
Janus Kinase Signaling: Oncogenic Criminal of Lymphoid Cancers

1
College of Pharmaceutical Sciences, Southwest University, Chongqing 400715, China
2
Department of Haematology-Oncology, National University Cancer Institute of Singapore, Singapore 119074, Singapore
3
Cancer Science Institute of Singapore, National University of Singapore, Singapore 117599, Singapore
4
Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 119228, Singapore
*
Authors to whom correspondence should be addressed.
Cancers 2021, 13(20), 5147; https://doi.org/10.3390/cancers13205147
Submission received: 5 September 2021
/
Revised: 8 October 2021
/
Accepted: 11 October 2021
/
Published: 14 October 2021
Abstract
:Simple Summary
Janus kinases (JAKs) are transmembrane receptors that pass signals from extracellular ligands to downstream. Increasing evidence has suggested that JAK family aberrations promote lymphoid cancer pathogenesis and progression through mediating gene expression via the JAK/STAT pathway or noncanonical JAK signaling. We are here to review how canonical JAK/STAT and noncanonical JAK signalings are represented and deregulated in lymphoid malignancies and how to target JAK for therapeutic purposes.
Abstract
The Janus kinase (JAK) family are known to respond to extracellular cytokine stimuli and to phosphorylate and activate signal transducers and activators of transcription (STAT), thereby modulating gene expression profiles. Recent studies have highlighted JAK abnormality in inducing over-activation of the JAK/STAT pathway, and that the cytoplasmic JAK tyrosine kinases may also have a nuclear role. A couple of anti-JAK therapeutics have been developed, which effectively harness lymphoid cancer cells. Here we discuss mutations and fusions leading to JAK deregulations, how upstream nodes drive JAK expression, how classical JAK/STAT pathways are represented in lymphoid malignancies and the noncanonical and nuclear role of JAKs. We also summarize JAK inhibition therapeutics applied alone or synergized with other drugs in treating lymphoid malignancies.
1. Introduction
Lymphoid cancers are lethal malignancies, which include lymphomas, myeloma and lymphoid leukemias. The Janus kinase (JAK) family comprises four members: JAK1, JAK2, JAK3 and TYK2. Structurally, all JAKs contain a FERM domain, a SH2 domain, a pseudokinase domain and a catalytic kinase domain. The JAK tyrosine kinases are mainly located in the cytoplasm and transmit signals from cytokines and their γ-chain receptors to signal transducers and activators of transcription (STAT), and the phosphorylated, dimerized and activated STAT then binds to chromosome and trans-regulates gene expression (Figure 1). There are seven members in the mammalian STAT family: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6 [1]. The JAK/STAT pathway is evolutionarily conserved and directly affects developmental hematopoiesis and oncogenic proliferation and migration. JAK deregulates, either by mutations and translocations of itself or by upstream aberrance of other nodes, augmented disease pathogenesis, promoted tumor cell survival, and out-of-control cell cycling via classical cytoplasmic JAK signaling or the noncanonical nuclear JAK pathway, both of which rewrite the epigenome and prompt the expression of oncogenes.
In this article, we review activating mutations and fusions of JAKs that enhance JAK/STAT phosphorylation and lead to overexpression of STAT target oncogenes in a couple of lymphoid cancerous contexts, canonical JAK/STAT signaling and the nuclear role of JAKs that non-canonically bind to RNA polymerase II and phosphorylate histones [2] or chromatin modifiers [3,4]. We also summarize the effectiveness of JAK-targeting monotherapy and combinational therapy in curing lymphoid cancers, which induce programmed cell death and cell cycle arrest [5].
2. JAK Abnormalities
2.1. Abnormally Activating JAK Mutations
JAK1 mutations have been found in adult precursor T acute lymphoblastic leukemia (ATLL, 18%) [6], T-cell prolymphocytic leukemia (T-PLL, V658F, responding well to JAK1-inhibition therapy) [7], cutaneous T-cell lymphoma (CTCL) [8], anaplastic large cell lymphoma (ALCL, 18%) [9,10], plasmablastic lymphoma (PBL,14%) [11], peripheral T-cell lymphoma (PTCL, G1097D) [12] and enteropathy-associated T cell lymphoma (EATL) [13]. JAK2 mutations have been associated with poor prognosis in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL) [14]. Three different JAK2 mutations, R683G, H574R and I682T, were identified in T-cell lymphoblastic lymphoma (T-LBL), and two of these mutations constitutively activated JAK2/STAT signaling. In primary T-LBL samples harboring JAK2 mutations, LMO2 expression was also increased [15]. Moreover, TYK2 heterozygous mutations were discovered in two siblings who developed Epstein–Barr virus (EBV)-associated B-cell lymphoma. Additionally, under 35% of TYK2 deficiency, these patients responded normally to type I interferon (IFN), IL-6, IL-10 and IL-12, whereas they responded abnormally to IL-23 [16].
JAK3 mutations have been reported using next generation sequencing in natural killer/T-cell lymphoma (NKTCL) from cohorts in Singapore [17,18,19], Latin America (Mexico, Peru and Argentina) [20], Korea [21,22], Thailand, Japan [23] and France [24], which partly led to the constitutive phosphorylation of JAK3 [18,24], activation of JAK3/STAT signaling [17,20] and interleukin-independent NKTCL cell survival [21]. The allelic ratio of JAK3 mutations ranged from 3% to 35.4% [18,19,20,21,22,24]. The mutation hotspots were mostly in the JAK3 pseudokinase domain and involved exon 13 [18], A572V, A573V [19,24], H583Y, G589D [21] and V722I [24]. In addition, JAK3 mutations were also reported in CTCL (3%) [6,8,25], T-PLL (30%) [26], ATLL (5%) [6,25], epitheliotropic intestinal T-cell lymphoma (EITL, 35%) [27], EATL [13] and ocular adnexal marginal zone lymphomas (OAML) (11%) [28], resulting in the activation of key cell survival pathways, including JAK3/STAT, with some known gain-of-function mutational hotspots.
Furthermore, some studies have well described alterations affecting one to multiple cell fate-related nodes of the JAK/STAT pathway, including Hodgkin–Reed–Sternberg (HRS)-like “cells of NK phenotype” [29], primary cutaneous γδ T cell lymphoma (PCGDTL) [30], EITL [31], post-transplant lymphoproliferative disorder (LPD) [32] and CTCL [32], part of which led to upregulated JAK phosphorylation and activation. All the JAK mutations mentioned above are summarized in Table 1.
2.2. JAK-Associated Gene Translocation
The firstly identified and heavily studied phenomenon of JAK translocation is TEL-JAK2 fusion. This fusion protein was characterized in T-cell acute lymphoblastic leukemia (T-ALL) patients, which constitutively activated JAK2 tyrosine kinase activity, STAT phosphorylation and conferred cytokine-independent T-ALL cell proliferation [33,34]. The chimeric TEL-JAK protein promoted several downstream oncogenic signals, including ERK, SAPK-JNK, P38 [35], PI3K/PKB [36] and SOCS1 [37]. The TEL-JAK2 drove T-cell leukemia development alone [38] and in cooperation with pre-TCR signaling [39] or TEL-ABL fusion protein [40]. This activating TEL-JAK2 translocation was detected in 2 out of 16 T-ALL patient samples studied [15].
Additionally, a three-way t(9;13;16) (p24;q34;p11) chromosome translocation was detected in a cutaneous CD4 positive T-cell lymphoma case, in which JAK2 was fused to a novel gene ATXN2L. This fusion product contained the full ATXN2L protein and the catalytic domain of the JAK2 kinase, leading to constitutive activation of the JAK2/STAT signaling pathway, similar to the TEL-JAK2 chimeric protein [41]. In one case of classical Hodgkin lymphoma (cHL), the t(4;9)(q21;p24) translocation was observed, which resulted in a new oncogenic and enzymatically activated SEC1A-JAK2 fusion protein. Additionally, the fused protein was sensitive to JAK inhibitors [42]. Interestingly, by genetic profiling of breast implant associated anaplastic large cell lymphoma (BIA-ALCL), JAK2 was found to fuse with its downstream node STAT3, and this is also the first reported fusion fact in BIA-ALCL [43]. Utilizing whole-transcriptome sequencing in CD30+ LPD, a fusion involving NPM1 (5q35) and TYK2 (19p13) was observed. The fusion encoded an NPM1-TYK2 chimeric protein containing the oligomerization domain of NPM1 and an intact catalytic domain in TYK2. The NPM1-TYK2 fusions were found in 2 of 47 (4%) primary cases and functionally evoked activation of TYK2 and STAT1/3/5 [44]. A recurrent chimera combining transcription factor NFkB2 and TYK2 was also discovered in WT JAK1/STAT3 ALK(-) ALCL [10]. Moreover, JAK chimeric aberrations were also identified in BCR-ABL1-like pediatric BCP-ALL [14], CTCL [45] and pediatric cHL [46].
3. JAK Signaling
3.1. Upstream Drivers for JAK Activation
This section describes how JAKs are deregulated by kinase/phosphatase, non-cytokine stimulus and trans-modulated by other factors. As members of the class I nonreceptor protein tyrosine phosphatase family, PTPN proteins are ubiquitously expressed with high levels in immune cells [47]. In cHL, splice variants of PTPN1, which missed one or more exon sequence and were catalytically inactive, augmented downstream JAK/STAT signaling [48,49]. As a tumor suppressor capable of inhibiting the JAK/STAT pathway, PTPN2 suppressed T cell proliferation. Therefore, bi-allelically inactivated PTPN2 identified in 2 out of 39 cases of PTCL led to JAK/STAT activation [50]. Similarly, PTPN6 loss-of-function N225K and A550V mutants exhibited reduced tyrosine phosphatase activity and caused the deregulated JAK3/STAT3 pathway in diffused large B cell lymphoma (DLBCL) [51]. Moreover, the PIM serine/threonine kinase aberrant expression and activation appeared in several cancerous contexts, including primary mediastinal large B-cell lymphoma and cHL, promoting cancer cell survival and immune surveillance escape partly via modulating JAK/STAT activity [52,53]. Abnormal suppression of SHP1/2 and SOCS-1 in multiple myeloma (MM) plasma cells significantly correlated with the sustained activation of the JAK/STAT3 pathway [54]. A double kinase fusion ITK-SYK was identified in PTLC, which drove cellular transformation and progression of this malignancy. Additionally, through microarray data analysis, JAK3/STAT5 activation was discovered as a downstream effect of ITK-SYK aberrance, and pharmacological inhibition of JAK3 abrogated STAT5 phosphorylation, suppressed cell survival and induced G1/S phase arrest [5].
Several non-cytokine upstream stimuli have been recounted to directly affect JAK/STAT signaling. By exploiting the IL-10/JAK pathway, the human T-cell leukemia virus type 1 (HTLV-1) viral protein HBZ induced an increased IL-10 level, suppressed host immune response and therefore upgraded HTLV-1 proliferation in infected T leukemia cells [55]. In cHL, lymphotoxin-α was characterized as one of the factors that promotes JAK2/STAT6 activation, as dissected by chromatography coupling with mass spectrometry [56]. In MM cells, hypoxia-dependent erythropoietin (EPO)-receptor was shown to be upstream of the JAK signaling pathway. JAK2 could be phosphorylated by recombinant EPO in kinase assay and EPO exposure intriguingly reduced myeloma cell survivals [57].
Trans-mediation of JAK family proteins was also reported in recent years. In high-grade B-cell lymphoma, BCL6 was characterized as a transcription factor, which directly bound to the JAK2 promoter, as evidenced by ChIP-seq [58]. In DLBCL and follicular lymphoma (FL), the histone methyltransferase KMT2D has been shown as a bona fide tumor suppressor and one of the most frequently mutated genes. KMT2D directly mediated histone H3K4 methylation and thereby perturbs expression of a set of genes, including JAK/STAT [59]. miR-155, associated with poor prognosis, has been implicated in the progression of CTCL. This microRNA simultaneously modulated multiple survival-associated pathways, including JAK/STAT. Cobomarsen, a locked nucleic-acid-modified oligonucleotide inhibitor of miR-155, effectively saved expression of these survival cascades [60]. The JAK signaling pathway could be driven by MALT1 [61], MYD88 [62], HSP90 [63] and SOD [64] via undescribed mechanisms.
3.2. Classical JAK/STAT Pathway
The cytokine/JAK/STAT pathway starts when a cytokine binds to its cognate receptor and induces the dimerization and phosphorylation of the receptor on its intracellular domain. These receptors contain a common γ chain and a unique α chain. Specifically, IL-2 and IL-15 receptors share an additional IL-2/IL-15Rβ subunit [1]. The receptor activation further causes JAKs protein phosphorylation, creating docking sites for STATs phosphorylation and dimerization. The dimerized STAT then transfers to the nucleus and trans-regulates gene expression via binding to DNA consensus sequences [65].
STAT3, firstly identified in 1993 in a biochemical study, has been the most-studied member within the STAT family [65]. The JAK/STAT3 cascade was mutated and aberrantly activated [10,66,67,68] in a number of lymphoid cancers, rendering cytokine-independent activation [69], immunosuppression- and tumor growth-related gene expressions (MCL1, SOX11, CD38, PD-L1, MUC1, MCL1, MYC and GTPase RhoU) [17,70,71,72,73,74,75,76], sustained tumor cell survival [71], prompted cell migration [76], differentiation advantage towards terminally differentiated B-cell lymphoma [77], resistance to cytotoxic and biological agents [74], disease progression [78] and shorter event-free survival [79]. Moreover, other STAT family members, such as STAT1, STAT5 and STAT6 were also mutated, upregulated, phosphorylated and activated in lymphoid disease subsets [69,71,80,81,82,83,84,85], resulting in increased expression of downstream nodes, such as BATF3 and MYC [86]. The JAK/STAT1/5/6 signaling was enriched in disease cohorts [87,88,89,90,91], which drove pathogenesis [89] and neoangiogenesis [85] and was associated with elevated frequencies of lymphoid malignancies [92].
3.3. Newly Identified Nuclear JAK Signaling
In addition to the traditional JAK/STAT signaling cascade, non-STAT phosphorylation and the nuclear role of JAKs have been proposed, which strongly relate to the pathogenesis and progression of lymphomas. In primary mediastinal B cell lymphoma (PMBL) and cHL, JAK2-mediated H3Y41 phosphorylation co-operated with JMJD2C-modulated H3K9 demethylation, thereby silencing the myc oncogene, promoting heterochromatin formation and remodeling epigenome [2] (Figure 2A). The H3Y41 locus may also be phosphorylated by JAK1, thus regulating nearly 3000 proliferation- and survival-associated genes in activated B cell-like diffuse large B cell lymphoma (ABC-DLBCL), including IRF4, MYD88 and MYC [93] (Figure 2A). Nuclear JAK3 has also been observed in CTCL cells, which interacted with the catalytic subunit of RNA polymerase II and phosphorylated histone H3 on its tyrosine residue [94] (Figure 2B). Epigenetic phosphorylation by JAK family members occurs on histone modifiers as well. We have shown that in NKTCL, JAK3 transferred to the nucleus and phosphorylated PRC2 methyltransferase EZH2 at Y244, switching EZH2 from an epigenetic silencer to a transcriptional activator (Figure 2B). The downstream activated genes were related to stemness, invasiveness, DNA replication, cell cycle, oncogenesis and proliferation [3]. Similarly, JAK2 also site-specifically phosphorylated EZH2 at Y641, and rendered EZH2 to avoid β-TRCP-mediated proteosomal degradation [4] (Figure 2B). Apart from JAK-catalyzed phosphorylation, JAK3 and SUZ12 mutations orchestrated to drive T-cell transformation and T-ALL development [95].
4. JAK-Based Targeted Therapeutics
4.1. Monotherapy
The most widely known JAK inhibitor tested in lymphoma trials is Ruxolitinib. This potent compound selectively inhibits JAK1 and JAK2 and is administrated orally. Ruxolitinib has been approved for the treatment of myelofibrosis (MF) by the US Food and Drug Administration (FDA) in 2011 and by the European Medicines Agency (EMA) in 2012, followed by the approval for treatment of hydroxyurea (HU)-resistant or -intolerant polycythemia vera (PV) in 2014 [96]. The drug is not only specific for the mutated form of JAK2 but also inhibits the wild-type JAK2 [97]. In cHL, Ruxolitinib has been seen to induce anti-proliferative effects and programmed cell death in vitro and significantly inhibited tumor progression and improved survival in vivo [98]. Effects of Ruxolitinib in cHL have also been validated in clinical trials, with a disease control rate of 54% (7/13) and a median response duration of 5.6 months [99], or an overall response rate of 9.4% (3/32) after six cycles of dosing for relapsed/refractory cases [100]. In MM, Ruxolitinib treatment decreased expression of genes including JAK2, TYK2, IL-6 and IL-18, driving disease progression and inducing antophagosome accumulation [101]. In a phase I clinical trial, Ruxolitinib was able to overcome lenalidomide and steroid resistance for relapsed/refractory MM patients, with a clinical benefit rate of 46% and an overall response rate of 38%, respectively [102]. Hypersensitivity of Ruxolitinib was noted in one patient with CSF3R T618I mutation, in which there were decreased white cell numbers and neutrophil counts as well as a normalization of the platelet count [103]. Effectiveness of Ruxolitinib was also seen in primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma [104], BCP-ALL [14] and ALCL [105], in which the JAK/STAT pathway played a vital role. However, whether Ruxolitinib is effective in treating PMBL remains controversial [98,99]. This medication has been approved to enter clinical trial phase I/II/III for the treatment of lymphoma, lymphoblastic leukemia or MM alone or together with other agents (NCT01877005, NCT01965119, NCT02164500, NCT02974647, NCT03117751, NCT03041636, NCT02723994, NCT03613428, NCT01712659, NCT03878524, NCT01914484, NCT01620216, NCT00674479, NCT00639002 and NCT03773107). The immunosuppressive side effects of Ruxolitinib have been reviewed extensively before [97].
Tofacitinib, an oral and small molecule compound, inhibits all four JAKs but preferentially inhibits JAK1 and JAK3 [106]. In EBV+ T and NK lymphoma cell lines and patient samples which displayed JAK3/STAT5 activation, Tofacitinib treatment effectively reduced p-STAT5 levels, suppressed proliferation, induced G1 cell cycle arrest and decreased EBV viro-protein LMP1 and EBNA1 expression [107]. In CTCL cells, Tofacitinib inhibited the level of aberrantly expressed anti-apoptotic miR-21 by blocking JAK3/STAT5 signaling, and STAT5 could directly bind to miR-21 promoter [108]. This drug reversed the majority of pro-survival signals modulated by JAK-STAT cascade in MM [109]. In PTCL, as mentioned above, the JAK3/STAT5 signaling program was identified to be downstream of ITK/SYK via Signal Net and cluster analyses of microarray data. JAK3 selective inhibitor tofacitinib abrogated the phosphorylation of STAT5, suppressed cell growth, induced cell apoptosis and arrested the cell cycle at the G1/S phase [5]. As JAK3-activating mutation was frequent in NKTCL pathogenesis, the pan-JAK inhibitor Tofacitinib efficiently reduced phosphorylated STAT5 and cell viability in JAK3-mutant and wild-type NKTCL cell lines and mouse xenografts [19,24]. However, in one case of relapsed T-ALL with two JAK3 activating mutations, Tofacitinib failed to induce a positive clinical response following failure of salvage chemotherapy, indicating that the presence of activating JAK3 mutations did not necessarily guarantee sensitivity to Tofacitinib treatment [110].
Moreover, several JAK-targeting new compounds or derivatives as well as JAK upstream inhibitor have been reported in recent years. Here I summarize these inhibitors based on the types of malignancy. In DLBCL, a natural osalmid derivative DCZ0858 blocked JAK2/STAT3 signaling and inhibited B lymphoma cell survival in a concentration- and time-dependent manner while causing no significant toxicity to normal B cells [111]. Additionally, upstream IRAK4 inhibition by highly selective novel small molecule inhibitors, ND-2158 and ND-2110, impeded survival of DLBCL cells by downregulating survival signals, including IL6/IL10/JAK/STAT3 [112]. In another lethal and skin-attacking lymphoma CTCL, a retinoic acid derivative, ECPIRM, induced cell apoptosis and induced G0/G1 phase arrest via inhibiting the JAK/STAT rather than the RAR/RXR pathway and exhibited little cytotoxicity in normal lymphoid counterparts [113]. Besides, a vitamin A derivative, 9-cis-RA, induced CTCL cellular apoptosis dose- and time-dependently via decreasing JAK1/STAT3/STAT5 phosphorylation, Bcl-xL and cyclin D1 levels [114]. A novel taspine derivate TPD7 was able to bind to the IL-2 receptor in CTCL and therefore suppressed the downstream cascade, including JAK/STAT and PI3K/AKT/mTOR [115] Additionally, another compound ONC201 exerted time-dependent cell survival inhibition in CTCL cell lines and patient-derived primary CD4+ malignant T cells, and the JAK/STAT pathway was downregulated with ONC201 treatment [116]. These derivatives or inhibitors demonstrated effectiveness and selectivity in harnessing JAK/STAT in order to treat CTCL. In NKTCL, frequent STAT3/5B activating mutations were detected in primary patient samples and cell lines, and JAK1/2/3 inhibitors potently suppressed cellular proliferation, inhibited tumor growth and induced apoptosis via abrogation of JAK/STAT program [117,118]. Moreover, NKTCL is known for EBV infection, which is also one of the criteria for NKTCL diagnosis, and LMP1 was a viro- and onco-protein generated by EBV. In NKTCL, a constructed human anti-LMP1 antibody successfully inhibited cell proliferation, induced apoptosis and activated antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity at least partly via inhibiting JAK3/STAT3 [119]. Even classic cytotoxic agents also exhibited anti-JAK/STAT properties. Doxorubicin inhibited c-myc and PIM1 expression by repressing JAK/STAT3 and promoted NKTCL cell death [120]. In MM, compounds including Icarrin, 3-formylchromone, TM-233, Auranofin, AZD1480, thalidomide analogs and tetracyclic pyridone 6, inhibited upstream JAK1/2, thereby blocking constitutive STAT3 phosphorylation and its nuclear translocation, downregulating downstream STAT3 target genes, such as Bcl-2, Bcl-xl, survivin, COX-2, VEGF, Mcl-1, Cyclin D2 and MMP-9 and inducing programmed cell death [121,122,123,124,125,126,127]. Similarly, two novel and highly selective JAK inhibitors, INCB20 and INCB16562, effectively suppressed IL-6 dependent growth of MM cell lines and primary bone marrow-derived plasma cells [128,129]. In addition, several natural product extracts blocked JAK/STAT as well and exerted anti-myeloma effects. Leelamine from pine’s bark attenuated phosphorylation of upstream JAK1/JAK2/Scr macromolecules and downstream STAT3, hence evoking myeloma cell cycle arrest and apoptosis [130]. A Scutellaria radix component, Baicalein, suppressed myeloma cell survival and proliferation by blocking IκB-α degradation, followed by downregulating IL-6/JAK/STAT3 and XIAP gene levels [131]. These findings demonstrated possibilities to inhibit myeloma cell survival, proliferation and invasiveness via targeting JAK/STAT using synthesized compounds and natural extracts. Moreover, in waldenström macroglobulinemia (WM), the pan-FGF trap molecule NSC12 significantly inhibited cellular growth and provoked apoptosis through halting JAK/STAT3, MAPK and PI3K-AKT pathways [132]. All the JAK-based monotherapies are summarized in Table 2.
4.2. Combinational Therapy
The most heavily studied and JAK-related dual inhibitor should be Cerdulatinib. This orally available compound demonstrates activities against JAK1/3 and SYK with limited inhibition of JAK2. Cerdulatinib did not inhibit phorbol-mediated signaling or activation in normal B and T cells, or T-cell receptor mediated signaling in T cells, showing selectivity and safety [133]. This inhibitor exerted potent antitumor activities in a subset of B-cell lymphomas, including ABC-DLBCL, germinal center-diffuse large B cell lymphoma (GC-DLBCL), mantle cell lymphoma (MCL), FL and small lymphocytic lymphoma (SLL) [133,134,135]. In CLL, the dual JAK/SYK inhibitor Cerdulatinib was a promising therapeutic agent that overcame the support of the microenvironment [136] and targeted critical survival pathways, used either alone or combined with Venetoclax [137]. This compound also displayed efficacies in ATLL [138]. Activities of Cerdulatinib against lymphoid tumors were evaluated in clinical trial phase I/II (NCT01994382 and NCT04757259). Another notable JAK-associated dual inhibitor is SB1518, which co-targets JAK2 and FLT3. This compound was selected as a development candidate and progressed into clinical trials for lymphomas [139]. SB1518 demonstrated safety and efficacy in various types of lymphomas, including refractory cases, and a phase I clinical trial demonstrated that an escalating dose of SB1518 led to significant tumor reduction of 4–46% among enrolled patients of relapsed/refractory lymphomas with well-tolerated toxicities [140,141] (NCT01263899 and NCT00741871).
The most widely known JAK inhibitor, Ruxolitinib, as mentioned above, has been applied in synergism with several different compounds. In ABC-DLBCL, JAK1/STAT3 was activated by autocrine IL-6/10 signaling, and Ruxolitinib synergized well with type I IFN inducer lenalidomide in vitro and in vivo [142]. In MM, both JAK1 and JAK2 presented overexpression in a proportion of patients, and Ruxolitinib treatment in combination with Bortezomib, Itacitinib or Daratumumab inhibited JAK/STAT3 phosphorylation, upregulated CD38 expression, inhibited in vitro and in vivo myeloma cell growth and induced cell apoptosis and subG0 arrest [73,143,144]. In NKTCL, Ruxolitinib and CDK4/6 inhibitor LEE011 treatment demonstrated synergistic growth inhibitory effects [145]. Ruxolitinib and Bcl-2/Bcl-xl inhibitor Navitoclax well synergized with each other, augmenting the expression of Bik, puma and Bax expression in cHL cells [146], lowering tumor burden and prolonging survival in an ATLL mouse model [147]. In CTCL cell lines, Ruxolitinib and Resminostat (HDAC inhibition) together exhibited substantial anti-cancer effects [148]. In relapsed/refractory T-ALL, Ruxolitinib and Venetoclax treatment reduced cell survival and proliferation in vitro [149].
The combination between JAK inhibitor and PI3K inhibitor showed significance in a few lymphoid malignancies. In relapsed/refractory B cell lymphoma, JAK1 inhibitor itacitinib+ PI3Kδ inhibitor INCB040093 demonstrated efficacy and few toxicities, presenting a promising treatment option [150]. In MM, JAK2 inhibitor TG101209 and PI3K inhibitor LY194002 combination displayed synergistic cytotoxicity against myeloma cells [151]. In PI3K inhibitor-resistant B-cell and T-cell lymphoma cell lines, the addition of JAK inhibitor BSK805 circumvented well with PI3K inhibitor acquired resistance in lymphomas, and simultaneous inhibition of these two pathways produced combined effects [152].
Successful combinations were also observed for inhibitors against JAK and BTK, a major target for B-cell malignancies [153]. The bromodomain and extra-terminal (BET) inhibitor OTX015 targeted different pathways including JAK/STAT in mature B-cell lymphoid cancer cell lines, and it presented in vitro synergism with BTK inhibitor [154]. The JAK/STAT inhibitor + BTK inhibitor Ibrutinib in combination bypassed survival stimuli from bone marrow mesenchymal stromal cells to induce cell death in CLL [155] and induced IRF4 levels to synergistically kill ABC-DLBCL cells [93].
A couple of studies have evaluated the combination between JAK inhibitors and the anti-apoptotic macromolecule BCL inhibitors. Combined inhibition of JAK and BCL2 demonstrated strong potentiation of cytotoxicity in CTCL cells, driven by intrinsic and extrinsic apoptosis pathways [156]. In Burkitt lymphoma (BL), BCL6 deficiency induced JAK2 expression and STAT3 phosphorylation, and a JAK2 inhibitor, Lestaurtinib, repressed survival of BCL6-deficient cells and tumor xenografts, demonstrating the significance of co-suppressing BCL6 and JAK2, which was considered as synthetic lethality [58]. In cHL, Decitabine inhibited cell growth but concurrently upregulated pro-survival signals, such as MEK/ERK, JAK/STAT and NF-κB, demonstrating a rationale for combining Decitabine with BCL/BCL2L1 inhibitor ABT263, JAK-STAT inhibitors Fedratinib and SH-4-54, AKT inhibitor KP372-1, NF-κB inhibitor QNZ, as well as the BET family proteins inhibitor JQ1 [157].
Investigators also tried to combine JAK inhibitor with conventional therapies in order to ameliorate clinical outcomes. In MCL, anti-JAK/STAT3 agent Degrasyn was considered as a useful therapy administered together with Bortezomib [158]. In MM, selective JAK1 inhibitor INCB052793 in combination with carfilzomib, bortezomib, dexamethasone or lenalidomide effectively reduced tumor volume in tumor-bearing mice [159]; another novel and orally available JAK1/2 inhibitor, CYT387, was able to prevent IL-6-induced STAT3 phosphorylation and was synergized in killing myeloma cells with traditional therapies Melphalan and Bortezomib [160]. JAK inhibitors combined with the cytotoxic anti-folic-acid agent methotrexate significantly suppressed lymphoma cell growth and prolonged survival of tumor xenografts, resulting in better clinical outcomes [161,162]. In CML, targeting JAK/STAT3 cascade by JAK inhibitor in combination with classical BCR-ABL inhibitor promoted cell death and eliminated minimal residual disease located in the bone marrow, representing a hopeful therapeutic strategy [163,164].
In addition, as JAK/STAT3 mutations promoted STAT3-based transcription activation and directly regulated NF-κB and CD30 levels in NIK+/ALK- ALCL, combined NIK and JAK inhibitor therapy could be applied to benefit patients [165]. JAK inhibitor AZD1480 treatment potently blocked STAT phosphorylation but yielded no anti-proliferative effects in cHL, as it led to ERK1/2 phosphorylation upregulation. Therefore, inhibiting ERK activities by MEK inhibitors along with JAK inhibition resulted in enhanced cyto-toxicities [166]. Histone deacetylase (HDAC) inhibitors represent an encouraging class of antitumor therapies, and these inhibitors induce minimal toxicity to normal cells [167]. The orally administered HDAC6 inhibitor Citarinostat was used together with JAK/STAT3 inhibitor Momelotinib, resulting in reduced mitochondrial membrane potential, decreased Bcl-2 and Bcl-xl and activated caspase 3/9, indicating extrinsic apoptosis [167]. In Sézary syndrome, an aggressive and diffused form of CTCL, the HDAC inhibitor Romidepsin showed remarkable but transient activity, and the add-in of JAK inhibitor in combination led to markedly increased therapeutic responses [168]. In LPD, constitutive JAK/STAT3 significantly contributed to disease progression, and combinations including JAK, HSP90 and mTOR inhibitors yielded satisfactory effects on repressing cell viability [169]. All the JAK-based combinational therapies are summarized in Table 3.
5. Conclusions and Future Directions
Accumulating evidence in this review demonstrates how JAKs are aberrantly expressed in lymphoid cancerous contexts and how JAKs connect with upstream and downstream signaling. JAK abnormalities, either mutation or translocation, were found in a few but not all cases in a variety of lymphoid cancers. These abnormalities augment the signals of the cytokine/JAK/STAT pathways, but do not necessarily support lymphoid tumor survival. In a majority of contexts, JAKs signal through STAT-based activation and transcriptional regulation, whereas in a few contexts, the tyrosine kinase JAKs may phosphorylate histone H3 or EZH2 and reprogram transcription profiles [3,4,93,94]. These findings contribute to the importance of the nuclear role of JAKs.
In the recent decade, a couple of specific small-molecule JAKs inhibitors have been developed and utilized to target JAK abnormalities in lymphoid malignancies, such as Ruxolitinib and Tofacitinib. Ruxolitinib has entered more than 10 clinical trials for lymphoid disease treatment. Several natural product derivatives and traditional medications have also been reported to be able to block JAK/STAT signaling and impede cancer cell survival [111,124]. Combinational JAK inhibition, either through a dual inhibitor or through several agents, exhibits better cell killing effects than monotherapy. These results demonstrate an indispensable role of JAK-targeting in treating lymphoid cancers, and future studies are needed to compare the effects of these JAK inhibition therapies over conventional therapeutics.
Author Contributions
B.L., Z.L. and W.-J.C. reviewed the literature and wrote the manuscript. B.L. and Q.W. made figures and tables for this review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science foundation of China, grant number 82100208 and Skagen Animal Health Products (Shangqiu) Co., Ltd., grant number 2021073.
Acknowledgments
We thank all authors in the references of this review. We apologize to those authors whose work was not cited due to space restraints.
Conflicts of Interest
The authors declare no competing interest.
References
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Im-munotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Rui, L.; Emre, N.T.; Kruhlak, M.J.; Chung, H.-J.; Steidl, C.; Slack, G.; Wright, G.W.; Lenz, G.; Ngo, V.N.; Shaffer, A.L.; et al. Cooperative Epigenetic Modulation by Cancer Amplicon Genes. Cancer Cell 2010, 18, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.-H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.-B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahasrabuddhe, A.A.; Chen, X.; Chung, F.; Velusamy, T.; Lim, M.S.; Elenitoba-Johnson, K.S. Oncogenic Y641 mutations in EZH2 prevent Jak2/β-TrCP-mediated degradation. Oncogene 2015, 34, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Pan, H.X.; Wang, Y.X.; Guo, T.; Liu, L. Genome profiling revealed the activation of IL2RG/JAK3/STAT5 in peripheral T-cell lymphoma expressing the ITK-SYK fusion gene. Int. J. Oncol. 2019, 55, 1077–1089. [Google Scholar] [CrossRef]
- Kameda, T.; Shide, K.; Shimoda, H.K.; Hidaka, T.; Kubuki, Y.; Katayose, K.; Taniguchi, Y.; Sekine, M.; Kamiunntenn, A.; Maeda, K.; et al. Absence of gain-of-function JAK1 and JAK3 mutations in adult T cell leukemia/lymphoma. Int. J. Hematol. 2010, 92, 320–325. [Google Scholar] [CrossRef]
- Greenplate, A.; Wang, K.; Tripathi, R.M.; Palma, N.; Ali, S.M.; Stephens, P.J.; Miller, V.A.; Shyr, Y.; Guo, Y.; Reddy, N.M.; et al. Genomic Profiling of T-Cell Neoplasms Reveals Frequent JAK1 and JAK3 Mutations with Clonal Evasion from Targeted Therapies. JCO Precis. Oncol. 2018, 2018, 1–16. [Google Scholar] [CrossRef]
- Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.C.M.; Velusamy, T.; Chung, F.; Schaller, M.; Bailey, N.G.; Betz, B.L.; Miranda, R.N.; Porcu, P.; et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK–STAT pathway in Sézary syndrome. Nat. Commun. 2015, 6, 8470. [Google Scholar] [CrossRef]
- Laurent, C.; Nicolae, A.; Laurent, C.; Le Bras, F.; Haioun, C.; Fataccioli, V.; Amara, N.; Adélaïde, J.; Guille, A.; Schiano, J.-M.; et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood 2020, 135, 360–370. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Filip, I.; Gomez, K.; Engelbrecht, D.; Meer, S.; Lalloo, P.N.; Patel, P.; Perner, Y.; Zhao, J.; Wang, J.; et al. Genomic characterization of HIV-associated plasmablastic lymphoma identifies pervasive mutations in the JAK-STAT pathway. Blood Cancer Discov. 2020, 1, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Fiore, D.; Cappelli, L.V.; Zumbo, P.; Phillips, J.M.; Liu, Z.; Cheng, S.; Yoffe, L.; Ghione, P.; Di Maggio, F.; Dogan, A.; et al. A Novel JAK1 Mutant Breast Implant-Associated Anaplastic Large Cell Lymphoma Patient-Derived Xenograft Fostering Pre-Clinical Discoveries. Cancers 2020, 12, 1603. [Google Scholar] [CrossRef]
- Moffitt, A.B.; Ondrejka, S.L.; McKinney, M.; Rempel, R.E.; Goodlad, J.R.; Teh, C.H.; Leppa, S.; Mannisto, S.; Kovanen, P.E.; Tse, E.; et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J. Exp. Med. 2017, 214, 1371–1386. [Google Scholar] [CrossRef]
- Steeghs, E.M.P.; Jerchel, I.S.; De Goffau-Nobel, W.; Hoogkamer, A.Q.; Boer, J.M.; Boeree, A.; Van De Ven, C.; Koudijs, M.J.; Besselink, N.J.M.; De Groot-Kruseman, H.A.; et al. JAK2 aberrations in childhood B-cell precursor acute lymphoblastic leukemia. Oncotarget 2017, 8, 89923–89938. [Google Scholar] [CrossRef] [Green Version]
- Roncero, A.M.; López-Nieva, P.; Cobos-Fernández, M.A.; Villa-Morales, M.; Gonzalez-Sanchez, L.; López-Lorenzo, J.L.; Llamas, P.; Ayuso, C.; Rodriguezpinilla, S.M.; Arriba, M.C.; et al. Contribution of JAK2 mutations to T-cell lymphoblastic lymphoma development. Leukemia 2016, 30, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemoto, M.; Hattori, H.; Maeda, N.; Akita, N.; Muramatsu, H.; Moritani, S.; Kawasaki, T.; Maejima, M.; Ode, H.; Hachiya, A.; et al. Compound heterozygous TYK2 mutations underlie primary immunodeficiency with T-cell lymphopenia. Sci. Rep. 2018, 8, 6956. [Google Scholar] [CrossRef]
- Song, T.L.; Nairismägi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Arakawa, F.; Miyoshi, H.; Niino, D.; Kawano, R.; Ohshima, K. Activated janus kinase 3 expression not by acti-vating mutations identified in natural killer/T-cell lymphoma. Pathol. Int. 2014, 64, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Koo, G.C.; Tan, S.-Y.; Tang, T.; Poon, S.L.; Allen, G.E.; Tan, L.; Chong, S.C.; Ong, W.S.; Tay, K.; Tao, M.; et al. Janus Kinase 3–Activating Mutations Identified in Natural Killer/T-cell Lymphoma. Cancer Discov. 2012, 2, 591–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes-Mojarro, I.A.; Chen, B.J.; Ramirez-Ibarguen, A.F.; Quezada-Fiallos, C.M.; Pérez-Báez, W.B.; Dueñas, D.; Casavil-ca-Zambrano, S.; Ortiz-Mayor, M.; Rojas-Bilbao, E.; García-Rivello, H.; et al. Mutational profile and EBV strains of ex-tranodal NK/T-cell lymphoma, nasal type in Latin America. Mod. Pathol. 2020, 33, 781–791. [Google Scholar] [CrossRef]
- Sim, S.H.; Kim, S.; Kim, T.M.; Jeon, Y.K.; Nam, S.J.; Ahn, Y.-O.; Keam, B.; Park, H.H.; Kim, D.-W.; Kim, C.W.; et al. Novel JAK3-Activating Mutations in Extranodal NK/T-Cell Lymphoma, Nasal Type. Am. J. Pathol. 2017, 187, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, H.Y.; Kang, S.Y.; Kim, S.J.; Hwang, J.; Lee, S.; Kwak, S.H.; Park, K.S.; Yoo, H.Y.; Kim, W.S.; et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget 2015, 6, 17764–17776. [Google Scholar] [CrossRef] [Green Version]
- Polprasert, C.; Takeuchi, Y.; Makishima, H.; Wudhikarn, K.; Kakiuchi, N.; Tangnuntachai, N.; Assanasen, T.; Sitthi, W.; Muhamad, H.; Lawasut, P.; et al. Frequent mutations in HLA and related genes in extranodal NK/T cell lymphomas. Leuk. Lymphoma 2021, 62, 95–103. [Google Scholar] [CrossRef]
- Bouchekioua, A.; Scourzic, L.; de Wever, O.; Zhang, Y.; Cervera, P.; Aline-Fardin, A.; Mercher, T.; Gaulard, P.; Nyga, R.; Jeziorowska, D.; et al. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal na-sal-type natural killer cell lymphoma. Leukemia 2014, 28, 338–348. [Google Scholar] [CrossRef]
- Pérez, C.; Mondéjar, R.; García-Díaz, N.; Cereceda, L.; León, A.; Montes, S.; Durán Vian, C.; Pérez Paredes, M.G.; Gon-zález-Morán, A.; Alegre de Miguel, V.; et al. Advanced-stage mycosis fungoides: Role of the signal transducer and activator of transcription 3, nuclear factor-κB and nuclear factor of activated T cells pathways. Br. J. Dermatol. 2020, 182, 147–155. [Google Scholar] [CrossRef]
- López, C.; Bergmann, A.K.; Paul, U.; Murga Penas, E.M.; Nagel, I.; Betts, M.J.; Johansson, P.; Ritgen, M.; Baumann, T.; Aymerich, M.; et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br. J. Haematol. 2016, 173, 265–273. [Google Scholar] [CrossRef]
- Nairismägi, M.-L.; Tan, J.; Lim, J.Q.; Nagarajan, S.; Ng, C.C.; Rajasegaran, V.; Huang, D.; Lim, W.K.; Laurensia, Y.; Wijaya, G.C.; et al. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia 2016, 30, 1311–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, P.; Klein-Hitpass, L.; Budeus, B.; Kuhn, M.; Lauber, C.; Seifert, M.; Roeder, I.; Pförtner, R.; Stuschke, M.; Dührsen, U.; et al. Identifying Genetic Lesions in Ocular Adnexal Extranodal Marginal Zone Lymphomas of the MALT Subtype by Whole Genome, Whole Exome and Targeted Sequencing. Cancers 2020, 12, 986. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, W.; Zhang, W.; Ye, Y.; Guan, P.; Gao, L.; Zhao, S. Chronic Active Epstein-Barr Virus Infection of T/NK-Cell Type Mimicking Classic Hodgkin Lymphoma: Clinicopathologic and Genetic Features of 8 Cases Supporting a Variant With “Hodgkin/Reed-Sternberg-like” Cells of NK Phenotype. Am. J. Surg. Pathol. 2019, 43, 1611–1621. [Google Scholar] [CrossRef]
- Daniels, J.; Doukas, P.G.; Escala, M.E.M.; Ringbloom, K.G.; Shih, D.J.H.; Yang, J.; Tegtmeyer, K.; Park, J.; Thomas, J.J.; Selli, M.E.; et al. Cellular origins and genetic landscape of cutaneous gamma delta T cell lymphomas. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Kikuti, Y.Y.; Carreras, J.; Sakai, R.; Takata, K.; Yoshino, T.; Bea, S.; Campo, E.; Missiaglia, E.; Bouilly, J.; et al. Monomorphic Epitheliotropic Intestinal T-Cell Lymphoma in Asia Frequently Shows SETD2 Alterations. Cancers 2020, 12, 3539. [Google Scholar] [CrossRef] [PubMed]
- Margolskee, E.; Jobanputra, V.; Jain, P.; Chen, J.; Ganapathi, K.; Nahum, O.; Levy, B.; Morscio, J.; Murty, V.; Tousseyn, T.; et al. Genetic landscape of T- and NK-cell post-transplant lymphoproliferative disorders. Oncotarget 2016, 7, 37636–37648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacronique, V.; Boureux, A.; Della Valle, V.; Poirel, H.; Quang, C.T.; Mauchauffeé, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 Fusion Protein with Constitutive Kinase Activity in Human Leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.M.; Beattie, B.K.; Squire, J.A.; Frank, D.A.; Barber, D.L. Fusion of the ets transcription factor TEL to Jak2 results in constitutive Jak-Stat signaling. Blood 1999, 93, 4354–4364. [Google Scholar] [CrossRef]
- Ho, J.M.; Nguyen, M.H.; Dierov, J.K.; Badger, K.M.; Beattie, B.K.; Tartaro, P.; Haq, R.; Zanke, B.W.; Carroll, M.P.; Barber, D.L. TEL-JAK2 constitutively activates the extracellular signal-regulated kinase (ERK), stress-activated protein/Jun kinase (SAPK/JNK), and p38 signaling pathways. Blood 2002, 100, 1438–1448. [Google Scholar]
- Nguyen, M.H.; Ho, J.M.; Beattie, B.K.; Barber, D.L. TEL-JAK2 Mediates Constitutive Activation of the Phosphatidylinositol 3′-Kinase/Protein Kinase B Signaling Pathway. J. Biol. Chem. 2001, 276, 32704–32713. [Google Scholar] [CrossRef] [Green Version]
- Monni, R.; Santos, S.C.; Mauchauffe, M.; Berger, R.; Ghysdael, J.; Gouilleux, F.; Gisselbrecht, S.; Bernard, O.; Penard-Lacronique, V. The TEL-Jak2 oncoprotein induces Socs1 expression and altered cytokine response in Ba/F3 cells. Oncogene 2001, 20, 849–858. [Google Scholar] [CrossRef] [Green Version]
- Carron, C.; Cormier, F.; Janin, A.; Lacronique, V.; Giovannini, M.; Daniel, M.T.; Bernard, O.; Ghysdael, J. TEL-JAK2 transgenic mice develop T-cell leukemia. Blood 2000, 95, 3891–3899. [Google Scholar] [CrossRef]
- Dos Santos, N.R.; Rickman, D.S.; De Reynies, A.; Cormier, F.; Williame, M.; Blanchard, C.; Stern, M.-H.; Ghysdael, J. Pre-TCR expression cooperates with TEL-JAK2 to transform immature thymocytes and induce T-cell leukemia. Blood 2007, 109, 3972–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinge, S.; Monni, R.; Bernard, O.; Penard-Lacronique, V. Activation of the NF-kappaB pathway by the leukemogenic TEL-Jak2 and TEL-Abl fusion proteins leads to the accumulation of antiapoptotic IAP proteins and involves IKKalpha. Oncogene 2006, 25, 3589–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulos, I.; Gorunova, L.; Spetalen, S.; Bassarova, A.; Beiske, K.; Micci, F.; Heim, S. Fusion of the genes ataxin 2 like, ATXN2L, and Janus kinase 2, JAK2, in cutaneous CD4 positive T-cell lymphoma. Oncotarget 2017, 8, 103775–103784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roosbroeck, K.; Cox, L.; Tousseyn, T.; Lahortiga, I.; Gielen, O.; Cauwelier, B.; De Paepe, P.; Verhoef, G.; Marynen, P.; Vandenberghe, P.; et al. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood 2011, 117, 4056–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesada, A.E.; Zhang, Y.; Ptashkin, R.; Ho, C.; Horwitz, S.; Benayed, R.; Dogan, A.; Arcila, M.E. Next generation se-quencing of breast implant-associated anaplastic large cell lymphomas reveals a novel STAT3-JAK2 fusion among other activating genetic alterations within the JAK-STAT pathway. Breast J. 2021, 27, 314–321. [Google Scholar] [CrossRef]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Rabionet, R.; Espinet, B.; Zapata, L.; Puiggros, A.; Melero, C.; Puig, A.; Sarria-Trujillo, Y.; Ossowski, S.; Garcia-Muret, M.P.; et al. Identification of Gene Mutations and Fusion Genes in Patients with Sézary Syndrome. J. Investig. Dermatol. 2016, 136, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Desch, A.-K.; Hartung, K.; Botzen, A.; Brobeil, A.; Rummel, M.; Kurch, L.; Georgi, T.; Jox, T.; Bielack, S.; Burdach, S.; et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 2020, 34, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Arimura, Y.; Yagi, J. Comprehensive Expression Profiles of Genes for Protein Tyrosine Phosphatases in Immune Cells. Sci. Signal. 2010, 3, rs1. [Google Scholar] [CrossRef]
- Zahn, M.; Marienfeld, R.; Melzner, I.; Heinrich, J.; Renner, B.; Wegener, S.; Mießner, A.; Barth, T.F.; Dorsch, K.; Brüderlein, S.; et al. A novel PTPN1 splice variant upregulates JAK/STAT activity in classical Hodgkin lymphoma cells. Blood 2017, 129, 1480–1490. [Google Scholar] [CrossRef] [Green Version]
- Zahn, M.; Kaluszniak, B.; Möller, P.; Marienfeld, R. The PTP1B mutant PTP1B∆2–4 is a positive regulator of the JAK/STAT signalling pathway in Hodgkin lymphoma. Carcinogenesis 2021, 42, 517–527. [Google Scholar] [CrossRef]
- Kleppe, M.; Tousseyn, T.; Geissinger, E.; Kalender Atak, Z.; Aerts, S.; Rosenwald, A.; Wlodarska, I.; Cools, J. Mutation analysis of the tyrosine phosphatase PTPN2 in Hodgkin’s lymphoma and T-cell non-Hodgkin’s lymphoma. Haematologica 2011, 96, 1723–1727. [Google Scholar] [CrossRef] [Green Version]
- Demosthenous, C.; Han, J.J.; Hu, G.; Stenson, M.; Gupta, M. Loss of function mutations in PTPN6 promote STAT3 dereg-ulation via JAK3 kinase in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 44703–44713. [Google Scholar] [CrossRef] [Green Version]
- Szydłowski, M.; Dębek, S.; Prochorec-Sobieszek, M.; Szołkowska, M.; Tomirotti, A.M.; Juszczyński, P.; Szumera-Ciećkiewicz, A. PIM Kinases Promote Survival and Immune Escape in Primary Mediastinal Large B-Cell Lymphoma through Modulation of JAK-STAT and NF-κB Activity. Am. J. Pathol. 2021, 191, 567–574. [Google Scholar] [CrossRef]
- Szydłowski, M.; Prochorec-Sobieszek, M.; Szumera-Ciećkiewicz, A.; Derezińska, E.; Hoser, G.; Wasilewska, D.; Szymańska-Giemza, O.; Jabłońska, E.; Białopiotrowicz, E.; Sewastianik, T.; et al. Expression of PIM kinases in Reed-Sternberg cells fosters immune privilege and tumor cell survival in Hodgkin lymphoma. Blood 2017, 130, 1418–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beldi-Ferchiou, A.; Skouri, N.; Ben Ali, C.; Safra, I.; Abdelkefi, A.; Ladeb, S.; Mrad, K.; Ben Othman, T.; Ben Ahmed, M. Abnormal repression of SHP-1, SHP-2 and SOCS-1 transcription sustains the activation of the JAK/STAT3 pathway and the progression of the disease in multiple myeloma. PLoS ONE 2017, 12, e0174835. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, Y.; Yasunaga, J.-I.; Mitagami, Y.; Tsukamoto, H.; Nakashima, K.; Ohshima, K.; Matsuoka, M. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 13740–13749. [Google Scholar] [CrossRef] [PubMed]
- von Hoff, L.; Kärgel, E.; Franke, V.; McShane, E.; Schulz-Beiss, K.W.; Patone, G.; Schleussner, N.; Kolesnichenko, M.; Hübner, N.; Daumke, O.; et al. Autocrine LTA signaling drives NF-κB and JAK-STAT activity and myeloid gene expression in Hodgkin lymphoma. Blood 2019, 133, 1489–1494. [Google Scholar] [CrossRef]
- Våtsveen, T.K.; Sponaas, A.-M.; Tian, E.; Zhang, Q.; Misund, K.; Sundan, A.; Børset, M.; Waage, A.; Brede, G. Erythropoietin (EPO)-receptor signaling induces cell death of primary myeloma cells in vitro. J. Hematol. Oncol. 2016, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Beck, D.; Zobel, J.; Barber, R.; Evans, S.; Lezina, L.; Allchin, R.L.; Blades, M.; Elliott, R.; Lord, C.J.; Ashworth, A.; et al. Synthetic Lethal Screen Demonstrates That a JAK2 Inhibitor Suppresses a BCL6-dependent IL10RA/JAK2/STAT3 Pathway in High Grade B-cell Lymphoma. J. Biol. Chem. 2016, 291, 16686–16698. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Molina, A.; Boss, I.W.; Canela, A.; Pan, H.; Jiang, Y.; Zhao, C.; Jiang, M.; Hu, D.; Agirre, X.; Niesvizky, I.; et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 2015, 21, 1199–1208. [Google Scholar] [CrossRef]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [Green Version]
- Fontán, L.; Qiao, Q.; Hatcher, J.M.; Casalena, G.; Us, I.; Teater, M.; Durant, M.; Du, G.; Xia, M.; Bilchuk, N.; et al. Specific covalent inhibition of MALT1 paracaspase suppresses B cell lymphoma growth. J. Clin. Investig. 2018, 128, 4397–4412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoof, N.; Von Bonin, F.; Trümper, L.; Kube, D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin lymphoma cells. Cell Commun. Signal. 2009, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Integrated molecular profiling of SOD2 expression in multiple myeloma. Blood 2007, 109, 3953–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Wang, K.B.; Rui, L. STAT3 Activation and Oncogenesis in Lymphoma. Cancers 2019, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Yabe, M.; Miranda, R.N.; Medeiros, L.J. Hepatosplenic T-cell Lymphoma: A review of clinicopathologic features, pathogenesis, and prognostic factors. Hum. Pathol. 2018, 74, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.; Dai, B.; Erdmann, T.; Berdel, W.E.; Lenz, G. The molecular pathogenesis of mantle cell lymphoma. Leuk. Lymphoma 2017, 58, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- DeCoster, R.C.; Clemens, M.W.; Di Napoli, A.; Lynch, E.B.; Bonaroti, A.R.; Rinker, B.D.; Butterfield, T.A.; Vasconez, H.C. Cellular and Molecular Mechanisms of Breast Implant–Associated Anaplastic Large Cell Lymphoma. Plast. Reconstr. Surg. 2021, 147, 30e–41e. [Google Scholar] [CrossRef] [PubMed]
- Netchiporouk, E.; Litvinov, I.V.; Moreau, L.; Gilbert, M.; Sasseville, D.; Duvic, M. Deregulation in STAT signaling is im-portant for cutaneous T-cell lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle 2014, 13, 3331–3335. [Google Scholar] [CrossRef]
- Herrmann, A.; Lahtz, C.; Nagao, T.; Song, J.Y.; Chan, W.C.; Lee, H.; Yue, C.; Look, T.; Mülfarth, R.; Li, W.; et al. CTLA4 Promotes Tyk2-STAT3–Dependent B-cell Oncogenicity. Cancer Res. 2017, 77, 5118–5128. [Google Scholar] [CrossRef] [Green Version]
- Prutsch, N.; Gurnhofer, E.; Suske, T.; Liang, H.C.; Schlederer, M.; Roos, S.; Wu, L.C.; Simonitsch-Klupp, I.; Alva-rez-Hernandez, A.; Kornauth, C.; et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia 2019, 33, 696–709. [Google Scholar] [CrossRef]
- Mohanty, A.; Sandoval, N.; Phan, A.; Nguyen, T.V.; Chen, R.W.; Budde, E.; Mei, M.; Popplewell, L.; Pham, L.V.; Kwak, L.W.; et al. Regulation of SOX11 expression through CCND1 and STAT3 in mantle cell lymphoma. Blood 2019, 133, 306–318. [Google Scholar] [CrossRef] [Green Version]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.-T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Stroopinsky, D.; Luptakova, K.; Coll, M.D.; Apel, A.; Rajabi, H.; Pyzer, A.R.; Palmer, K.; Reagan, M.R.; Nahas, M.R.; et al. Bone marrow stroma protects myeloma cells from cytotoxic damage via induction of the oncoprotein MUC1. Br. J. Haematol. 2017, 176, 929–938. [Google Scholar] [CrossRef]
- Qian, T.; Cui, L.; Liu, Y.; Cheng, Z.; Quan, L.; Zeng, T.; Huang, W.; Dai, Y.; Chen, J.; Liu, L.; et al. High expression of chaperonin-containing TCP1 subunit 3 may induce dismal prognosis in multiple myeloma. Pharm. J. 2020, 20, 563–573. [Google Scholar] [CrossRef]
- Canovas Nunes, S.; Manzoni, M.; Pizzi, M.; Mandato, E.; Carrino, M.; Quotti Tubi, L.; Zambello, R.; Adami, F.; Visentin, A.; Barilà, G.; et al. The small GTPase RhoU lays downstream of JAK/STAT signaling and mediates cell migration in multiple myeloma. Blood Cancer J. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Scherger, A.K.; Al-Maarri, M.; Maurer, H.C.; Schick, M.; Maurer, S.; Öllinger, R.; Gonzalez-Menendez, I.; Martella, M.; Thaler, M.; Pechloff, K.; et al. Activated gp130 signaling selectively targets B cell differentiation to induce mature lymphoma and plasmacytoma. JCI Insight 2019, 4, e128435. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, L.; Thakur, C.; Lu, Y.; Zhang, X.; Yi, Z.; Chen, F. Proteomic Characterization of the World Trade Center dust-activated mdig and c-myc signaling circuit linked to multiple myeloma. Sci. Rep. 2016, 6, 36305. [Google Scholar] [CrossRef]
- Gupta, M.; Han, J.J.; Stenson, M.; Maurer, M.; Wellik, L.; Hu, G.; Ziesmer, S.; Dogan, A.; Witzig, T.E. Elevated serum IL-10 levels in diffuse large B-cell lymphoma: A mechanism of aberrant JAK2 activation. Blood 2012, 119, 2844–2853. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Assouline, S.; Alcaide, M.; Mohajeri, A.; Johnston, R.L.; Chong, L.; Grewal, J.; Yu, S.; Fornika, D.; Bushell, K.; et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin. Cancer Res. 2016, 22, 2290–2300. [Google Scholar] [CrossRef] [Green Version]
- Collignon, A.; Wanquet, A.; Maitre, E.; Cornet, E.; Troussard, X.; Aurran-Schleinitz, T. Prolymphocytic Leukemia: New In-sights in Diagnosis and in Treatment. Curr. Oncol. Rep. 2017, 19, 29. [Google Scholar] [CrossRef]
- Lees, C.; Keane, C.; Gandhi, M.K.; Gunawardana, J. Biology and therapy of primary mediastinal B-cell lymphoma: Current status and future directions. Br. J. Haematol. 2019, 185, 25–41. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Wienand, K.; Kamburov, A.; Griffin, G.K.; Chen, P.-H.; Lako, A.; Redd, R.A.; et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood 2019, 134, 2369–2382. [Google Scholar] [CrossRef]
- Viganò, E.; Gunawardana, J.; Mottok, A.; Van Tol, T.; Mak, K.; Chan, F.C.; Chong, L.; Chavez, E.; Woolcock, B.; Takata, K.; et al. Somatic IL4R mutations in primary mediastinal large B-cell lymphoma lead to constitutive JAK-STAT signaling activation. Blood 2018, 131, 2036–2046. [Google Scholar] [CrossRef] [Green Version]
- Annese, T.; Tamma, R.; De Giorgis, M.; Ruggieri, S.; Maiorano, E.; Specchia, G.; Ribatti, D. RNAscope dual ISH–IHC technology to study angiogenesis in diffuse large B-cell lymphomas. Histochem. Cell Biol. 2020, 153, 185–192. [Google Scholar] [CrossRef]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiß, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.-L.; Küppers, R.; et al. An oncogenic axis of STAT-mediated BATF3 upregulation causing MYC activity in classical Hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.; Kopp, N.; Layer, J.V.; Redd, R.A.; Tschuri, S.; Haebe, S.; Van Bodegom, D.; Bird, L.; Christie, A.L.; Christodoulou, A.; et al. HSP90 inhibition overcomes ibrutinib resistance in mantle cell lymphoma. Blood 2016, 128, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Ramis-Zaldivar, J.E.; Gonzalez-Farre, B.; Nicolae, A.; Pack, S.; Clot, G.; Nadeu, F.; Mottok, A.; Horn, H.; Song, J.Y.; Fu, K.; et al. MAP-kinase and JAK-STAT pathways dysregulation in plasmablastic lymphoma. Haematologica 2021, 106, 2682–2693. [Google Scholar] [CrossRef]
- Kato, H.; Karube, K.; Yamamoto, K.; Takizawa, J.; Tsuzuki, S.; Yatabe, Y.; Kanda, T.; Katayama, M.; Ozawa, Y.; Ishitsuka, K.; et al. Gene expression profiling of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014, 105, 537–544. [Google Scholar] [CrossRef]
- Karube, K.; Tsuzuki, S.; Yoshida, N.; Arita, K.; Kato, H.; Katayama, M.; Ko, Y.-H.; Ohshima, K.; Nakamura, S.; Kinoshita, T.; et al. Comprehensive gene expression profiles of NK cell neoplasms identify vorinostat as an effective drug candidate. Cancer Lett. 2013, 333, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Drennan, A.C.; Ceribelli, M.; Zhu, F.; Wright, G.W.; Huang, D.W.; Xiao, W.; Li, Y.; Grindle, K.M.; Lu, L.; et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7260–E7267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadivel, C.K.; Gluud, M.; Torres-Rusillo, S.; Boding, L.; Willerslev-Olsen, A.; Buus, T.B.; Nielsen, T.K.; Persson, J.L.; Bonefeld, C.M.; Geisler, C.; et al. JAK3 Is Expressed in the Nucleus of Malignant T Cells in Cutaneous T Cell Lymphoma (CTCL). Cancers 2021, 13, 280. [Google Scholar] [CrossRef] [PubMed]
- Broux, M.; Prieto, C.; Demeyer, S.; Bempt, M.V.; Alberti-Servera, L.; Lodewijckx, I.; Vandepoel, R.; Mentens, N.; Gielen, O.; Jacobs, K.; et al. Suz12 inactivation cooperates with JAK3 mutant signaling in the development of T-cell acute lymphoblastic leukemia. Blood 2019, 134, 1323–1336. [Google Scholar] [CrossRef] [Green Version]
- Ajayi, S.; Becker, H.; Reinhardt, H.; Engelhardt, M.; Zeiser, R.; von Bubnoff, N.; Wäsch, R. Ruxolitinib. Recent Results Cancer Res. 2018, 212, 119–132. [Google Scholar]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Shah, T.; Yin, C.; Hochberg, J.; Ayello, J.; Morris, E.; Van De Ven, C.; Cairo, M.S. Ruxolitinib significantly enhances in vitro apoptosis in Hodgkin lymphoma and primary mediastinal B-cell lymphoma and survival in a lymphoma xenograft murine model. Oncotarget 2018, 9, 9776–9788. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Yoon, D.H.; Kang, H.J.; Hong, J.Y.; Lee, H.S.; Oh, S.Y.; Shin, H.J.; Kong, J.H.; Yi, J.H.; Sakamoto, K.; et al. Rux-olitinib shows activity against Hodgkin lymphoma but not primary mediastinal large B-cell lymphoma. BMC Cancer 2019, 19, 1080. [Google Scholar] [CrossRef] [Green Version]
- Van Den Neste, E.; André, M.; Gastinne, T.; Stamatoullas, A.; Haioun, C.; Belhabri, A.; Reman, O.; Casasnovas, O.; Ghesquieres, H.; Verhoef, G.; et al. A phase II study of the oral JAK1/JAK2 inhibitor ruxolitinib in advanced re-lapsed/refractory Hodgkin lymphoma. Haematologica 2018, 103, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Kusoglu, A.; Bagca, B.G.; Ay, N.P.O.; Saydam, G.; Avci, C.B. Ruxolitinib Regulates the Autophagy Machinery in Multiple Myeloma Cells. Anti-Cancer Agents Med. Chem. 2020, 20, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Berenson, J.R.; To, J.; Spektor, T.M.; Martinez, D.; Turner, C.; Sanchez, A.; Ghermezi, M.; Eades, B.M.; Swift, R.A.; Schwartz, G.; et al. A Phase I Study of Ruxolitinib, Lenalidomide, and Steroids for Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2346–2353. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E.; et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.N.B.; Cats, D.; Out-Luiting, J.J.; Fanoni, D.; Mei, H.; Venegoni, L.; Willemze, R.; Vermeer, M.H.; Berti, E.; Tensen, C.P. Deregulation of JAK2 signaling underlies primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. Haematologica 2021. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine receptor signaling is required for the survival of ALK- anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; Niezychowski, W. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2017, 376, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Kawada, J.-I.; Watanabe, T.; Suzuki, M.; Sato, Y.; Torii, Y.; Asai, M.; Goshima, F.; Murata, T.; Shimizu, N.; et al. Tofacitinib induces G1 cell-cycle arrest and inhibits tumor growth in Epstein-Barr virus-associated T and natural killer cell lymphoma cells. Oncotarget 2016, 7, 76793–76805. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, L.M.; Fredholm, S.; Joseph, C.; Nielsen, B.S.; Jønson, L.; Willerslev-Olsen, A.; Gluud, M.; Blümel, E.; Petersen, D.L.; Sibbesen, N.; et al. STAT5 induces miR-21 expression in cutaneous T cell lymphoma. Oncotarget 2016, 7, 45730–45744. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.; Ferguson, I.D.; Mariano, M.C.; Lin, Y.T.; Murnane, M.; Liu, H.; Smith, G.A.; Wong, S.W.; Taunton, J.; Liu, J.O.; et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow mi-croenvironment. Haematologica 2018, 103, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Wall, M.; Corboy, G.P.; Taubenheim, N.; Gregory, G.P.; Opat, S.; Shortt, J. Failure of tofacitinib to achieve an objective response in a DDX3X-MLLT10 T-lymphoblastic leukemia with activating JAK3 mutations. Mol. Case Stud. 2020, 6, a004994. [Google Scholar] [CrossRef]
- Lu, K.; Li, B.; Zhang, H.; Xu, Z.; Song, D.; Gao, L.; Sun, H.; Li, L.; Wang, Y.; Feng, Q.; et al. A novel silicone derivative of natural osalmid (DCZ0858) induces apoptosis and cell cycle arrest in diffuse large B-cell lymphoma via the JAK2/STAT3 pathway. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef]
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L., III; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–21201. [Google Scholar] [CrossRef]
- Yang, H.; Ma, P.; Cao, Y.; Zhang, M.; Li, L.; Wei, J.; Tao, L.; Qian, K. ECPIRM, a Potential Therapeutic Agent for Cutaneous T-Cell Lymphoma, Inhibits Cell Proliferation and Promotes Apoptosis via a JAK/STAT Pathway. Anti-Cancer Agents Med. Chem. 2018, 18, 401–411. [Google Scholar] [CrossRef]
- Yang, H.; Tao, Y.; Zhang, M.; Ma, P.; Li, L.; Diao, Q. Effects of 9-cis-retinoic acid on the proliferation and apoptosis of cutaneous T-cell lymphoma cells. Anti-Cancer Drugs 2019, 30, 56–64. [Google Scholar] [CrossRef]
- Zhu, M.; Yang, L.; Shi, X.; Gong, Z.; Yu, R.; Zhang, D.; Zhang, Y.; Ma, W. TPD7 inhibits the growth of cutaneous T cell lymphoma H9 cell through regulating IL-2R signalling pathway. J. Cell. Mol. Med. 2020, 24, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Zhang, X.; Hu, C.H.; Langridge, T.; Tarapore, R.S.; Allen, J.E.; Oster, W.; Duvic, M. ONC201 selectively induces apoptosis in cutaneous T-cell lymphoma cells via activating pro-apoptotic integrated stress response and inactivating JAK/STAT and NF-κB pathways. Oncotarget 2017, 8, 61761–61776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nairismägi, M.; Gerritsen, M.E.; Li, Z.M.; Wijaya, G.C.; Chia, B.K.H.; Laurensia, Y.; Lim, J.Q.; Yeoh, K.W.; Yao, X.S.; Pang, W.L.; et al. Oncogenic activation of JAK3-STAT signaling confers clinical sensitivity to PRN371, a novel selective and potent JAK3 inhibitor, in natural killer/T-cell lymphoma. Leukemia 2018, 32, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Ac-tivating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Wang, J.; Zhang, M.; Fan, W.; Tang, Q.; Xiong, S.; Tang, X.; Xu, J.; Wang, L.; Yang, S.; et al. A neutralized human LMP1-IgG inhibits ENKTL growth by suppressing the JAK3/STAT3 signaling pathway. Oncotarget 2017, 8, 10954–10965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, X.; Yuan, D.; Liu, Y.; Li, J.; Wen, Q.; Kong, P.; Gao, L.; Zhang, C.; Gao, L.; Peng, X.; et al. PIM1 overexpression in T-cell lymphomas protects tumor cells from apoptosis and confers doxorubicin resistance by upregulating c-myc expression. Acta Biochim. Biophys. Sin. 2018, 50, 800–806. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Lee, J.H.; Nam, D.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Um, J.-Y.; Sethi, G.; Ahn, K.S. Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade. Front. Pharmacol. 2018, 9, 531. [Google Scholar] [CrossRef]
- Ko, J.-H.; Ho Baek, S.; Nam, D.; Chung, W.-S.; Lee, S.-G.; Lee, J.; Mo Yang, W.; Um, J.-Y.; Ahn, K.S. 3-Formylchromone inhibits proliferation and induces apoptosis of multiple myeloma cells by abrogating STAT3 signaling through the induction of PIAS3. Immunopharmacol. Immunotoxicol. 2016, 38, 334–343. [Google Scholar] [CrossRef]
- Sagawa, M.; Tabayashi, T.; Kimura, Y.; Tomikawa, T.; Nemoto-Anan, T.; Watanabe, R.; Tokuhira, M.; Ri, M.; Hashimoto, Y.; Iida, S.; et al. TM-233, a novel analog of 1′-acetoxychavicol acetate, induces cell death in myeloma cells by inhibiting both JAK/STAT and proteasome activities. Cancer Sci. 2015, 106, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, A.; Sagawa, M.; Muto, A.; Uchida, H.; Ikeda, Y.; Kizaki, M. The gold compound auranofin induces apoptosis of human multiple myeloma cells through both down-regulation of STAT3 and inhibition of NF-κB activity. Leuk. Res. 2011, 35, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Scuto, A.; Krejci, P.; Popplewell, L.; Wu, J.; Wang, Y.; Kujawski, M.; Kowolik, C.; Xin, H.; Chen, L.; Kretzner, L.; et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia 2011, 25, 538–550. [Google Scholar] [CrossRef]
- Kumar, S.; Raje, N.; Hideshima, T.; Ishitsuka, K.; Roccaro, A.; Shiraishi, N.; Hamasaki, M.; Yasui, H.; Munshi, N.C.; Richardson, P.; et al. Antimyeloma activity of two novel N-substituted and tetraflourinated thalidomide analogs. Leukemia 2005, 19, 1253–1261. [Google Scholar] [CrossRef]
- Pedranzini, L.; Dechow, T.; Berishaj, M.; Comenzo, R.; Zhou, P.; Azare, J.; Bornmann, W.; Bromberg, J. Pyridone 6, A Pan-Janus-Activated Kinase Inhibitor, Induces Growth Inhibition of Multiple Myeloma Cells. Cancer Res. 2006, 66, 9714–9721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, R.; le Gouill, S.; Tai, Y.-T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Favata, M.; Kelley, J.A.; Caulder, E.; Thomas, B.; Wen, X.; Sparks, R.B.; Arvanitis, A.; Rogers, J.D.; Combs, A.P.; et al. INCB16562, a JAK1/2 Selective Inhibitor, Is Efficacious against Multiple Myeloma Cells and Reverses the Protective Effects of Cytokine and Stromal Cell Support. Neoplasia 2010, 12, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.Y.; Um, J.-Y.; Nasif, O.; Alharbi, S.A.; Sethi, G.; Ahn, K.S. Blockage of the JAK/STAT3 signaling pathway in multiple myeloma by leelamine. Phytomedicine 2021, 87, 153574. [Google Scholar] [CrossRef]
- Liu, S.; Ma, Z.; Cai, H.; Li, Q.; Rong, W.; Kawano, M. Inhibitory effect of baicalein on IL-6-mediated signaling cascades in human myeloma cells. Eur. J. Haematol. 2010, 84, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Federico, C.; Giacomini, A.; Caprio, C.; Maccarinelli, F.; Todoerti, K.; Favasuli, V.; Anastasia, A.; Motta, M.; Russo, D.; et al. Halting the FGF/FGFR axis leads to antitumor activity in Waldenström macroglobulinemia by silencing MYD88. Blood 2021, 137, 2495–2508. [Google Scholar] [CrossRef] [PubMed]
- Coffey, G.; Betz, A.; DeGuzman, F.; Pak, Y.; Inagaki, M.; Baker, D.C.; Hollenbach, S.J.; Pandey, A.; Sinha, U. The Novel Kinase Inhibitor PRT062070 (Cerdulatinib) Demonstrates Efficacy in Models of Autoimmunity and B-Cell Cancer. J. Pharmacol. Exp. Ther. 2014, 351, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Xing, W.; Coffey, G.; Dresser, K.; Lu, K.; Guo, A.; Raca, G.; Pandey, A.; Conley, P.; Yu, H.; et al. Cerdulatinib, a novel dual SYK/JAK kinase inhibitor, has broad anti-tumor activity in both ABC and GCB types of diffuse large B cell lymphoma. Oncotarget 2015, 6, 43881–43896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, G.P.; Feng, J.; Betz, A.; Pandey, A.; Birrell, M.; Leeds, J.M.; Der, K.; Kadri, S.; Lu, P.; Segal, J.P.; et al. Cerdulatinib Pharmacodynamics and Relationships to Tumor Response Following Oral Dosing in Patients with Relapsed/Refractory B-cell Malignancies. Clin. Cancer Res. 2019, 25, 1174–1184. [Google Scholar] [CrossRef] [Green Version]
- Guo, A.; Lu, P.; Coffey, G.; Conley, P.; Pandey, A.; Wang, Y.L. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: Cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget 2017, 8, 12953–12967. [Google Scholar] [CrossRef] [Green Version]
- Blunt, M.D.; Koehrer, S.; Dobson, R.C.; Larrayoz, M.; Wilmore, S.; Hayman, A.; Parnell, J.; Smith, L.D.; Davies, A.; Johnson, P.W.M.; et al. The Dual Syk/JAK Inhibitor Cerdulatinib Antagonizes B-cell Receptor and Microenvironmental Signaling in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 2313–2324. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, C.; Senba, M.; Mori, N. Anti-adult T-cell leukemia/lymphoma activity of cerdulatinib, a dual SYK/JAK kinase inhibitor. Int. J. Oncol. 2018, 53, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- William, A.D.; Lee, A.C.; Blanchard, S.; Poulsen, A.; Teo, E.L.; Nagaraj, H.; Tan, E.; Chen, D.; Williams, M.; Sun, E.T.; et al. Discovery of the macrocycle 11-(2-pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[1 9.3.1.1(2,6).1(8,12)]heptacosa-1,2,3,5,8,10,12,16,21,23-decaene (SB1518), a potent Janus kinase 2/fms-like tyrosine kinase-3 (JAK2/FLT3) inhibitor for the treatment of myelofibrosis and lymphoma. J. Med. Chem. 2011, 54, 4638–4658. [Google Scholar]
- Younes, A.; Romaguera, J.; Fanale, M.; McLaughlin, P.; Hagemeister, F.; Copeland, A.; Neelapu, S.; Kwak, L.; Shah, J.; de Castro Faria, S.; et al. Phase I Study of a Novel Oral Janus Kinase 2 Inhibitor, SB1518, in Patients with Relapsed Lymphoma: Evidence of Clinical and Biologic Activity in Multiple Lymphoma Subtypes. J. Clin. Oncol. 2012, 30, 4161–4167. [Google Scholar] [CrossRef] [Green Version]
- Derenzini, E.; Younes, A. Targeting the JAK-STAT pathway in lymphoma: A focus on pacritinib. Expert Opin. Investig. Drugs 2013, 22, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhu, F.; Zhang, M.; Li, Y.; Drennan, A.C.; Kimpara, S.; Rumball, I.; Selzer, C.; Cameron, H.; Kellicut, A.; et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2018, 115, E498–E505. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, M.B.; Fook-Alves, V.L.; Eugenio, A.I.P.; Fernando, R.C.; Sanson, L.F.G.; de Carvalho, M.F.; Braga, W.M.T.; Davies, F.E.; Colleoni, G.W.B. Anti-myeloma effects of ruxolitinib combined with bortezomib and lenalidomide: A rationale for JAK/STAT pathway inhibition in myeloma patients. Cancer Lett. 2017, 403, 206–215. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Burn, T.C.; Sparks, R.; Maduskuie, T.; Diamond, S.; Rupar, M.; Wen, X.; Volgina, A.; Zolotarjova, N.; Waeltz, P.; et al. The Novel Bromodomain and Extraterminal Domain Inhibitor INCB054329 Induces Vulnerabilities in Myeloma Cells That Inform Rational Combination Strategies. Clin. Cancer Res. 2019, 25, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Hee, Y.T.; Yan, J.; Nizetic, D.; Chng, W.-J. LEE011 and ruxolitinib: A synergistic drug combination for natural killer/T-cell lymphoma (NKTCL). Oncotarget 2018, 9, 31832–31841. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Zhang, M.; Wilson, K.M.; Petrus, M.N.; Bamford, R.N.; Zhang, X.; Guha, R.; Ferrer, M.; Thomas, C.J.; Waldmann, T.A. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or Navitoclax in a murine model of human Hodgkin’s lymphoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1624–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Mathews Griner, L.A.; Ju, W.; Duveau, D.Y.; Guha, R.; Petrus, M.N.; Wen, B.; Maeda, M.; Shinn, P.; Ferrer, M.; et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc. Natl. Acad. Sci. USA 2015, 112, 12480–12485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagianni, F.; Piperi, C.; Mpakou, V.; Spathis, A.; Foukas, P.G.; Dalamaga, M.; Pappa, V.; Papadavid, E. Ruxolitinib with resminostat exert synergistic antitumor effects in Cutaneous T-cell Lymphoma. PLoS ONE 2021, 16, e0248298. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.L.; Rinella, S.P.; Hess, N.J.; Turicek, D.P.; Kabakov, S.A.; Zhu, F.; Bouchlaka, M.N.; Olson, S.L.; Cho, M.M.; Quamine, A.E.; et al. CXCR4 allows T cell acute lymphoblastic leukemia to escape from JAK1/2 and BCL2 inhibition through CNS infiltration. Leuk. Lymphoma 2021, 62, 1167–1177. [Google Scholar] [CrossRef]
- Phillips, T.J.; Forero-Torres, A.; Sher, T.; Diefenbach, C.S.; Johnston, P.; Talpaz, M.; Pulini, J.; Zhou, L.; Scherle, P.; Chen, X.; et al. Phase 1 study of the PI3Kδ inhibitor INCB040093 ± JAK1 inhibitor itacitinib in relapsed/refractory B-cell lymphoma. Blood 2018, 132, 293–306. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Kimlinger, T.; Haug, J.; Timm, M.; Wellik, L.; Halling, T.; Pardanani, A.; Tefferi, A.; Rajkumar, S.V.; Kumar, S. TG101209, a novel JAK2 inhibitor, has significant in vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. Am. J. Hematol. 2010, 85, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, W.S.; Park, C. Interleukin-6 mediates resistance to PI3K-pathway–targeted therapy in lymphoma. BMC Cancer 2019, 19, 936. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, R.; London, N. A Fast and Clean BTK Inhibitor. J. Med. Chem. 2020, 63, 5100–5101. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Syn-ergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severin, F.; Frezzato, F.; Visentin, A.; Martini, V.; Trimarco, V.; Carraro, S.; Tibaldi, E.; Brunati, A.M.; Piazza, F.; Semenzato, G.; et al. In Chronic Lymphocytic Leukemia the JAK2/STAT3 Pathway Is Constitutively Activated and Its Inhibition Leads to CLL Cell Death Unaffected by the Protective Bone Marrow Microenvironment. Cancers 2019, 11, 1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumeen, S.; Mirza, F.N.; Lewis, J.M.; King, A.L.O.; Kim, S.R.; Carlson, K.R.; Umlauf, S.R.; Surovtseva, Y.V.; Foss, F.M.; Girardi, M. JAK inhibition synergistically potentiates BCL2, BET, HDAC, and proteasome inhibition in advanced CTCL. Blood Adv. 2020, 4, 2213–2226. [Google Scholar] [CrossRef]
- Swerev, T.M.; Wirth, T.; Ushmorov, A. Activation of oncogenic pathways in classical Hodgkin lymphoma by decitabine: A rationale for combination with small molecular weight inhibitors. Int. J. Oncol. 2017, 50, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Pham, L.V.; Tamayo, A.T.; Li, C.; Bornmann, W.; Priebe, W.; Ford, R.J. Degrasyn Potentiates the Antitumor Effects of Bortezomib in Mantle Cell Lymphoma Cells In vitro and In vivo: Therapeutic Implications. Mol. Cancer Ther. 2010, 9, 2026–2036. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.; Li, M.; Patil, S.; Soof, C.M.; Nosrati, J.D.; Schlossberg, R.E.; Vidisheva, A.; Tanenbaum, E.J.; Hekmati, T.; Zahab, B.; et al. The anti-myeloma effects of the selective JAK1 inhibitor (INCB052793) alone and in combination in vitro and in vivo. Ann. Hematol. 2019, 98, 691–703. [Google Scholar] [CrossRef]
- Monaghan, K.A.; Khong, T.; Burns, C.J.; Spencer, A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia 2011, 25, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Chang, C.-H.; Huang, Z.-N.; Su, Y.-C.; Chang, S.-J.; Jan, J.-S. The JAK inhibitor antcin H exhibits direct anticancer activity while enhancing chemotherapy against LMP1-expressed lymphoma. Leuk. Lymphoma 2019, 60, 1193–1203. [Google Scholar] [CrossRef]
- Gremese, E.; Alivernini, S.; Tolusso, B.; Zeidler, M.P.; Ferraccioli, G. JAK inhibition by methotrexate (and csDMARDs) may explain clinical efficacy as monotherapy and combination therapy. J. Leukoc. Biol. 2019, 106, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.R.; Tolentino, J.H.; Argilagos, R.F.; Zhang, L.; Pinilla-Ibarz, J.; Hazlehurst, L.A. Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML. Leuk. Res. 2012, 36, 756–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, R.R.; Tolentino, J.H.; Hazlehurst, L.A. Role of STAT3 in Transformation and Drug Resistance in CML. Front. Oncol. 2012, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wei, W.; Zhang, J.P.; Song, Z.; Li, Y.; Xiao, W.; Liu, Y.; Zeng, M.S.; Petrus, M.N.; Thomas, C.J.; et al. A novel model of alternative NF-κB pathway activation in anaplastic large cell lymphoma. Leukemia 2021, 35, 1976–1989. [Google Scholar] [CrossRef]
- Derenzini, E.; Lemoine, M.; Buglio, D.; Katayama, H.; Ji, Y.; Davis, R.E.; Sen, S.; Younes, A. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. 2011, 1, e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosenza, M.; Civallero, M.; Marcheselli, L.; Sacchi, S.; Pozzi, S. Citarinostat and Momelotinib co-target HDAC6 and JAK2/STAT3 in lymphoid malignant cell lines: A potential new therapeutic combination. Apoptosis 2020, 25, 370–387. [Google Scholar] [CrossRef]
- Cortes, J.R.; Patrone, C.C.; Quinn, S.A.; Gu, Y.; Sanchez-Martin, M.; Mackey, A.; Cooke, A.J.; Shih, B.B.; Laurent, A.P.; Trager, M.H.; et al. Jak-STAT Inhibition Mediates Romidepsin and Mechlorethamine Synergism in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2021. [Google Scholar] [CrossRef]
- Kuusanmäki, H.; Dufva, O.; Parri, E.; Van Adrichem, A.J.; Rajala, H.; Majumder, M.M.; Yadav, B.; Parsons, A.; Chan, W.C.; Wennerberg, K.; et al. Drug sensitivity profiling identifies potential therapies for lymphoproliferative disorders with overactive JAK/STAT3 signaling. Oncotarget 2017, 8, 97516–97527. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic model indicating the JAK/STAT pathway.

Figure 2.
Schematic model showing the nuclear role of JAKs. (A) Phosphorylation maintained by JAK1/2 and de-methylation maintained by JMJD2C leads to gene repression. (B) JAK2 phosphorylates EZH2 and blocks EZH2 ubiquitination. JAK3 phosphorylates histone and EZH2, therefore promoting gene expression.
Figure 2.
Schematic model showing the nuclear role of JAKs. (A) Phosphorylation maintained by JAK1/2 and de-methylation maintained by JMJD2C leads to gene repression. (B) JAK2 phosphorylates EZH2 and blocks EZH2 ubiquitination. JAK3 phosphorylates histone and EZH2, therefore promoting gene expression.


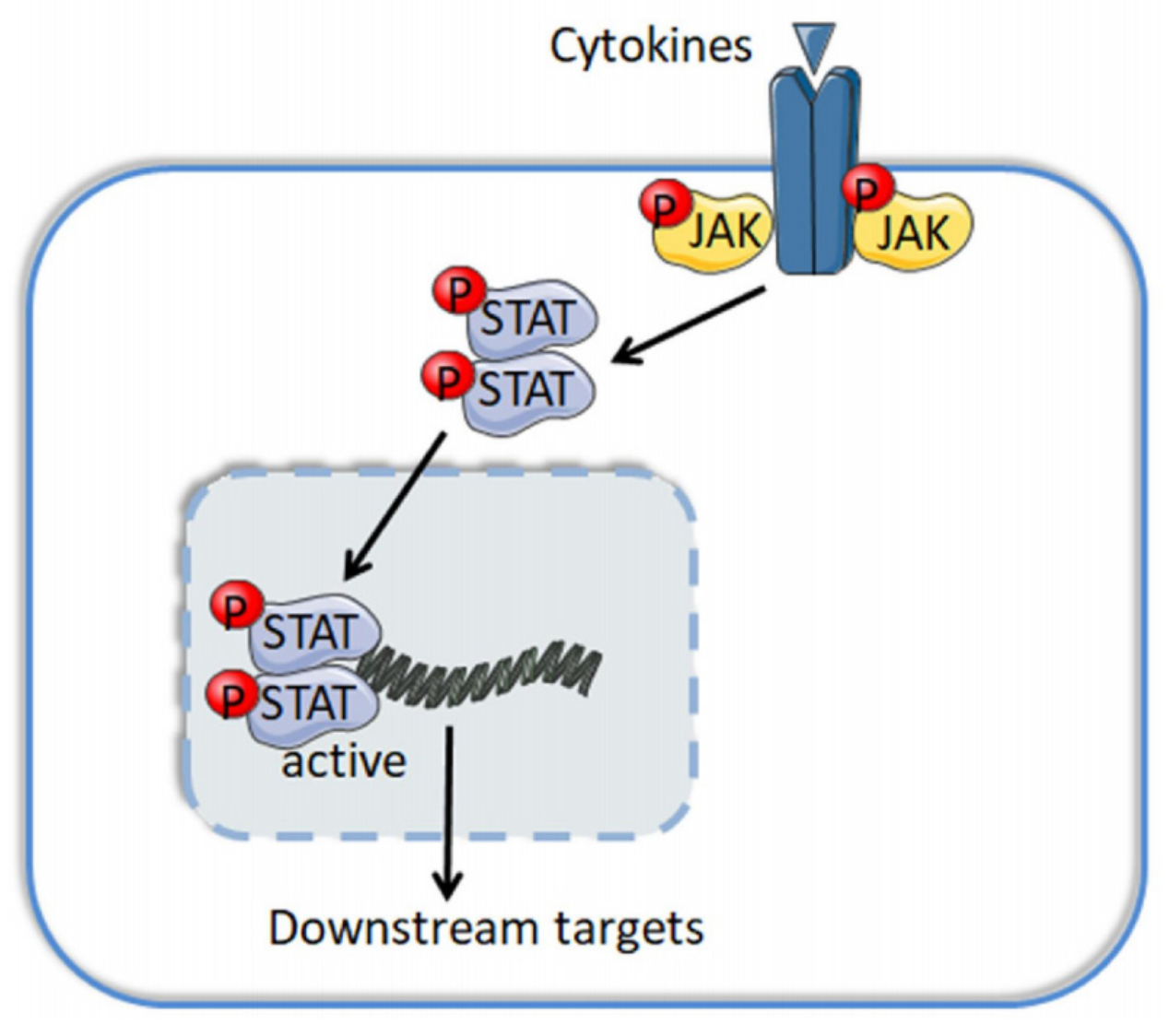{kind=link}
{kind=link}
Table 1.
JAK mutations in lymphoid cancers.
| Malignancy | Protein(s) | Mutation Site(s) | References |
|---|---|---|---|
| ATLL | JAK3 | -- | [6] |
| T-PLL | JAK1 | V658F | [7] |
| CTCL | JAK1 | Y654F, L710V | [8] |
| ALCL | JAK1 | R174 *, G1097D/S | [9,10] |
| PBL | JAK1 | G1097D/V | [11] |
| PTCL | JAK1 | G1097D | [12] |
| EATL | JAK1 | -- | [13] |
| Pediatric BCP-ALL | JAK2 | R873N, R683T/G/S, D873N, T875N, R923H, K914E | [14] |
| T-LBL | JAK2 | H574R, R683G, I682T | [15] |
| EBV-associated B-cell lymphoma | TYK2 | R231W | [16] |
| NKTCL | JAK3 | M511I, A572V, A573V, H583Y, G589D, R657Q, V722I, | [17,18,19,20,21,22,23,24] |
| CTCL | JAK3 | S989I, Y1023H | [8,25] |
| T-PLL | JAK3 | M511I, A657G, G491S, V674A, V678L, A573V, G507P | [26] |
| EITL | JAK3 | V674A, M511I | [27] |
| EATL | JAK3 | -- | [13] |
| OAML | JAK3 | -- | [28] |
| PCGDTL | JAK3 | R657W | [30] |
| EITL | JAK1, JAK3 | JAK1: L1026G, S703I, L783P; JAK3: M511I, A573V, V674A | [31] |
Abbreviations: ATLL: adult precursor T acute lymphoblastic leukemia, T-PLL: T-cell prolymphocytic leukemia, CTCL: cutaneous T-cell lymphoma, ALCL: anaplastic large cell lymphoma, PBL: plasmablastic lymphoma, PTCL: peripheral T-cell lymphoma, EATL: enteropathy-associated T cell lymphoma, BCP-ALL: B-cell precursor acute lymphoblastic leukemia, T-LBL: T-cell lymphoblastic lymphoma, EBV: Epstein–Barr virus, NKTCL: natural killer/T-cell lymphoma, EITL: epitheliotropic intestinal T-cell lymphoma, OAML: ocular adnexal marginal zone lymphomas, HRS: Hodgkin–Reed–Sternberg, PCGDTL: primary cutaneous γδ T cell lymphoma, LPD: lymphoproliferative disorder. * indicated mutated amino acid not identified.
Table 2.
JAK inhibition in lymphoid malignancies.
| Inhibitor(s) | Malignancies | Mechanism of Action | References |
|---|---|---|---|
| Ruxolitinib | MPN, HL, PMBL, MM, RRMM, CNL, pcAECyTCL, BCP-ALL, ALCL | Inhibit JAK1/2 | [14,96,97,98,99,100,101,102,103,104,105] |
| Tofacitinib | EBV-associated T and NK cell lymphoma, CTCL, MM, PTCL, NKTCL | Inhibit JAK1/3 | [5,19,24,107,108,109] |
| DCZ0858 | DLBCL | Inhibit JAK2/STAT3 | [111] |
| ND-2158 and ND-2110 | DLBCL | Inhibit IRAK4 | [112] |
| ECPIRM | CTCL | Inhibit JAK/STAT | [113] |
| 9-cis-retinoic acid | CTCL | Inhibit JAK1/STAT3/5 | [114] |
| TPD7 | CTCL | Bind to IL-2 receptor | [115] |
| ONC201 | CTLC | Inhibit JAK/STAT | [116] |
| PRN371 | NKTCL | Inhibit JAK3/STAT | [117] |
| AZD1480 | NKTCL | Inhibit JAK1/2 | [118] |
| LMP1-IgG | ENKTL | Inhibit JAK3/STAT3 | [119] |
| Doxorubicin | TCL | Inhibit JAK/STAT3 | [120] |
| Icariin | MM | Inhibit JAK1/2/STAT3 | [121] |
| 3-Formylchromone | MM | Inhibit JAK1/2/STAT3 | [122] |
| TM-233 | MM | Inhibit JAK2/STAT3 | [123] |
| Auranofin | MM | Inhibit JAK2/STAT3 | [124] |
| AZD1480 | MM | Inhibit JAK2/STAT3 | [125] |
| CPS11/CPS49 | MM | Inhibit JAK/STAT | [126] |
| Pyridone 6 | MM | Inhibit JAK/STAT | [127] |
| INCB20 | MM | Inhibit JAKs | [128] |
| INCB16562 | MM | Inhibit JAKs | [129] |
| leelamine | MM | Inhibit JAK1/2 | [130] |
| Baicalein | MM | Inhibit IL6/JAK/STAT3 | [131] |
| NSC12 | WM | Inhibit JAK/STAT3 | [132] |
Abbreviations: MPN: myeloproliferative neoplasm, HL: Hodgkin lymphoma, PMBL: primary mediastinal B-cell lymphoma, MM: multiple myeloma; RRMM: relapsed/refractory multiple myeloma, CNL: chronic neutrophilic leukemia, pcAECyTCL: primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma, BCP-ALL: B-cell precursor acute lymphoblastic leukemia, ALCL: anaplastic large cell lymphomas, CTCL: cutaneous T cell lymphomas, PTCL: peripheral T-cell lymphoma, NKTCL: natural killer/T-cell lymphoma, DLBCL: diffuse large B-cell lymphoma, ENKTL: extranodal nasal-type natural killer (NK)/T-cell lymphoma, TCL: T-cell lymphomas, WM: waldenström macroglobulinemia.
Table 3.
Combining JAK inhibitors with other chemo-agents in lymphoid malignancies.
| Regimen | Malignancies | Mechanism of Action | References |
|---|---|---|---|
| Cerdulatinib | B-cell malignancies, ABC-DLBCL, GC-DLBCL, MCL, FL, SLL, CLL, ATLL | Inhibit JAK1/3 and SYK | [133,134,135,136,138] |
| Cerdulatinib and Venetoclax | CLL | Inhibit JAK1/3, SYK and Bcl-2 | [137] |
| SB1518 | Relapsed/refractory lymphoma | Inhibit JAK2 and FLT3 | [139,140,141] |
| Ruxolitinib and Lenalidomide | ABC-DLBCL | Inhibit JAK1/2 and induce type I IFN | [142] |
| Ruxolitinib and Daratumumab | MM | Inhibit JAK1/2 and upregulate CD38 | [73] |
| Ruxolitinib, Bortezomib and Itacitinib | MM | Inhibit JAK1/2 and proteasome | [143] |
| INCB054329 and Ruxolitinib/Itacitinib | MM | Inhibit JAK1/2 and BET | [144] |
| Ruxolitinib and LEE011 | NKTCL | Inhibit JAK1/2 and CDK4/6 | [145] |
| Brentuximab Vedotin and Ruxolitinib/Navitoclax | HL | Inhibit JAK1/2 and Bcl-2/Bcl-x, anti-CD30 | [146] |
| Ruxolitinib and Navitoclax | ATL | Inhibit JAK1/2 and Bcl-2/Bcl-xl | [147] |
| Ruxolitinib and Resminostat | CTCL | Inhibit JAK1/2 and HDAC1/3/6 | [148] |
| Ruxolitinib and Venetoclax | Relapsed/refractory T-ALL | Inhibit JAK1/2 and Bcl-2 | [149] |
| Itacitinib and INCB040093 | Relapsed/refractory BCL | Inhibit JAK1 and PI3Kδ | [150] |
| TG101209 and LY194002 | MM | Inhibit JAK2 and PI3K | [151] |
| BSK805 and Copanlisib/Duvelisib | B-cell and T-cell lymphoma | Inhibit JAKs and PI3K | [152] |
| Ibrutinib and OTX015 | B cell lymphoma | Inhibit JAK/STAT and BTK | [154] |
| Ibrutinib and AG490/Stattic | CLL | Inhibit JAK/STAT and BTK | [155] |
| Ibrutinib and AZD1480 | ABC-DLBC | Inhibit JAK2 and BTK | [93] |
| Ruxolitinib and Venetoclax | CTCL | Inhibit JAK1/2 and Bcl-2 | [156] |
| ABT263/Fedratinib/SH-4-54/KP372-1/QNZ/JQ1 and Decitabine | cHL | Inhibit JAK/STAT, BCL/BCL2L1, NFκB, AKT and BET | [157] |
| Degrasyn and Bortezomib | MCL | Inhibit JAK/STAT3 and proteasome | [158] |
| Carfilzomib/Bortezomib/Dexamethasone/Lenalidomide and INCB052793 | MM | Inhibit JAK1 and proteasome, induce type I IFN | [159] |
| CYT387 and Melphalan/Bortezomib | MM | Inhibit JAK1/2 and proteasome | [160] |
| Antcin H and Methotrexate | BCL | Inhibit JAK and folic acid | [161] |
| csDMARDs and Methotrexate | NSHL, AML | Inhibit JAK and folic acid | [162] |
| Nilotinib and INC424 | CML | Inhibit JAK and Bcl-Abl | [163] |
| INK inhibitor and JAK inhibitor | ALCL | Inhibit JAK and INK | [165] |
| AZD1480 and UO126/PD98059 | HL | Inhibit JAK and MEK | [166] |
| Citarinostat and Momelotinib | Lymphoid malignancies | Inhibit JAK/STAT3 and HDAC6 | [167] |
| Romidepsin and Mechlorethamine | CTCL | Inhibit JAK and HDAC | [168] |
| INK128/Temsirolimus/Ruxolitinib and Luminespib | LPD | Inhibit JAK/STAT3, HSP90 and mTOR | [169] |
Abbreviations: ABC-DLBCL: activated B cell-like diffuse large B cell lymphoma, GC-DLBCL: germinal center-diffuse large B cell lymphoma, MCL: mantle cell lymphoma, FL: follicular lymphoma, SLL: small lymphocytic lymphoma, CLL: chronic lymphocytic leukemia, ATLL: adult precursor T acute lymphoblastic leukemia, MM: multiple myeloma, BCL: B cell lymphoma, NKTCL: natural killer/T-cell lymphoma, HL: Hodgkin lymphoma, ATL: adult T-cell leukemia, CTCL: cutaneous T-cell lymphoma, T-ALL: T cell acute lymphoblastic leukemia, cHL: classical Hodgkin lymphoma, MCL: mantle cell lymphoma, NSHL: nodular sclerosis Hodgkin’s lymphoma, AML: acute myeloid leukemia, CML: chronic myeloid leukemia, ALCL: anaplastic large cell lymphomas, LPD: lymphoproliferative disorder.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, B.; Wan, Q.; Li, Z.; Chng, W.-J. Janus Kinase Signaling: Oncogenic Criminal of Lymphoid Cancers. Cancers 2021, 13, 5147. https://doi.org/10.3390/cancers13205147
AMA Style
Li B, Wan Q, Li Z, Chng W-J. Janus Kinase Signaling: Oncogenic Criminal of Lymphoid Cancers. Cancers. 2021; 13(20):5147. https://doi.org/10.3390/cancers13205147
Chicago/Turabian StyleLi, Boheng, Qin Wan, Zhubo Li, and Wee-Joo Chng. 2021. "Janus Kinase Signaling: Oncogenic Criminal of Lymphoid Cancers" Cancers 13, no. 20: 5147. https://doi.org/10.3390/cancers13205147
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.