Won, J.-K.; Shin, J.-Y.; et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS Genet. 2016, 12, e1006239. [Google Scholar] [CrossRef]
- Romei, C.; Elisei, R. A Narrative Review of Genetic Alterations in Primary Thyroid Epithelial Cancer. Int. J. Mol. Sci. 2021, 22, 1726. [Google Scholar] [CrossRef]
- Paulsson, J.O.; Rafati, N.; Dilorenzo, S.; Chen, Y.; Haglund, F.; Zedenius, J.; Juhlin, C.C. Whole-Genome Sequencing of Follicular Thyroid Carcinomas Reveal Recurrent Mutations in MicroRNA Processing Subunit DGCR8. J. Clin. Endocrinol. Metab. 2021, dgab471. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn Ii, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS Point Mutations and PAX8-PPAR Rearrangement in Thyroid Tumors: Evidence for Distinct Molecular Pathways in Thyroid Follicular Carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakabashi, C.C.D.; Guimarães, G.S.; Michaluart, P.J.; Ward, L.S.; Cerutti, J.M.; Maciel, R.M.B. The expression of PAX8-PPARgamma rearrangements is not specific to follicular thyroid carcinoma. Clin. Endocrinol. 2004, 61, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, A.U.; de Jesus, A.C.; Cerutti, J.M. ETV6-NTRK3 and STRN-ALK kinase fusions are recurrent events in papillary thyroid cancer of adult population. Eur. J. Endocrinol. 2018, 178, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.U.; Oler, G.; Nozima, B.H.N.; Moysés, R.A.; Cerutti, J.M. BRAF V600E and decreased NIS and TPO expression are associated with aggressiveness of a subgroup of papillary thyroid microcarcinoma. Eur. J. Endocrinol. 2015, 173, 525–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, T.N.R.; Bim, L.V.; Buzatto, V.C.; Galdeno, V.; Asprino, P.F.; Lee, E.A.; Galante, P.A.F.; Cerutti, J.M. Evidence of Cooperation between Hippo Pathway and RAS Mutation in Thyroid Carcinomas. Cancers 2021, 13, 2306. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venselaar, H.; te Beek, T.A.H.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.J.; Beltrán, J.F.; Liang, S.; Fragoza, R.; Rumack, A.; Liang, J.; Wei, X.; Yu, H. Interactome INSIDER: A structural interactome browser for genomic studies. Nat. Methods 2018, 15, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and accurate fusion transcript detection from RNA-Seq. bioRxiv 2017. pre-print. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef] [Green Version]
- De Jesus Paniza, A.C.; Mendes, T.B.; Viana, M.D.B.; Thomaz, D.M.D.; Chiappini, P.B.O.; Colozza-Gama, G.A.; Lindsey, S.C.; de Carvalho, M.B.; Alves, V.A.F.; Curioni, O.; et al. Revised criteria for diagnosis of NIFTP reveals a better correlation with tumor biological behavior. Endocr. Connect. 2019, 8, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Brock, G.; Pihur, V.; Datta, S.; Datta, S. clValid: An R Package for Cluster Validation. J. Stat. Softw. 2008, 25, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. Available online: https://rpkgs.datanovia.com/factoextra/index.html (accessed on 1 October 2018).
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Kim, J.H.; Gurumurthy, C.B.; Naramura, M.; Zhang, Y.; Dudley, A.T.; Doglio, L.; Band, H.; Band, V. Role of Mammalian Ecdysoneless in Cell Cycle Regulation. J. Biol. Chem. 2009, 284, 26402–26410. [Google Scholar] [CrossRef] [Green Version]
- Bele, A.; Mirza, S.; Zhang, Y.; Ahmad Mir, R.; Lin, S.; Kim, J.H.; Gurumurthy, C.B.; West, W.; Qiu, F.; Band, H.; et al. The cell cycle regulator ecdysoneless cooperates with H-Ras to promote oncogenic transformation of human mammary epithelial cells. Cell Cycle 2015, 14, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Mirza, S.; Alshareeda, A.; Zhang, Y.; Gurumurthy, C.B.; Bele, A.; Kim, J.H.; Mohibi, S.; Goswami, M.; Lele, S.M.; et al. Overexpression of a novel cell cycle regulator ecdysoneless in breast cancer: A marker of poor prognosis in HER2/neu-overexpressing breast cancer patients. Breast Cancer Res. Treat. 2012, 134, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Rachagani, S.; Chakraborty, S.; Singh, P.K.; Zhao, X.; Gurumurthy, C.B.; Anderson, J.M.; Lele, S.; Hollingsworth, M.A.; Band, V.; et al. Overexpression of Ecdysoneless in Pancreatic Cancer and Its Role in Oncogenesis by Regulating Glycolysis. Clin. Cancer Res. 2012, 18, 6188–6198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleem, I.; Mirza, S.; Sarkar, A.; Raza, M.; Mohapatra, B.; Mushtaq, I.; Kim, J.H.; Mishra, N.K.; Alsaleem, M.A.; Rakha, E.A.; et al. The Mammalian Ecdysoneless Protein Interacts with RNA Helicase DDX39A To Regulate Nuclear mRNA Export. Mol. Cell. Biol. 2021, 41, e0010321. [Google Scholar] [CrossRef]
- Singer, S.; Zhao, R.; Barsotti, A.M.; Ouwehand, A.; Fazollahi, M.; Coutavas, E.; Breuhahn, K.; Neumann, O.; Longerich, T.; Pusterla, T.; et al. Nuclear Pore Component Nup98 Is a Potential Tumor Suppressor and Regulates Posttranscriptional Expression of Select p53 Target Genes. Mol. Cell 2012, 48, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michmerhuizen, N.L.; Klco, J.M.; Mullighan, C.G. Mechanistic Insights and Potential Therapeutic Approaches for NUP98-Rearranged Hematologic Malignancies. Blood 2020, 136, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [Green Version]
- Prazeres, H.; Torres, J.; Rodrigues, F.; Pinto, M.; Pastoriza, M.C.; Gomes, D.; Cameselle-Teijeiro, J.; Vidal, A.; Martins, T.C.; Sobrinho-Simões, M.; et al. Chromosomal, epigenetic and microRNA-mediated inactivation of LRP1B, a modulator of the extracellular environment of thyroid cancer cells. Oncogene 2011, 30, 1302–1317. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.C.; Tucker, M.D.; Sedhom, R.; Schwartz, E.B.; Zhu, J.; Kao, C.; Labriola, M.K.; Gupta, R.T.; Marin, D.; Wu, Y.; et al. LRP1B mutations are associated with favorable outcomes to immune checkpoint inhibitors across multiple cancer types. J. Immunother. Cancer 2021, 9, e001792. [Google Scholar] [CrossRef]
- Chen, H.; Chong, W.; Wu, Q.; Yao, Y.; Mao, M.; Wang, X. Association of LRP1B Mutation With Tumor Mutation Burden and Outcomes in Melanoma and Non-small Cell Lung Cancer Patients Treated With Immune Check-Point Blockades. Front. Immunol. 2019, 10, 1113. [Google Scholar] [CrossRef] [Green Version]
- Tabouret, E.; Labussière, M.; Alentorn, A.; Schmitt, Y.; Marie, Y.; Sanson, M. LRP1B deletion is associated with poor outcome for glioblastoma patients. J. Neurol. Sci. 2015, 358, 440–443. [Google Scholar] [CrossRef]
- Lima, T.I.; Guimarães, D.S.; Oliveira, A.G.; Araujo, H.; Sponton, C.H.G.; Souza-Pinto, N.C.; Saito, Â.; Figueira, A.C.M.; Palameta, S.; Bajgelman, M.C.; et al. Opposing action of NCoR1 and PGC-1α in mitochondrial redox homeostasis. Free Radic. Biol. Med. 2019, 143, 203–208. [Google Scholar] [CrossRef]
- Liu, F.; Hou, W.; Liang, J.; Zhu, L.; Luo, C. LRP1B mutation: A novel independent prognostic factor and a predictive tumor mutation burden in hepatocellular carcinoma. J. Cancer 2021, 12, 4039–4048. [Google Scholar] [CrossRef]
- Hong, J.Y.; Cho, H.J.; Kim, S.T.; Park, Y.S.; Shin, S.H.; Han, I.W.; Lee, J.; Heo, J.S.; Park, J.O. Comprehensive molecular profiling to predict clinical outcomes in pancreatic cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211038478. [Google Scholar] [CrossRef]
- Shimizu, H.; Astapova, I.; Ye, F.; Bilban, M.; Cohen, R.N.; Hollenberg, A.N. NCoR1 and SMRT Play Unique Roles in Thyroid Hormone Action In Vivo. Mol. Cell. Biol. 2015, 35, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Furuya, F.; Guigon, C.J.; Zhao, L.; Lu, C.; Hanover, J.A.; Cheng, S. Nuclear receptor corepressor is a novel regulator of phosphatidylinositol 3-kinase signaling. Mol. Cell. Biol. 2007, 27, 6116–6126. [Google Scholar] [CrossRef] [Green Version]
- Fozzatti, L.; Park, J.W.; Zhao, L.; Willingham, M.C.; Cheng, S.-Y. Oncogenic Actions of the Nuclear Receptor Corepressor (NCOR1) in a Mouse Model of Thyroid Cancer. PLoS ONE 2013, 8, e67954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Jean, S.; De Castro, A.C.; Lecours, M.; Jones, C.; Rivard, N.; Rodier, F.; Perreault, N.; Boudreau, F. NCOR1 Sustains Colorectal Cancer Cell Growth and Protects against Cellular Senescence. Cancers 2021, 13, 4414. [Google Scholar] [CrossRef]
- Ritter, M.J.; Amano, I.; Imai, N.; Soares De Oliveira, L.; Vella, K.R.; Hollenberg, A.N. Nuclear Receptor CoRepressors, NCOR1 and SMRT, are required for maintaining systemic metabolic homeostasis. Mol. Metab. 2021, 53, 101315. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, R.-C.; Zhang, Q.; Guo, L.-L. ATM mutations as an independent prognostic factor and potential biomarker for immune checkpoint therapy in endometrial cancer. Pathol. Res. Pract. 2020, 216, 153032. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, Y.; Pan, C.; Yan, L.; Wang, Z.; Zhu, X. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol. Cancer 2020, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.J.; Otero, J.H.; Scott, D.C.; Szulc, E.; Martin, E.W.; Sabri, N.; Granata, D.; Marzahn, M.R.; Lindorff-Larsen, K.; Salvatella, X.; et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell 2018, 72, 19–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmud, I.; Liao, D. DAXX in cancer: Phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 2019, 47, 7734–7752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Zhou, X.; Huang, F.; Wang, W.; Qi, Y.; Xu, H.; Shu, Y.; Shen, L.; Fei, X.; Xie, J.; et al. The genetic landscape of benign thyroid nodules revealed by whole exome and transcriptome sequencing. Nat. Commun. 2017, 8, 15533. [Google Scholar] [CrossRef]
- Jung, S.-H.; Kim, M.S.; Jung, C.K.; Park, H.-C.; Kim, S.Y.; Liu, J.; Bae, J.-S.; Lee, S.H.; Kim, T.-M.; Lee, S.H.; et al. Mutational burdens and evolutionary ages of thyroid follicular adenoma are comparable to those of follicular carcinoma. Oncotarget 2016, 7, 69638–69648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon, G.S.T.; Chang, K.T.E.; Kuick, C.H.; Petersson, F. A case of nasal low-grade non-intestinal-type adenocarcinoma with aberrant CDX2 expression and a novel SYN2-PPARG gene fusion in a 13-year-old girl. Virchows Arch. 2019, 474, 619–623. [Google Scholar] [CrossRef]
- Viaene, A.N.; Zhang, B.; Martinez-Lage, M.; Xiang, C.; Tosi, U.; Thawani, J.P.; Gungor, B.; Zhu, Y.; Roccograndi, L.; Zhang, L.; et al. Transcriptome signatures associated with meningioma progression. Acta Neuropathol. Commun. 2019, 7, 67. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Moon, H.-G.; Han, W.; Noh, D.-Y. Perspective Insight into Future Potential Fusion Gene Transcript Biomarker Candidates in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 502. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.X.; Yang, X.; Ning, S.; Wang, L.; Wang, K.; Zhang, Y.; Yuan, F.; Li, F.; Zhuo, D.D.; Tang, L.; et al. Identification of KANSARL as the first cancer predisposition fusion gene specific to the population of European ancestry origin. Oncotarget 2017, 8, 50594–50607. [Google Scholar] [CrossRef] [Green Version]
- Tuna, M.; Amos, C.I.; Mills, G.B. Molecular mechanisms and pathobiology of oncogenic fusion transcripts in epithelial tumors. Oncotarget 2019, 10, 2095–2111. [Google Scholar] [CrossRef] [Green Version]
- Faustino, A.; Couto, J.P.; Pópulo, H.; Rocha, A.S.; Pardal, F.; Cameselle-Teijeiro, J.M.; Lopes, J.M.; Sobrinho-Simões, M.; Soares, P. mTOR Pathway Overactivation in BRAF Mutated Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, E1139–E1149. [Google Scholar] [CrossRef]
- Zhang, W.; Yin, L.; Song, G.; Han, X.; Yin, Z.; Luo, D. LKB1 loss cooperating with BRAF V600E promotes melanoma cell invasion and migration by up-regulation MMP-2 via PI3K/Akt/mTOR pathway. Oncotarget 2017, 8, 113847–113857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; Correa, A.J.; LoPresti, J.S.; Epstein, A.L. BRAF V600E in Papillary Thyroid Carcinoma Is Associated with Increased Programmed Death Ligand 1 Expression and Suppressive Immune Cell Infiltration. Thyroid 2014, 24, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Li, X.; He, Y.; Wu, S.; Wang, S.; Sun, J.; He, Y.; Lun, Y.; Zhang, J. Immune Cell Confrontation in the Papillary Thyroid Carcinoma Microenvironment. Front. Endocrinol. 2020, 11, 570604. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takács, T.; Kudlik, G.; Kurilla, A.; Szeder, B.; Buday, L.; Vas, V. The effects of mutant Ras proteins on the cell signalome. Cancer Metastasis Rev. 2020, 39, 1051–1065. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tumor Type | Mutational Status | Expression Classifier Group | Gender | Age at Surgery | Tumor Size | Multifocality | ETE | Vascular Invasion | Lymph Node Metastasis | Stage | Risk |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FVPTC | Negative | Negative RL | F | 45 | 3.0 | N | N | N | N | I | Low |
| 2 | FVPTC | Negative | Negative RL | F | 31 | 3.2 | N | N | Y | Y | I | High |
| 3 | FVPTC | Negative | Negative BL | F | 76 | 1.0 | Y | N | N | Y | II | High |
| 4 | FVPTC | Negative | - | F | 68 | 8.0 | Y | Y | Y | N | IVA | High |
| 5 | FVPTC | Negative | Negative RL | F | 52 | 0.9 | N | N | N | N | I | Low |
| 6 | FVPTC | Positive | RAS/PAX8-PPARg | M | 68 | 7.5 | N | N | N | N | II | High |
| 7 | FVPTC | Positive | RAS/PAX8-PPARg | F | 53 | 4.0 | Y | N | N | N | I | Low |
| 8 | FVPTC | Negative | Negative BL | M | 40 | 8.5 | Y | N | Y | Y | I | High |
| 9 | FVPTC | Negative | Negative RL | F | 73 | 1.2 | Y | N | N | N | I | Low |
| 10 | CVPTC | Negative | Negative BL | F | 59 | 2.5 | Y | N | N | N | I | Low |
| 11 | CVPTC | Positive | BRAF V600E | F | 46 | 1.7 | N | Y | Y | Y | I | High |
| 12 | CVPTC | Positive | BRAF V600E | F | 41 | 1.3 | N | N | N | N | I | Low |
| 13 | CVPTC | Negative | Negative RL | F | 56 | 3.0 | Y | Y | N | Y | III | High |
| 14 | FVPTC | Positive | RAS/PAX8-PPARg | M | 32 | 5.0 | N | Y | N | N | I | High |
| 15 | FVPTC | Positive | RAS/PAX8-PPARg | M | 40 | 4.0 | N | N | N | N | I | Low |
| 16 | FVPTC | Positive | RAS/PAX8-PPARg | F | 28 | 5.0 | Y | N | N | N | I | Low |
| 17 | FVPTC | Positive | RAS/PAX8-PPARg | F | 55 | 1.7 | Y | N | N | N | I | Low |
| 18 | FVPTC | Positive | RAS/PAX8-PPARg | F | 36 | 3.0 | N | N | N | N | I | Low |
| 19 | FVPTC | Positive | RAS/PAX8-PPARg | F | 45 | 1.1 | N | N | N | N | I | Low |
| 20 | FTC | Positive | RAS/PAX8-PPARg | F | 45 | 3.5 | Y | Y | N | N | II | High |
| 21 | FTC | Positive | RAS/PAX8-PPARg | F | 48 | 3.2 | N | N | Y | N | I | High |
| 22 | FTC | Positive | RAS/PAX8-PPARg | F | 48 | 1.6 | N | Y | Y | N | I | High |
| 23 | FTC | Negative | Negative BL | F | 35 | 4.5 | N | N | N | N | I | Low |
| 24 | FTC | Positive | RAS/PAX8-PPARg | F | 76 | 7.5 | N | Y | Y | Y | IVA | High |
| 25 | FTC | Positive | RAS/PAX8-PPARg | M | 70 | 10.3 | N | Y | Y | Y | IVA | High |
| 26 | FTC | Positive | RAS/PAX8-PPARg | F | 70 | 10.0 | N | N | N | N | II | High |
| 27 | FTC | Negative | Negative RL | F | 60 | 2.5 | Y | N | Y | N | IVA | High |
| 28 | HCC | Negative | Negative (HCC) | M | NA | NA | NA | NA | NA | NA | NA | NA |
| 29 | HCC | Negative | Negative (HCC) | F | 70 | 5.0 | N | N | Y | Y | IVA | High |
| 30 | HCC | Negative | Negative (HCC) | F | 63 | 6.0 | N | N | Y | N | IVA | High |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bim, L.V.; Carneiro, T.N.R.; Buzatto, V.C.; Colozza-Gama, G.A.; Koyama, F.C.; Thomaz, D.M.D.; de Jesus Paniza, A.C.; Lee, E.A.; Galante, P.A.F.; Cerutti, J.M. Molecular Signature Expands the Landscape of Driver Negative Thyroid Cancers. Cancers 2021, 13, 5184. https://doi.org/10.3390/cancers13205184
Bim LV, Carneiro TNR, Buzatto VC, Colozza-Gama GA, Koyama FC, Thomaz DMD, de Jesus Paniza AC, Lee EA, Galante PAF, Cerutti JM. Molecular Signature Expands the Landscape of Driver Negative Thyroid Cancers. Cancers. 2021; 13(20):5184. https://doi.org/10.3390/cancers13205184
Chicago/Turabian StyleBim, Larissa Valdemarin, Thaise Nayane Ribeiro Carneiro, Vanessa Candiotti Buzatto, Gabriel Avelar Colozza-Gama, Fernanda C. Koyama, Debora Mota Dias Thomaz, Ana Carolina de Jesus Paniza, Eunjung Alice Lee, Pedro Alexandre Favoretto Galante, and Janete Maria Cerutti. 2021. "Molecular Signature Expands the Landscape of Driver Negative Thyroid Cancers" Cancers 13, no. 20: 5184. https://doi.org/10.3390/cancers13205184