
Small Extracellular Vesicles and Metastasis—Blame the Messenger


Department for Internal Medicine, University Clinic Ulm, 89081 Ulm, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this paper as senior authors.
Cancers 2021, 13(17), 4380; https://doi.org/10.3390/cancers13174380
Submission received: 30 July 2021
/
Revised: 23 August 2021
/
Accepted: 26 August 2021
/
Published: 30 August 2021
(This article belongs to the Special Issue Exosome Biology for Nucleic Acid Medicine—From Bench to Bed)
Abstract
:Simple Summary
Due to their systemic nature, metastatic lesions are a major problem for curative cancer treatment. According to a common model for metastasis, tumor cells disseminate by local invasion, survival in the blood stream and extravasation into suitable tissue environments. At secondary sites, metastatic cells adapt, proliferate and foster vascularization to satisfy nutrient and oxygen demand. In recent years, tumors were shown to extensively communicate with cells in the local microenvironment and future metastatic sites by secreting small extracellular vesicles (sEVs, exosomes). sEVs deliver bioactive cargos, e.g., proteins, and in particular, several nucleic acid classes to reprogram target cells, which in turn facilitate tumor growth, cell motility, angiogenesis, immune evasion and establishment of pre-metastatic niches. sEV-cargos also act as biomarkers for diagnosis and prognosis. This review discusses how tumor cells utilize sEVs with nucleic acid cargos to progress through metastasis, and how sEVs may be employed for prognosis and treatment.
Abstract
Cancer is a complex disease, driven by genetic defects and environmental cues. Systemic dissemination of cancer cells by metastasis is generally associated with poor prognosis and is responsible for more than 90% of cancer deaths. Metastasis is thought to follow a sequence of events, starting with loss of epithelial features, detachment of tumor cells, basement membrane breakdown, migration, intravasation and survival in the circulation. At suitable distant niches, tumor cells reattach, extravasate and establish themselves by proliferating and attracting vascularization to fuel metastatic growth. These processes are facilitated by extensive cross-communication of tumor cells with cells in the primary tumor microenvironment (TME) as well as at distant pre-metastatic niches. A vital part of this communication network are small extracellular vesicles (sEVs, exosomes) with a size of 30–150 nm. Tumor-derived sEVs educate recipient cells with bioactive cargos, such as proteins, and in particular, major nucleic acid classes, to drive tumor growth, cell motility, angiogenesis, immune evasion and formation of pre-metastatic niches. Circulating sEVs are also utilized as biomarker platforms for diagnosis and prognosis. This review discusses how tumor cells facilitate progression through the metastatic cascade by employing sEV-based communication and evaluates their role as biomarkers and vehicles for drug delivery.
1. Tumor Metastasis and Nucleic Acid Cargo in Small Extracellular Vesicles
Cancer is a complex disease, that is driven both by the acquisition of genetic defects and environmental cues. The development of metastatic lesions is a major obstacle impeding curative treatment. The dissemination of cancer cells from a primary lesion to distant organs is referred to as the invasion-metastasis cascade. Due to their systemic nature and the limited treatment options, more than 90% of cancer deaths can be attributed to metastases and not to the primary tumor [1]. The metastatic cascade is a sequence of local and distant events, starting with invasion of primary tumor cells into the blood circulation by the loss of epithelial features, detachment of tumor cells, breakdown of the basement membrane (BM), migration and intravasation. Subsequently, tumor cells need to survive during transport in the circulation, followed by re-attachment and extravasation at the parenchyma of distant tissues [2]. Upon establishment in secondary sites, metastatic cells must adapt and proliferate in a new microenvironment, fostering vascularization to sustain nutrient and oxygen supply during the growth of macroscopic metastases. Each of these events is initiated by the deregulation of signaling cascades due to genetic alterations in cancer cells, but also by signaling from cells in the tumor microenvironment (TME), requiring a coordinated communication between the resident cell types [3]. Besides, tumors were shown to communicate over long distances via the circulation with future metastatic sites to facilitate establishment of suitable pre-metastatic niches (PMNs) [4]. On-going research has attributed a vital part of the coordinated crosstalk in the TME and during the formation of PMNs to small extracellular vesicles (sEVs) and the main, highly abundant sEV-population: exosomes [5,6,7]. sEVs are secreted in large quantities from cancer cells and were originally described as vehicles for cellular waste removal. Further research has indicated that sEVs can also function as vital mediators of intercellular communication by transferring a wide array of bioactive cargos, including proteins, lipids and many different nucleic acids species [8,9]. Two major cargo classes are predominantly involved in the regulation of the metastatic cascade by sEVs: proteins and nucleic acids. In particular, miRNAs and other non-coding RNAs (ncRNAs) were described as the major RNA species in sEVs, but transfer of functional full-length mRNAs was also reported in some instances [10,11]. Intercellular communication utilizing RNA species has been demonstrated during metastasis-associated processes in the TME, such as activation of tumor-associated fibroblasts, tumor cell migration, tumor progression, immunosuppression, angiogenesis and the establishment of organ-specific PMNs [12,13,14,15]. Nucleic acids in sEVs further have promising potential as cancer biomarkers. This includes analysis of sEV-RNAs, but also DNA fragments [16,17,18]. Thus, this review will summarize and discuss the role of sEVs during metastasis in general, including a potential role as diagnostic and prognostic biomarkers, as well as describe the vital contribution of prominent nucleic acid cargos, such as miRNAs during the metastatic cascade.
2. sEVs and Their Highly Abundant Exosome-sEV Sub-Population
sEVs are lipid bilayer-engulfed extracellular vesicles with a diameter of 30–150 nm [19]. Based on their size, sEVs are made up mainly by exosomes, but also a sub-population of microvesicles, such as arrestin domain-containing protein 1 (ARRDC1)-mediated microvesicles (ARMMs), with a diameter that reaches below 100 nm, was described [20]. Due to the high abundance of exosomes in the sEV group, this review is mainly focused on the role of exosomes in cancer progression and metastasis. For the sake of an easy communication with the reader, we have however attributed biological effects and in vivo functions to the broader specification “sEVs”, which is often used instead of the term “exosomes” in the literature [21]. The exosome-sEV population is formed as intraluminal vesicles (ILVs) in endosomal-derived multivesicular bodies (MVBs) and released at the plasma membrane. sEVs are present in various body fluids, such as blood, saliva or urine [22]. Omics characterization identified various sEV cargo subtypes, including proteins, lipids and nucleic acids [8,16,17,18,23]. Due to the endosomal origin of respective sEVs, cargo proteins implicated in biogenesis of ILVs, such as tetraspanins (CD9, CD63, CD81, CD82, CD53 and CD37), ALG-2 interacting protein X (ALIX) or the tumor susceptibility gene 101 protein (TSG101), are utilized as markers, e.g., tetraspanins are enriched around 100-fold in sEVs when compared to their parental cells [8,23]. It has to be noted that not all of these proteins are exclusively found in sEVs, and some markers can be detected in microvesicles as well [24]. Moreover, not all tetraspanins are always expressed in a specific cancer cell line. Therefore, the Kalluri group recently performed broad-spectrum mass spectrometry of sEVs across different cancer lines and reported that synthenin-1 was the ideal global marker for sEVs released from cancer cells [25]. sEVs are also enriched in annexins, small Rab-GTPases and lipid-raft-associated factors (e.g., flotillin), as well as major histocompatibility complex class I and II receptors (MHC I and MHC II), heat shock proteins (e.g., Hsp70 and Hsp90), cytoskeleton components (myosin, actin and tubulin), cell membrane receptors, such as integrins, and epidermal growth factor receptor (EGFR), but also cytokines or cytosolic components (e.g., signaling proteins, metabolic enzymes) [26,27]. In addition, sEVs were shown to have a conserved lipid composition that is required for sEV biogenesis, morphology and homeostasis upon uptake. They are enriched in cholesterol, sphingomyelin, glycosphingolipids, phosphatidylserine and ceramide [28,29]. sEVs also contain a large number of nucleic acid classes: mRNAs, miRNAs, long-non-coding RNAs (lncRNAs), circular RNAs (circRNAs), ribosomal RNAs (rRNAs), transfer RNAs (tRNAs) and other RNA subclasses, but DNA was also described [9,26,27]. Thus, sEVs are generated by almost every cell type using multiple biogenesis pathways under physiological and pathophysiological conditions, which vitally determines their respective cargo profiles [30,31].
2.1. sEV Biogenesis and Cargo Loading
Biogenesis of exosome-sEVs starts at the endosomal compartment by maturing early endosomes into late endosomes or MVBs, where membranes invaginate to generate ILVs [8,32]. Subsequently, MVBs can either fuse with lysosomes and degrade their content, or are transported along microtubules to the cell periphery, where they fuse with the plasma membrane to release ILVs as sEVs [33]. The biogenesis of ILVs can be facilitated by two major pathways: (1) The endosomal sorting complex required for transport (ESCRT) mediates ILV formation by the ESCRT machinery, which is assembled into four larger complexes, ESCRT-0, -I, -II and -III, and aided by the associated proteins AAA-ATPase VPS4, VTA1 and ALIX [34] (Figure 1). To facilitate packaging by the ESCRT complex, cargo proteins need to display post-translational modifications, such as ubiquitination [35]. The ESCRT-0 complex then binds these tagged cargos, segregates the cargo proteins into microdomains and coordinates the additional interaction of ESCRT-1 with these cargos [36,37]. ESCRT-1 in turn recruits ESCRT-2 to facilitate ILV formation and loading of cytosolic proteins or RNAs into ILVs. Subsequently, ESCRT-2 recruits ESCRT-3 to the newly formed vesicles to promote scission of the cargo-laden ILVs together with VPS4, while ubiquitin and ESCRT subunits are released in the cytosol for recycling [36,37,38].
(2) Additional studies have suggested that MVB biogenesis can also work without the ESCRT complex. It was reported that ILVs formed in MVBs even when vital ESCRT subunits were silenced, indicating ESCRT-independent biogenesis [39]. These mechanisms are dependent on tetraspanins and lipids, such as ceramide that is generated by neutral sphingomyelinase 2 (nSMase2). Thus, an inhibitor of nSMase, GW4869, was able to effectively reduce sEV-release in several studies [32,40,41,42]. Mechanistically, lipid-mediated ILV biogenesis is facilitated by spontaneous budding of limited membranes due to incorporation of ceramide, lysophospho- or glycosphingo-lipids. Interestingly, enzymatic conversion of ceramide to sphingosine and sphingosin1-phosphate (S1P), and thus activation of sphingosine1-phosphate receptors on limiting membranes, was also implicated in the sorting of tetraspanins into ILVs [43]. Tetraspanins are major sEV membrane markers, characterized by four transmembrane domains. At the plasma membrane, they are sequestered in tetraspanin-enriched microdomains (TEMs) and interact with a wide variety of associated factors, that can be recycled together with their tetraspanin binding partners into MVBs and eventually ILVs [44] (see Figure 1).
RNA is also loaded in ILVs by a lipid-mediated mechanism. To this end, specific RNA sequences increase the affinity for lipid structures, such as lipid rafts, hydrophobic lipids or sphingosine [45]. In addition, a number of RNA-binding proteins were reported to load mRNAs or miRNAs into sEVs [46]. Concerning miRNAs, Teng et al. demonstrated removal of the tumor-suppressor miR-193 from cells during colon cancer progression by packaging in sEVs, utilizing the miRNA-binding protein, major vault protein (MVP) [47]. Thus, sEVs are used by cancer cells in order to rebalance their content of tumor-suppressive and oncogenic miRNA populations [47].
Upon completion of ILV biogenesis, MVBs are transported along microtubules to the plasma membrane, where ILVs are released [48,49]. Here, several factors are involved on the way, which include small Rab family GTPases as molecular switches, microtubules and their associated regulatory proteins, molecular motors that transport the MVBs and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) that mediate final membrane fusion. In particular, the Rab-GTPases are vital regulators of vesicular trafficking and ILV budding or release, e.g., Rab27a/b or Rab11 regulate different aspects of ESCRT-dependent or independent sEV-release [9,50,51] (Figure 1). The efficient fusion and sEV-release at the plasma membrane also requires the presence of branched actin filaments. Branched actin is either stabilized or debranched by the actin-regulatory proteins Cortactin and Coronin-1, respectively [52]. Cortactin also facilitates Arp2/3-complex-dependent synergistic nucleation of branched actin filaments together with nucleation-promoting factors [53]. Our group has recently shown that Cortactin together with WAVE2 is required for sEV-release from pancreatic ductal adenocarcinoma (PDAC) cells. In this study, we have also described Protein Kinase D1 (PRKD1) as a vital inhibitory upstream regulator, that phosphorylates Cortactin at S298 [54,55]. When phosphorylation of this site was abrogated, Cortactin induced synergistic nucleation of branched filaments required for sEV-release, thus drastically increasing the respective sEV-secretion [54]. Interestingly, PRKD1 expression is reduced in a majority of PDACs and other invasive cancer cells [54,56]. In summary, sEV biogenesis can occur in an ESCRT-dependent and independent manner in a highly regulated process that involves the coordinated action of multiple cellular compartments as well as tight spatio-temporal control.
2.2. sEV-Uptake in Recipient Cells
Once released into the extracellular milieu or the circulation, sEVs can interact with recipient cells by different mechanisms. They can either bind to membrane receptors and activate specific signaling pathways, or cargo is transferred upon uptake into the respective cells. Mechanisms for uptake and delivery of active biomolecules include direct membrane fusion, clathrin- or lipid raft (Caveolae/caveolin-1)-mediated endocytosis, macro-pinocytosis and phagocytosis [57] (see Figure 1). In addition, molecular and cellular stress conditions were shown to modify sEV-release and -uptake rates, as well as their respective cargo composition [9].
2.3. Modifiers of sEV-Release in Tumor Cells
Tumor cells are associated with strongly increased sEV-release [58,59,60,61], which further display a significantly altered cargo profile to act as signaling hubs for intercellular communication during tumor metastasis. Thus, quantitative and qualitative changes in sEV populations were identified in the blood circulation of cancer patients, corroborating a function of sEVs as diagnostic and prognostic markers [62,63,64,65]. The molecular mechanisms involved in sEV trafficking and release are complex and not fully understood. However, it has become evident that cellular and molecular stresses, such as environmental cues and oncogenic transformation, have a crucial role in triggering sEV secretion [66,67]. One hallmark of the TME is low pH, which was shown to be a key factor for sEV-release, but also sEV-uptake in recipient cells [66]. Similar observations were made for hypoxic conditions, where the low oxygen concentrations not only quantitatively alter sEV-release, but also facilitate qualitative changes in the sEV cargo content to promote vascularization and cell proliferation [67]. Since ESCRT-dependent and independent pathways are major regulators of sEV biogenesis, interference with the respective pathways was able to potently modulate secretion [68,69,70,71]. A recent study has demonstrated that oncogenes found in many cancer entities, such as MYC, aurora kinase B (AURKB) and HRAS, trigger hyperactivation of ESCRT and ceramide pathways, as well as the inhibition of lysosome genes causing abundant sEV secretion. Again, the oncogenes mediated a shift in cargo composition, in particular for proteins and miRNA, thus promoting a pro-tumorigenic phenotype [71]. In another setting, oncogenic HRAS induced considerable release of sEVs from epithelial cell lines, which were shown to contain the whole cancer cell genome, including the mutant HRAS oncogene [72,73]. Moreover, several lines of evidence suggest that the KRAS oncogene was able to enhance sEV-release and modulate their functional cargo compared to wild-type KRAS. Mutated, oncogenic KRAS-sEVs were characterized by tumor-promoting proteins, including mutant KRAS, and an altered miRNA content, enabling oncogenic transfer and metabolic reprograming in recipient cells [74,75,76]. Different mechanisms are thought to be involved in RAS-dependent regulation of sEV-release, e.g., the activation of syndecan-1, as well as the RHO pathway, which have both functions in sEV biogenesis [77,78,79]. In addition, the Wnt protein WNT5A, an oncogene correlating with metastasis and poor prognosis, was able to trigger Ca2+-dependent release of sEVs with pro-angiogenic and immunosuppressive features [80]. There are opposing observations and hypotheses regarding the miRNA content of oncogene-induced sEVs. Several studies report the release of pro-tumor miRNAs through sEVs by cancer cells to reprogram recipient malignant and non-malignant cells, enabling cancer progression [81]. On the other hand, oncogene-induced sEVs were found to be enriched in miRNAs with tumor-suppressor functions, supporting the hypotheses that tumor cells eliminate undesirable cellular miRNAs as cargo in these sEVs [71,75].
Since nucleic acids and in particular miRNAs have vital functions during tumor progression and metastasis, we were prompted to focus this review on delineating the contribution of sEV-based nucleic acid transfer during progression through the metastatic cascade.
3. Role of sEVs in Local Stroma Invasion
The first steps of cancer metastasis are dictated by the local invasion from the primary tumor site into the surrounding stroma. Tumor cells need to breach the BM, the extracellular matrix (ECM) which separates epithelial from stromal compartments and acts as a barrier to invasiveness [82]. The detachment from the BM is initialized by the loss of cell–ECM and cell–cell adhesive contacts, such as different adherens and tight junctions that maintain cellular connections, e.g., by E-cadherin and claudins, which under normal conditions facilitate the formation of a tight epithelium. During cancer progression and invasion, these structures are weakened or disassembled, as a consequence of mutations and dysregulated signaling in order to allow the invasion into the surrounding stroma [83]. A process implicated in the disassembly of epithelial cell–cell interactions and acquisition of invasive features is epithelial-to-mesenchymal transition (EMT) [84].
3.1. Epithelial-to-Mesenchymal Transition (EMT)
During EMT, cells lose epithelial traits and transform into a motile, mesenchymal state. EMT can be observed during embryonic development and wound-healing processes, where the loss of adhesion properties, polarity and basement anchoring are important physiological events. However, when cancer cells activate the EMT program, they gain stemness and migratory abilities, which contribute to invasiveness and ultimately metastasis [85]. On a molecular level, EMT includes loss of epithelial markers, such as E-cadherin, and upregulation of mesenchymal proteins, e.g., vimentin, fibronectin, N-cadherin, as well as the change to a fibroblast-like cell [3]. The downregulation of E-cadherin and upregulation of mesenchymal markers during EMT is orchestrated by zinc-finger transcription factors, such as SNAIL, SLUG, TWIST and ZEB-1/2, downstream of growth factor signaling induced by hepatocyte growth factor (HGF), epidermal growth factor (EGF) and transforming growth factor beta (TGFβ), or Wnt/β-catenin pathways [84,86,87,88]. However, the transcriptional deregulation and induction of EMT is also prominently facilitated by uptake of sEVs, which modulate signaling of the respective pathways, e.g., upon transfer of mir-301a in sEVs, targeting p63 and thus release/activation of ZEB1/2 [89]. EMT is also promoted by the transfer of constitutively active β-catenin or Wnt ligands (Wnt1 and Wnt3a), as well as sEV-resident miRNAs mir-92a [90], miR-191 [14] and miR-1260b [91], implicated in modulating Wnt/β-catenin signaling. However, loss of epithelial characteristics and detachment from the ECM also pose a risk for cancer cells by triggering anoikis, a form of programmed cell death to prevent metastasis. Thus, cancer cells need to acquire anoikis resistance, which can occur via several mechanisms, such as the transfer of sEVs containing miRNA-210 and miR-222-3p, as shown for non-small-cell lung cancer (NSCLC) and gastric cancer cells (GC) [92,93].
3.2. ECM Degradation
The invasion of cancer cells is further supported by the degradation of the BM and other ECM structures in the tissue. Key players in ECM remodeling are matrix metalloproteinases (MMPs), whose activity is controlled by transcriptional and post-translational regulation. Cancer cells have acquired different mechanisms to deactivate this tight control of MMPs and render them highly active. Enhanced function of several MMPs was shown to regulate ECM stiffness via integrins and contributed to invasive phenotypes of cancer cells [94]. Moreover, MMP expression was shown to release growth factors, such as TGFβ, from the TME by influencing their bioavailability or functionality, thus enabling cell proliferation [95,96]. Interestingly, a study by Yokoi and colleagues in 2017 established that sEVs may play a crucial role in fostering MMP activity. Here, no MMP protein was found in sEVs, as shown in various studies [97,98,99], but the selective packaging of MMP1 mRNA into sEVs derived from ovarian cancer (OC) cells was found. These sEVs then triggered destruction of the peritoneal mesothelium and BM in vitro and in vivo, ultimately promoting metastatic behavior [100]. Several studies have found miR-21 to be overexpressed in different cancer types and demonstrated its transfer via sEVs to recipient cells [101,102,103]. miR-21 positively correlated with the expression of ECM mediators MMP-2, MMP-9 and MMP-11, due to the potent inhibition of phosphatase and tensin homolog (PTEN) and inhibitor of metalloproteinases 3 (TIMP-3) [102,104]. Similar effects have been established for miR-181b during carcinogenesis in hepatocellular carcinomas (HCC) and esophageal cancer (ESCC) [105,106].
4. sEV-Based Communication in the Tumor Microenvironment (TME)
Once cancer cells have breached the BM, they invade the stroma, where they are exposed to stromal cells. These non-malignant cells comprise fibroblasts, myofibroblasts, vascular and lymphatic endothelial cells, adipocytes and infiltrating immune cells [107]. The immediate ECM and cellular components of the stroma make up the TME. The classical theory suggests that upon cancer initiation, adjacent non-transformed stromal cells are recruited and reprogrammed, which is accompanied by extensive intercellular communication via cytokines, chemokines and vesicles to generate a dedicated favorable TME [108]. Chronic inflammation and wound-healing processes in an aberrant microenvironment also promote tumorigenesis [109,110,111]. Research in recent years has reinforced the crucial role of crosstalk between cancer cells and cellular components of their TME to create a supportive environment, thus enhancing the aggressive and invasive behaviors of tumors. Reprogrammed stroma cells enable the supply with essential nutrients in order to meet the high demand of proliferating tumor cells, but also to remodel the ECM and escape immune surveillance [107]. This communication is largely dependent on soluble factors and the transfer of sEVs. Tumor-sEVs or sEV-based crosstalk have been described for many cell types in the TME, such as cancer-associated fibroblasts (CAFs), which facilitate cancer progression either in an autocrine or paracrine fashion, e.g., by inducing EMT or stemness [112]. Infiltrating innate and adaptive immune cells, such as tumor-associated macrophages or Tregulatory cells (Treg), also generate an immunosuppressive TME [113]. Moreover, tumor angiogenesis is induced by tumor-derived-sEVs [114,115]. Once sEVs have entered the circulation, they were recently also described to foster metastasis in distant organs by establishing favorable PMNs [5,54,116].
4.1. sEV-Based Crosstalk with Cancer-Associated Fibroblasts (CAFs)
CAFs are the most common constituent of the TME. Their crosstalk with tumor cells and the extended tumor stroma influences invasion, metastasis as well as therapeutic responses. When normal fibroblasts (NFs) are converted into CAFs, they acquire the expression of specific protein markers, including fibroblast activation protein (FAP), alpha-smooth muscle actin (α-SMA) and fibronectin. Activated CAFs in turn secrete growth factors, e.g., vascular endothelial growth factor (VEGF), TGFβ, cytokines (IL6, IL10, IL1β), collagen and ECM-modifying enzymes, but also sEVs. These secreted mediators then act on tumor cells in a paracrine manner to alter the ECM and further facilitate tumor invasion across tissues [117]. sEVs have shown the ability to act on the TME and convert NFs into CAFs. A mechanism involved in cellular differentiation of fibroblasts to CAFs by sEVs is activation of the TGF-β/Smad pathway. This is either accomplished by direct transfer of growth factors on sEVs or by transcriptional reprogramming induced by the introduction of ncRNAs. In prostate cancer (PC) cells, the direct delivery of TGFβ to fibroblasts by sEVs initiated a phenotype resembling stromal cells from cancerous prostate tissue and the elimination of sEVs in vivo abolished CAF formation [6]. Similar observations were made for breast cancer (BC) [118], OC [119] and GC [120]. Additional examples for a role of sEVs in CAF conversion include melanosomes, sEVs derived from melanocytes, containing mir-211, which targets the tumor suppressor insulin-like growth factor 2 receptor in normal fibroblasts, resulting in the activation of mitogen-activated protein kinase (MAPK), thus enabling CAF reprogramming [12]. MiR-9 is upregulated in several BC cell lines and can be transferred via sEVs to recipient breast fibroblasts, thereby inducing CAF-like properties [121], whereas in PDAC, miR-155 was shown to activate NFs [122]. Once CAFs are transformed and activated in the TME, they also secrete a plethora of functional non-tumor sEVs with altered ncRNA composition and the ability to promote cancer progression as well as metastasis (Table 1). For example, sEVs released from CAFs were shown to regulate BC cell migration and invasion by transfer of mir-181d-5p, targeting CDC2 and HOXA5 to enhance proliferation, EMT and aggressiveness [123]. In addition to CAFs, the TME is also characterized by the infiltration of immunosuppressive cells, which help the tumors to avoid immune surveillance.
4.2. sEVs in Immune Suppression
Tumor-derived sEVs are targeted towards different innate and adaptive immune cells in the TME or at metastatic sites to facilitate immune evasion. Usually, antitumor immunity is triggered by the release of tumor-associated antigens (TAAs) and the subsequent activation of innate and adaptive effector cells, e.g., natural killer cells (NKs) and CD8+ Teffector cells. In particular, the activation of CD8+ Teffector or CD4+ Thelper cells was shown to be suppressed by tumor-derived sEVs, e.g., upon targeting antigen-presenting dendritic cells (DC), thus impairing lymphocyte activation and survival [162].
In immunologically cold tumors, such as PDAC, the TME is also spiked with a large number of immunosuppressive regulatory T-cells (Tregs), M2-polarized tumor-associated macrophages (TAMs) and immature myeloid-derived suppressor cells (iMDSCs), which inhibit functional CD8+ T-cell responses and impede proper antigen presentation by DCs or anti-tumor responses by M1-polarized macrophages [163].
Besides, sEVs secreted by tumor cells often reflect the parental protein composition, and thus were shown to present TAAs on MHC class I and II receptors. These sEV-based TAAs are able to stimulate NK- and T-cell-dependent cytotoxicity, but were further shown to decoy anti-TAA-antibodies and thus prevent complement-mediated cytotoxicity, impairing functional anti-tumor B-cell responses [164].
To escape immune surveillance, tumors have acquired diverse mechanisms that target the innate and adaptive immune defense by sEVs. The respective concepts, targeted cells and the role of nucleic acids are summarized in the following section. A more extensive overview is available in Table 1.
4.2.1. Innate Antitumor Defense—Natural Killer Cells (NKs)
NKs mediate the innate defense against tumors or infected cells [165]. The antitumor cytotoxicity of NKs can be blocked by inhibiting the activating NK cell surface receptor NK group 2-member D (NKG2D). To this end, membrane-bound ligands MICA, MICB and ULBP against NKG2D are transferred by sEVs, thus alleviating NK-mediated cytotoxic responses [166,167]. Hypoxic tumor MVs, comprising of tumor sEVs and microvesicles (MVs), also impaired NK cell cytotoxicity against different tumor cells in vitro and in vivo. This was mediated by EV-resident TGF-β1, decreasing the expression of NKG2D, while miR-23a synergistically targeted CD107a expression in NK cells. Another mechanism to impair NK cell function is the sEV-mediated transfer of the circular RNA:ubiquitin-like PHD and ring finger domain 1 (circUHRF1), which was associated with the suppression of miR-449c-5p and enhanced expression of TIM-3. Interestingly, in HCC patients, increased levels of plasma-circUHRF1 were also correlated with resistance to anti-PD-1 therapy [131]. The cytotoxicity of NK cells was further inhibited by tumor sEVs from different cancer cell lines [133] by suppressing the secretion of granzyme-B via miR-378a-3p.
4.2.2. M2-Polarized Macrophages
In addition to NKs, tumor sEVs facilitate the transformation (polarization) of TAMs into immunosuppressive M2 subtypes, e.g., sEVs derived from hypoxic PDAC cells convert macrophages to the M2 phenotype in a HIF1a- or HIF2a-dependent manner. miR-301a-3p is highly expressed in the respective cells as well as sEVs under hypoxic conditions. Upon uptake by macrophages, M2-polarization is induced via the activation of PTEN/PI3Kγ signaling. The respective macrophages in turn also promote tumor-invasive behavior. The circulating exosomal miR-301a-3p levels were thus positively associated with depth of invasion, lymph node metastasis, late TNM stage and poor prognosis in patients [132].
4.2.3. Adaptive Antitumor Immunity—Targeting Antigen-Presenting Dendritic Cells (DCs) and Anti-Tumor T-Cells
Tumors can also bypass surveillance by the adaptive immune system utilizing sEVs. Major targets include antigen-presenting DCs, CD8+ Teffector and CD4+ Thelper cell types, as well as regulatory Tregs, which are immunosuppressive and promote tumor growth and metastasis. For example, in PDAC, T-cell activation by antigen-presenting cells was impaired upon uptake of PDAC-derived sEVs in DCs, e.g., uptake of miR-203-containing PDAC-sEVs inhibited the expression of toll-like receptor 4 (TLR4), tumor necrosis factor α (TNF-α) and interleukin IL-12, inducing DC dysfunction [136,168]. PDAC-sEVs can also specifically impact the activation of CD4+ T-cells by DCs. This was mediated by inhibiting the expression of the regulatory factor X-associated protein (RFXAP) with miR-212-3p, resulting in compromised MHCII expression. Such a phenotype could also be validated in the tissue of PDAC patients [134]. In addition to tumor cells, Tregs possess the ability to produce sEVs that induce DC tolerogenic phenotypes. Mechanistically, this was attributed to the sEV miRNA cargos miR-150-5p and miR-142-3p, that are transferred with Treg-derived sEVs, which trigger IL-10 and IL-6 production in tolerogenic DCs to limit T-cell responses [135].
Additionally, the different T-cell populations are impacted by various sEVs-dependent mechanisms. This includes induction of apoptosis, as well as inhibition of CD8+/CD4+ T-cell activation, expansion and conversion towards immunosuppressive Treg phenotypes [169]. Apoptosis in CD8+ T-cells is mediated by different proteins and nucleic acid cargos, e.g., the interaction between Fas ligand (FasL) on sEVs and Fas receptors (CD95) on the respective T-cell clones, resulting in caspase activation [170]. The presence of FasL on sEVs was reported for different cancer entities, such as melanoma [171], PC [172] as well as head and neck cancer [173]. In melanoma, tumor sEVs also induce the mitochondrial apoptotic pathway in CD4+ T-cells, and this is mediated by transfer of miRNA cargos miR-690, miR-677 as well as miR-29b [130]. Another mechanism of immune evasion involves binding of programmed death-ligand 1 (PD-L1) expressed on tumor cells and other cells in the TME to its receptor programmed cell death protein 1 (PD-1) on activated T-cells, thus triggering T-cell exhaustion by suppressing activation and expansion [174,175]. Interestingly, tumor-derived sEVs were also described to transfer PD-L1 to other cell populations in the TME, thereby amplifying immunosuppression. Metastatic melanomas release PD-L1-positive sEVs, which enable tumor growth due to the suppression of CD8+ T-cell cytotoxicity. The level of PD-L1 in circulating sEVs could even be utilized for the stratification of early-stage responders and non-responders after anti-PD-1 therapy [176]. Release of PD-L1-positive sEVs has been observed for BC [177], NSCLC [178], glioblastoma (GBM) [179], GC [180] as well as other cancer entities. In cervical cancer (CeC), increased PD-L1 levels can also be traced back to the upregulation of miR-18a, which targets PTEN and SOX6 [137], whereas in HCC, miR-23a-3p induced elevated PD-L1 levels in macrophages [138]. For both miR-18a and miR-23a-3p, transfer by tumor sEVs has been reported [139,181]. sEVs-based signaling further interfered with T-cell polarization and cytokine release. Here, sEV-resident miRNAs, hsa-miR-24-3p, hsa-miR-891a, hsa-miR-106a-5p, hsa-miR-20a-5p and hsa-miR-1908, are upregulated in the serum of patients with nasopharyngeal carcinomas. The respective sEVs impeded T-cell proliferation and Th1/Th17 differentiation in vitro, accompanied by decreased levels of IFNγ, IL-2 and IL-17, while on the other hand, immunosuppressive Tregs were promoted. This was facilitated by altered mitogen-activated protein kinase (MAPK) and STAT signaling [129].
4.2.4. Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs are immature myeloid cells with immunosuppressive features that are present in the TME of many tumors. MDSCs decrease the cytotoxicity of effector immune cells and increase Treg cell responses, thus contributing to cancer progression. In several cancer subtypes, circulating MDSC levels were shown to correlate with the clinical stage and therapeutic response in patients [182].
A study by Guo and colleagues established a role for glioma cell-derived sEVs in potentiating the activation of MDSCs, targeting RAR-related orphan receptor alpha (RORα) and PTEN with miR-10a and miR-21. These findings were further corroborated by inoculation experiments in mice, where glioma cells with miR-10a or miR-21 knockout generated a lower number of MDSCs than normal glioma cells [140]. In PDAC, loss of the tumor suppressor SMAD4 was associated with poor prognosis as well as production of sEVs containing miR-1260a and miR-494-3p. The respective sEVs were reported to reprogram granulocytic and monocytic g/mMDSCs to bolster proliferation and glycolysis, thus facilitating the establishment of an immunosuppressive TME [183].
In summary, tumor sEVs and nucleic acid cargos have vital functions in evading immunosurveillance by the innate and adaptive immune system, fostering tumor growth and progression towards metastasis.
4.3. sEVs in Angiogenesis
As tumors grow and disseminate, they are critically dependent on access to blood vessels. Angiogenesis is a multi-step process initiated during carcinogenesis to form new blood vessels from pre-existing ones. Angiogenesis facilitates the adequate supply of oxygen and nutrients, as well as the removal of waste products from cancer cells, and is therefore essential for tumor proliferation and metastasis [184]. Hypoxic and acidic conditions, encountered in the TME of most tumors, promote new vascularization due to the release of pro-angiogenic factors, such as VEGF [185]. Tumor-derived sEVs are further implicated in pro-angiogenetic signaling, e.g., sEVs from GBM stem-like cells were shown to transport VEGF-A to brain endothelial cells in order to activate the VEGF pathway, initiating blood vessel growth and enhanced vascular permeability [114]. The presence of VEGF has also been reported in melanoma-derived sEVs, thus enhancing the angiogenic capacity of respective tumors [80]. Besides VEGF, tumor-derived sEVs transfer many more proteins, e.g., MMPs [186], involved in blood vessels’ formation. Moreover, several miRNAs present in tumor-derived sEVs were found to prominently facilitate angiogenesis in endothelial cells (Table 1). miR-17-5p is loaded in sEVs of nasopharyngeal carcinoma cells suppressing the transmembrane protein BMP and activin membrane-bound inhibitor (BAMBI). As a consequence, expression of AKT/VEGF-A and thus angiogenetic activity of HUVEC cells was increased. A positive correlation between miR-17-5p in the serum of nasopharyngeal carcinoma patients and tumor angiogenic activity further demonstrated the importance of this miRNA in vivo [115]. In GC, sEV-resident miR-155 was show to target Forkhead box O3 (FOXO3a) as well as c-MYB, thus enhancing the expression of VEGF in vascular cells to induce angiogenesis and tumor growth [150]. In PDAC, mir-27a in sEVs induced vascularization through inhibition of BTG anti-proliferation factor 2 (BTG2) [13]. Interestingly, sEVs secreted under hypoxic conditions showed the greatest potential to promote blood vessel formation, when compared to normoxic sEVs due to their enrichment of pro-angiogenic miRNAs, such as miR-135b, miR-210, miR-21 and miR-23a [151,152,153,187]. In addition to miRNAs, other ncRNAs are also packed in sEVs to foster tumor angiogenesis. In OC, high expression of the lncRNA MALAT1 (metastasis-associated lung adenocarcinoma transcript-1) was correlated with angiogenesis and metastasis, and MALAT1 was found in sEVs, which induce pro-angiogenic gene expression upon uptake in HUVEC cells [154].
Additionally, lymphatic vessels play crucial roles in tissue homeostasis, fluid balance, immune function as well as transport of metabolic molecules, and are therefore important for tumor proliferation [188]. Furthermore, they lack the presence of a complete BM and are more permeable to tumor cells, thus fostering metastasis [189]. Tumor cells have been shown to secrete growth factors such as VEGF-C or FGF to induce the formation of new lymphatic vessels (lymph-angiogenesis), correlating with lymph node as well as distant metastasis [190,191,192]. Besides, tumor-derived ncRNAs were shown to influence lymphatic vessel growth. miR-221-3p packaged into sEVs of CeC was identified to promote expansion of lymphatic vessels by targeting vasohibin-1 (VASH-1), a negative regulator of lymph-angiogenesis [156]. Additionally, the lncRNA lymph node metastasis-associated transcript 2 (LNMAT2) in sEVs secreted from bladder cancer was reported to facilitate lymph-angiogenesis via VEGF-C-independent mechanisms [157].
5. Intravasation of Tumor Cells and Survival in the Circulation
One of the most crucial steps enabling metastasis is the intravasation of tumor cells into the blood circulation. Neo-angiogenesis and lymph-angiogenesis induced by tumors, as well as tumor-derived sEVs, favor this process, since the newly developed vasculature is more prone to leakiness [193]. Thus, a weak interaction between endothelial cells facilitates transmigration of cancer cells through the endothelial monolayer into the circulation. Additionally, intravasation is promoted by TGFβ, enhancing penetration of tumor cells through microvessel walls [194], as well as TAMs, which foster paracrine interactions with tumor cells and an altered microvessel density [195]. Moreover, sEV-derived miRNAs, which disrupt endothelial stability, promote the process of tumor cell intravasation (Table 1). For example, miR-939 transferred by BC-sEVs targeted vascular endothelial (VE) cadherin, the main constituent of adherens junctions, causing enhanced leakiness of blood vessels, whereas miR-105 downregulated the tight junction protein ZO-1 [147,148]. Upon their successful intravasation, cancer cells can disseminate widely in the blood stream as circulating tumor cells (CTCs) and travel to distant sites, where they seed metastatic lesions. However, CTCs must survive in the circulation, and this is challenged by a variety of stress conditions, such as risk of anoikis in the absence of anchorage, shear forces and immunologic surveillance [196]. CTCs can be detected as single cells or dense clusters from 2 up to >100 cells with strong cell–cell junctions [107]. The aggregation of CTCs is of advantage, since clustered cells are protected from shear stress and anoikis. Thus, the presence of CTC clusters is associated with a poor prognosis and an earlier onset of metastasis [197,198,199]. CTC clusters directly derive from the primary tumor or develop via intravascular aggregation of single CTCs [200,201]. In HCC, tumor-derived sEVs were shown to transfer SMAD Family Member 3 (SMAD3) protein and mRNA to detached HCC cells and promote their homotypic adhesion [202]. Furthermore, sEVs have the ability to indirectly facilitate CTC clustering via the regulation of adhesion proteins, as well as the conversion of fibroblasts into CAFs, which also play a major role in cluster formation [203,204,205,206]. Furthermore, CTCs directly interact with blood platelets, which then accompany cancer cells and protect them from recognition and lysis by NKs [207,208]. The importance of platelets was further corroborated, since their depletion inhibited the formation of metastasis in mice [209]. This phenomenon can also lead to an imbalance in the normal blood clotting, which is often observed in cancer patients with a poor prognosis [210,211,212]. Thrombotic events and the activation of platelets in cancer patients are strongly correlated with the overexpression of tissue factor (TF), which can be transferred to endothelial cells via tumor-derived sEVs. The ECs are then reprogrammed into a procoagulant phenotype and generate high levels of thrombin, one of the most important activators of platelets [213]. In turn, platelets secrete sEVs with pro-metastatic cargoes, such as TGFβ, integrins, P-selectin and glycoprotein IIb–IIIa, that reprogram endothelial cells, leukocytes and tumor cells [214,215,216]. Moreover, the aggregation of platelets and inhibition of coagulation inhibitors are triggered by the formation of neutrophil extracellular traps (NETs), that can also be induced by tumor-derived sEVs [217].
At the moment, the influence of sEV-derived miRNAs during the clustering of CTCs as well as the activation of platelets and NETs is unclear, and further research is needed to unravel potential mechanisms. However, several miRNAs contained in sEVs play a crucial role in protecting CTCs in the blood stream from being recognized by the immune surveillance due to suppression of immune cells or recruitment of Tregs, which is reviewed as part of Section 4.2.
6. Extravasation from the Circulation
Extravasation occurs when CTCs transmigrate through the endothelial wall in order to enter the parenchyma of distant tissues. This process is dependent on enhanced vascular permeability and the disruption of cell–cell junctions, which normally maintain a physical barrier for fluid proteins and cells. Several secreted factors from cancer cells are involved in the disruption of endothelia barriers, e.g., TGFβ, angiopoetin-like 4 (ANGPTL4), VEGF, MMPs and ADAM12 [218,219,220]. Additionally, the association of platelet-cancer cell hybrids to the vessel wall is a step in the extravasation of CTCs, and this is guided by interactions of platelet selectins with ECs [221]. As described for intravasation, several miRNAs in sEVs support transendothelial migration by weakening endothelial cell–cell junctions, thus enabling vascular permeability (Table 1). However, the question arises if extravasation occurs just randomly or if CTCs are specifically directed towards distinct organs niches, facilitating organotropic metastasis.
7. sEV-Mediated Organotropism and Formation of Pre-Metastatic Niches
The concept of organotropism describes the homing of circulating tumor cells to specific organs as a consequence of complex tumor–stroma interactions. Although organotropic metastasis was first hypothesized 1889 by Stephen Paget, the exact underlying mechanism is not yet fully unraveled. Recent studies have indicated that sEVs may play a crucial role in organotropism. sEVs prepare PMNs by recruiting bone marrow-derived cells (BMDCs), endothelial progenitor cells and mesenchymal cells, but also induce the upregulation of proinflammatory molecules and facilitate vascular leakiness to create a suitable niche environment [222,223]. These alterations in distant organs are already observed before the arrival of cancer cells. Interestingly, various studies indicate that sEVs from different types of tumors preferably migrate to distinct organs, e.g., melanoma sEVs to sentinel lymph nodes, BC-sEVs to the lung and PDAC-sEVs to the liver [224,225,226]. Thus, the question arises of how sEVs are directed to specific sites to enable organotropic metastasis. In 2015, Hoshino et al. established that the expression of integrin patterns in sEVs and their interaction with ECM molecules, such as laminin and fibronectin, is crucially important for governing the formation of organ-specific PMNs. Integrins α6β4 and α6β1 were shown to promote lung tropism, and their uptake by lung fibroblasts triggered expression of the pro-inflammatory S100A proteins, heat shock proteins (HSPs), laminin and fibronectin, as well as annexin A6 and CD44 [5,227,228,229]. sEVs positive for integrins avß5 and macrophage migration-inhibitory factor (MIF) favor liver organotropism, a common site for metastasis associated with a poor prognosis in many types of cancer [226]. These sEVs bind to the fibronectin-enriched ECM in the liver and educate liver-resident macrophages (Kupffer cells) to increase the expression of TGFβ. High TGFβ levels in turn activate the secretion of fibronectin and pro-inflammatory mediators from hepatic stellate cells, thus preparing the PMN for the tumors [5,226].
Our group has recently observed that in PDAC, PRKD1 expression is downregulated compared to non-tumor tissue. Impaired PRKD1 expression was associated with increased sEVs secretion from different PDAC cell lines as well as altered expression and loading of sEVs with integrin α6β4. The respective sEVs also displayed low levels of integrin β5, impairing the formation of functional αvβ5 dimers, due to transcriptional downregulation of integrin β5 expression upon PRKD1 loss. In line with the model of Hoshino et al., injection of PRKD1KO-sEVs from Panc-1 cells in subcutaneously xenografted mice promoted lung metastasis, and this was corroborated in an autochthonous Prkd1KOKC mouse model, were predominant lung and no macroscopic liver metastasis was detected [54].
Interestingly, another mechanism for sEV-mediated liver metastasis was recently established by Zhang et al. Here, the transfer of EGFR by GC-sEVs into the liver facilitated HGF activation via miR-26a/b suppression. HGF was shown to bind c-MET on cancer cells and thus rendered the liver a preferable metastatic site for multiple tumors [230].
The establishment of bone metastasis can be observed for many solid tumors and often occurs in advanced disease stages. In bone metastasis, the homeostatic balance between bone-producing osteoblasts and bone-resorbing osteoclasts is disrupted and metastatic lesions are either osteolytic or osteoblastic. There are various studies assigning a crucial role for sEVs in inducing said imbalance, e.g., via transfer of amphiregulin, causing osteoclastogenesis through upregulation of receptor-activator of NF-κB ligand (RANKL), a mechanism that is observed in NSCLC [231].
Although most studies have defined a crucial role for integrins and other protein cargos in the establishment of organ-specific PMNs, a few miRNAs are also known to contribute to organotropic metastasis. One important mediator is miR-141-3p, and its presence in sEVs was associated with metastatic PC, promoting osteoblast activity and bone metastasis [15]. Similarly, miR-940 from PC and BC cells was reported to facilitate osteoblastic lesions by targeting Rho-GTPase-activating protein 1 (ARHGAP1) and family with sequence-similarity 134, member A (FAM134A) [158]. In the PMN, sEV-derived miRNAs further reprogram non-tumor stromal cells, promoting a pro-tumor environment. Thus, alterations in glucose metabolism are frequently observed in the PMN in order to fuel nutrient demand of incoming cancer cells. Here, miR-122 from BC-sEVs was described to target and downregulate pyruvate kinase, in order to impair glucose uptake in lung- and brain-resident cell populations to supply cancer cells with the remaining glucose [116]. Similar to the primary tumor site, the sEV-induced conversion of fibroblasts into CAFs is also observed in the PMN. Relevant nucleic acid targets are listed in Table 1. One specifically important cargo for the lung PMN is miR-1247-3p, that is secreted in sEVs from highly metastatic HCC and induces the activation of NF due to the upregulation of NFκB signaling, enabling the release of pro-inflammatory cytokines to further niche formation [159].
In summary, PMN formation has emerged as a vital step during metastasis and is crucially determined by tumor-derived sEVs. Although most of the important sEV cargos involved in the regulation of niche establishment are proteins, some miRNAs have been described to facilitate or aid niche formation, and thus more research is needed to unlock the full potential of nucleic acids in the regulation of organotropic metastasis.
8. sEVs as Biomarker Platforms and Therapeutic Vehicles
Since sEVs are representative for their cells of origin and are released in large quantities by cancer cells in the bloodstream [22,64], they are ideal biomarker platforms for liquid biopsy to facilitate early detection and prognosis [62,63]. Different sEV cargo classes can be utilized as promising prognostic and diagnostic biomarkers upon liquid biopsy, e.g., proteins, lipids and nucleic acids are intensively investigated. For example, glypican-1 was identified as a promising diagnostic protein biomarker for PDAC [232], but the ratio of lipids in plasma or serum sEVs was also used for cancer diagnosis and prognosis (reviewed in [233]). To this end, specific ratios of lysophosphatidylcholine (LPC), phosphatidylcholine (PC) and phosphatidylethanolamine (PE) in sEVs were associated with the tumor stage of PDAC, and PE was even linked to overall patient survival [234]. However, this review mainly focuses on the role of nucleic acids, and in particular ncRNAs in sEVs, as markers for diagnosis and prognosis in different cancer entities. A major obstacle for the use of sEVs as reliable biomarkers in the clinical routine is reproducibility and specificity due to different isolation and detection methods. sEV isolation for biomarker studies so far is not standardized, and each method, such as ultracentrifugation, sucrose gradients, size exclusion chromatography, affinity-based purification, isolation by asymmetric flow field–flow fractionation or microfluidic devices, has its own advantages and downsides [235,236,237]. This has become evident in the most studied sEV marker for PDAC diagnosis, glypican-1. Glypican-1 was discovered by Melo et al. in animal and human cell lines and was described to demonstrate a sensitivity/specificity of 100%, upon detection by transmission electron microscopy on sEVs. Detection of glypican-1-positive sEVs by ELISA reduced the sensitivity and specificity to 82.14% and 75%, respectively [232]. A validation study with alternative sEV-purification techniques after sampling sEVs from peripheral or portal vein blood has reported a drastically reduced sensitivity of 64%, whereas the specificity was still at 90%. Nevertheless, this was more sensitive than fine-needle biopsy and the currently used clinical tumor marker CA19-9 [238]. However, another attempt as part of a study using ELISAs to detect sEVs identified no significant difference in glypican-1 for PDAC patients with respect to benign pancreatic conditions. Thus, further validation and standardization of sEV-purification as well as detection methods is vitally required to allow for routine clinical use of sEVs as diagnostic and prognostic biomarkers [239]. However, sEV-based analysis of biomarkers also has advantages. The nanovesicles protect proteins from proteolytic cleavage and prevent degradation of enclosed nucleic acids [30]. This is why a large number of miRNAs, but also other ncRNAs, in sEVs have been characterized as biomarkers for prognosis and treatment response during tumor progression and metastasis (see Table 2), e.g., mir-21 in sEVs has been associated with lymph node metastasis in PDAC [240], bone metastasis in BC [241], tumor spinal/ventricle metastasis in glioma [242], lymph node metastasis in laryngeal squamous cell carcinoma (LSCC) [243], metastasis in general in ESCC [243], as well as in multi-miR panels with peritoneal metastasis of GC [244], and metastasis in PC [245] or CRC [246]. In addition to RNA, sEV-DNA was also described as a biomarker for the detection of cancer-specific mutations, since sEV-DNA fragments were shown to stochastically represent the entire genome of cancer cells, including mitochondrial DNA [247,248]. Several studies indicated that PDAC-sEVs could be used to probe the mutational landscape of tumors, e.g., for KRAS or TP53 [17,18,249], and an increased mutational allelic frequency in the pool of sEV-DNAs was even correlated with poor prognosis and survival [18,249]. Moreover, to further increase specificity and sensitivity during EV analysis of biomarkers, a strategy for the tumor-specific enrichment of sEVs has been developed [250] for PDAC. Here, a panel of six surface markers on sEVs has been identified to immuno-purify PDAC-specific sEVs after liquid biopsy, thus enabling a more sensitive detection of mutated KRAS alleles [251]. Similar strategies may be applied to enrich sEVs from other cancer entities and different prognostic cargos. As shown in Table 2, nucleic acids, and in particular ncRNAs, in sEVs have a great potential to act as biomarkers for tumor metastatic behavior.
In addition to a role as biomarkers, sEVs have been proposed as therapeutic vehicles, but practical applications are still in their early development phase. sEVs have been explored as novel drug delivery agents due to their inherent non-toxic, biodegradable properties and their ability to cross endogenous barriers, such as the blood–brain barrier [252]. This was demonstrated using engineered sEVs loaded with siRNAs, which targeted the central nervous system (CNS) by expressing Lamp2-RVG on their surface to specifically knockdown genes in the CNS after systemic administration [253]. Another promising study by Kamerkar et al. in 2017 modified sEVs from fibroblast-like mesenchymal cells with siRNAs or shRNAs against mutated and wild-type KRASG12D (iExosomes), and the treatment of mice with PDAC tumors in a KrasG12D background demonstrated a significant reduction in tumor size, compared to the control. Interestingly, tumors treated with iExosomes also showed superior size reduction with respect to liposomes loaded with the same cargo [254]. A corresponding clinical study is currently on the way as part of a Phase I trial in PDAC patients with a KRASG12D mutation (NCT03608631). Cancer progression and metastasis in different cancer entities is often associated with downregulation of tumor-suppressor miRNAs. The re-introduction of such miRNAs as EV-based therapies may thus reduce tumor proliferation and invasion. For efficient treatment, the mode of delivery is also a crucial factor, e.g., intra-tumoral injection of miR-335-containing EVs caused cancer inhibition in hepatocellular carcinoma xenograft models [255]. EVs for therapeutic approaches can be obtained from different sources, such as fibroblasts [256], stromal cells [257], red blood cells [258] and umbilical cord stem cells [259], which are subsequently loaded with therapeutic miRNA. NK cell-derived sEVs with miR-186 were used to impair immune escape of neuroblastoma [260], whereas red blood cell-sEVs loaded with miR-125b were shown to inhibit cell proliferation of leukemia and BC in xenografted mice [258]. As discussed earlier, concepts for targeted drug-delivery using engineered sEVs are still in development. It will be necessary to further optimize sEVs to exploit natural tropisms for specific organs and cell populations, or modify sEVs with artificial targeting constructs to increase specificity and uptake in recipient cells. In addition, strategies for loading of sEVs with cargos, e.g., specific siRNAs by electroporation [261], have to be adapted and optimized to increase efficacy for future clinical applications.
9. Conclusions and Perspectives
Here, we have discussed the role of sEVs and the important major cargo class, nucleic acids, in the progression of tumors through different aspects of the metastatic cascade (Figure 2, Table 1), as well as their role as biomarker platforms and vehicles for treatment (Table 2). Tumors release large quantities of sEVs with an altered cargo profile, and this is directed by genetic alteration as well as environmental cues, such as low pH or hypoxia (Section 1 and Section 2). Oncogenes, e.g., mutated KRAS, not only trigger activation of ESCRT and ceramide synthesis pathways to drive enhanced sEV-release from tumor cells, but also change the sEV miRNA content, enabling oncogenic transfer and metabolic reprograming (Section 2.3). Thus, it would be interesting to systematically assess how cancer-relevant genes and mutations affect sEV secretion as well as alterations of cargo content. Nucleic acids, in particular ncRNA cargo, such as miRNAs are vital cargos for the education of recipient cells during metastasis (Table 1). Extensive research in the last years has indicated that tumor sEVs and sEV-based crosstalk with other cell populations in the TME influence local invasion of tumor cells by facilitating EMT, ECM remodeling, stroma reprogramming, immune evasion and angiogenesis (Section 3 and Section 4). In addition, sEVs have been implicated in the establishment of distant organ-specific PMNs with a pro-inflammatory microenvironment (Section 7, Figure 2, Table 1). The intercellular communication network enabled by sEVs, as well as their systemic distribution, are vital factors in determining whether tumor cells successfully metastasize. It has to be noted, however, that the same sEV-cargos, in particular miRNAs, may have different functions dependent on the investigated cancer entities, e.g., miR-21 drives various routes of metastasis, when compared to BC, PDAC or glioma [240,241,242]. Nevertheless, ample evidence has indicated that circulating sEVs can be employed as effective biomarker platforms for diagnosis or prognosis (Section 8, Table 2). sEVs are released in all body fluids and are thought to generally reflect the state of their parental cells [22]. Multiple liquid biopsy studies have indicated that sEVs, and in particular miRNAs, can be used as biomarkers for various cancer subtypes, disease progression, metastasis as well as treatment response (Section 8, Table 2). Moreover sEV-DNA fragments, which statistically cover the entire genome and mutational landscape, were used to assess mutated genes in tumors as prognostic markers (Section 8). sEVs are ideal vehicles in this context, since they protect nucleic acid cargos from degradation. However, there are also caveats, which have so far prevented the broad adoption of sEV profiling in the clinical routine. Different isolation techniques and detection methods have yielded drastically different results, and thus rigorous standardization will be required to promote clinical sEV analysis (Section 8). Another issue might be the heterogeneity of sEVs in liquid biopsy samples, which are not only derived from tumor cells but many other cell types [251]. Thus, to increase specificity and sensitivity, first, steps towards immuno-enrichment of tumor-specific sEVs have been conducted by identifying and utilizing a panel of surface markers to enrich PDAC-derived sEVs [251]. Interestingly, sEVs can also be engineered as vehicles for delivery of therapeutic agents, such as siRNAs and miRNAs to target cancer cells in order to downregulate oncogenes or tumor-promoting factors. For example, transfer of miR-122 was shown to induce chemosensitivity in HCC [280], whereas sEVs loaded with siRNAs against mutated KRASG12D impaired PDAC tumor growth in mice [254]. The latter study has even resulted in a first phase I clinical trial [254]. However, research on therapeutic sEVs is only at the beginning. Once established and transferred into the clinic, these approaches have immense potential and will open up new avenues to promising treatment options, as shown for sEVs loaded with drugs, such as paclitaxel or doxorubicin [281,282,283]. A major necessity for the design of new sEV-based therapeutic options is improved understanding of molecular mechanisms for sEV-mediated tumorigenesis and metastasis. Additionally, technical issues need to be optimized, such as modification of sEV with cell-specific targeting constructs [284,285], or how natural tropisms of different sEV populations may be exploited to further target and improve uptake in tumor cells [15,286,287]. Nevertheless, sEV research over the last years has greatly contributed to a better understanding of the complex mechanisms that drive tumor progression and metastatic dissemination, and these studies will hopefully help to translate sEV-based applications into clinical use.
Author Contributions
Conceptualization, T.S. (Thomas Seufferlein) and T.E.; collection and review of the literature, T.S. (Tanja Seibold), M.W. and T.E.; writing, T.S. (Tanja Seibold) and T.E.; review and editing, T.S. (Tanja Seibold), T.S. (Thomas Seufferlein) and T.E. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by DFG-GRK-2254 (HEIST project No. 288342734) to T.S. (Thomas Seufferlein) and T.E., as well as DFG project No. 380319649 (EI792/7-1 and BL-1186/5-1) to T.E.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Dudas, J. Supportive and rejective functions of tumor stroma on tumor cell growth, survival, and invasivity: The cancer evolution. Front. Oncol. 2015, 5, 44. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Thuma, F.; Zöller, M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin. Cancer Biol. 2014, 28, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D′Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Redis, R.S.; Calin, S.; Yang, Y.; You, M.J.; Calin, G.A. Cell-to-cell miRNA transfer: From body homeostasis to therapy. Pharmacol Ther. 2012, 136, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Dror, S.; Sander, L.; Schwartz, H.; Sheinboim, D.; Barzilai, A.; Dishon, Y.; Apcher, S.; Golan, T.; Greenberger, S.; Barshack, I.; et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol. 2016, 18, 1006–1017. [Google Scholar] [CrossRef]
- Shang, D.; Xie, C.; Hu, J.; Tan, J.; Yuan, Y.; Liu, Z.; Yang, Z. Pancreatic cancer cell-derived exosomal microRNA-27a promotes angiogenesis of human microvascular endothelial cells in pancreatic cancer via BTG2. J. Cell Mol. Med. 2020, 24, 588–604. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma cell-derived exosomes promote epithelial-mesenchymal transition in primary melanocytes through paracrine/autocrine signaling in the tumor microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Li, S.L.; Ma, Y.Y.; Diao, Y.J.; Yang, L.; Su, M.Q.; Li, Z.; Ji, Y.; Wang, J.; Lei, L.; et al. Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget 2017, 8, 94834–94849. [Google Scholar] [CrossRef] [Green Version]
- Thind, A.; Wilson, C. Exosomal miRNAs as cancer biomarkers and therapeutic targets. J. Extracell. Vesicles 2016, 5, 31292. [Google Scholar] [CrossRef]
- Allenson, K.; Castillo, J.; San Lucas, F.A.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol 2017, 28, 741–747. [Google Scholar] [CrossRef]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated with Outcomes of Patients with Pancreatic Cancer. Gastroenterology 2019, 156, 108.e104–118.e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Wolfram, J.; Srivastava, S. Extracellular Vesicles in Cancer Detection: Hopes and Hypes. Trends Cancer 2021, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nature Commun. 2018, 9, 960. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Théry, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer-implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugeratski, F.G.; Hodge, K.; Lilla, S.; McAndrews, K.M.; Zhou, X.; Hwang, R.F.; Zanivan, S.; Kalluri, R. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat. Cell Biol. 2021, 23, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. Exosomes as a Multicomponent Biomarker Platform in Cancer. Trends Cancer 2020, 6, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H. The ESCRT complexes. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.B.; Oestreich, A.J.; Winistorfer, S.; Nguyen, D.; Payne, J.A.; Katzmann, D.J.; Piper, R. ESCRT ubiquitin-binding domains function cooperatively during MVB cargo sorting. J. Cell Biol. 2009, 185, 213–224. [Google Scholar] [CrossRef]
- Meister, M.; Banfer, S.; Gartner, U.; Koskimies, J.; Amaddii, M.; Jacob, R.; Tikkanen, R. Regulation of cargo transfer between ESCRT-0 and ESCRT-I complexes by flotillin-1 during endosomal sorting of ubiquitinated cargo. Oncogenesis 2017, 6, e344. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Babst, M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr. Opin. Cell Biol. 2011, 23, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Tschuschke, M.; Kocherova, I.; Bryja, A.; Mozdziak, P.; Angelova Volponi, A.; Janowicz, K.; Sibiak, R.; Piotrowska-Kempisty, H.; Izycki, D.; Bukowska, D.; et al. Inclusion Biogenesis, Methods of Isolation and Clinical Application of Human Cellular Exosomes. J. Clin. Med. 2020, 9, 436. [Google Scholar] [CrossRef] [Green Version]
- Middleton, R.C.; Rogers, R.G.; De Couto, G.; Tseliou, E.; Luther, K.; Holewinski, R.; Soetkamp, D.; Van Eyk, J.E.; Antes, T.J.; Marbán, E. Newt cells secrete extracellular vesicles with therapeutic bioactivity in mammalian cardiomyocytes. J. Extracell. Vesicles 2018, 7, 1456888. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging. 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Yang, H.; Wang, J.; Ru, W.; Wu, J.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Exosome biogenesis, secretion and function of exosomal miRNAs in skeletal muscle myogenesis. Cell Prolif. 2020, 53, e12857. [Google Scholar] [CrossRef]
- Andreu, Z.; Yanez-Mo, M. Tetraspanins in extracellular vesicle formation and function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.; Rossello, C.A.; Fernandez-Garcia, P.; Llado, V.; Kakhlon, O.; Escriba, P.V. The Implications for Cells of the Lipid Switches Driven by Protein-Membrane Interactions and the Development of Membrane Lipid Therapy. Int. J. Mol. Sci. 2020, 21, 2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.; Ren, Y.; Hu, X.; Mu, J.; Samykutty, A.; Zhuang, X.; Deng, Z.; Kumar, A.; Zhang, L.; Merchant, M.L.; et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat. Commun. 2017, 8, 14448. [Google Scholar] [CrossRef] [PubMed]
- Ni, K.; Wang, C.; Carnino, J.M.; Jin, Y. The Evolving Role of Caveolin-1: A Critical Regulator of Extracellular Vesicles. Med. Sci. 2020, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Li, S.P.; Lin, Z.X.; Jiang, X.Y.; Yu, X.Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharm. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef] [Green Version]
- Di Giaimo, R.; Penna, E.; Pizzella, A.; Cirillo, R.; Perrone-Capano, C.; Crispino, M. Cross Talk at the Cytoskeleton-Plasma Membrane Interface: Impact on Neuronal Morphology and Functions. Int. J. Mol. Sci. 2020, 21, 9133. [Google Scholar] [CrossRef]
- Jin, H.; Tang, Y.; Yang, L.; Peng, X.; Li, B.; Fan, Q.; Wei, S.; Yang, S.; Li, X.; Wu, B.; et al. Rab GTPases: Central Coordinators of Membrane Trafficking in Cancer. Front. Cell Dev. Biol. 2021, 9, 648384. [Google Scholar] [CrossRef]
- Sinha, S.; Hoshino, D.; Hong, N.H.; Kirkbride, K.C.; Grega-Larson, N.E.; Seiki, M.; Tyska, M.J.; Weaver, A.M. Cortactin promotes exosome secretion by controlling branched actin dynamics. J. Cell Biol. 2016, 214, 197–213. [Google Scholar] [CrossRef]
- Helgeson, L.A.; Nolen, B.J. Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. eLife 2013, 2, e00884. [Google Scholar] [CrossRef] [PubMed]
- Armacki, M.; Polaschek, S.; Waldenmaier, M.; Morawe, M.; Ruhland, C.; Schmid, R.; Lechel, A.; Tharehalli, U.; Steup, C.; Bektas, Y.; et al. Protein Kinase D1, Reduced in Human Pancreatic Tumors, Increases Secretion of Small Extracellular Vesicles From Cancer Cells That Promote Metastasis to Lung in Mice. Gastroenterology 2020, 159, 1019.e22–1035.e22. [Google Scholar] [CrossRef]
- Eiseler, T.; Hausser, A.; De Kimpe, L.; Van Lint, J.; Pfizenmaier, K. Protein kinase D controls actin polymerization and cell motility through phosphorylation of cortactin. J. Biol. Chem. 2010, 285, 18672–18683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, S.; Döppler, H.; Perez, E.A.; Andorfer, C.A.; Sun, Z.; Anastasiadis, P.Z.; Thompson, E.; Geiger, X.J.; Storz, P. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013, 15, R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, A.; Lobb, R.J. The evolving translational potential of small extracellular vesicles in cancer. Nat. Rev. Cancer 2020, 20, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Messenger, S.W.; Woo, S.S.; Sun, Z.; Martin, T.F.J. A Ca(2+)-stimulated exosome release pathway in cancer cells is regulated by Munc13-4. J. Cell Biol. 2018, 217, 2877–2890. [Google Scholar] [CrossRef]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chen, C.K.; Ho, C.M.; Lee, S.S.; Chang, C.Y.; Chen, K.J.; Jou, Y.S. EIF3C-enhanced exosome secretion promotes angiogenesis and tumorigenesis of human hepatocellular carcinoma. Oncotarget 2018, 9, 13193–13205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin. Chim. Acta 2016, 454, 28–32. [Google Scholar] [CrossRef]
- Arbelaiz, A.; Azkargorta, M.; Krawczyk, M.; Santos-Laso, A.; Lapitz, A.; Perugorria, M.J.; Erice, O.; Gonzalez, E.; Jimenez-Agüero, R.; Lacasta, A.; et al. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2017, 66, 1125–1143. [Google Scholar] [CrossRef]
- Cumba Garcia, L.M.; Peterson, T.E.; Cepeda, M.A.; Johnson, A.J.; Parney, I.F. Isolation and Analysis of Plasma-Derived Exosomes in Patients With Glioma. Front. Oncol. 2019, 9, 651. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, E.M.; Schultz, S.W.; Schink, K.O.; Pedersen, N.M.; Nähse, V.; Carlson, A.; Brech, A.; Stenmark, H.; Raiborg, C. Concerted ESCRT and clathrin recruitment waves define the timing and morphology of intraluminal vesicle formation. Nat. Commun. 2018, 9, 2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattissek, C.; Teis, D. The role of the endosomal sorting complexes required for transport (ESCRT) in tumorigenesis. Mol. Membr. Biol. 2014, 31, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Li, X.; Wang, Y.; Dong, M.; Zhan, F.H.; Liu, J. The ceramide pathway is involved in the survival, apoptosis and exosome functions of human multiple myeloma cells in vitro. Acta Pharm. Sin. 2018, 39, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, S.; Paisner, R.; Camarda, R.; Gupta, S.; Momcilovic, O.; Kohnz, R.A.; Avsaroglu, B.; L’Etoile, N.D.; Perera, R.M.; Nomura, D.K.; et al. Oncogene-regulated release of extracellular vesicles. Dev. Cell 2021, 56, 1989.e6–2006.e6. [Google Scholar] [CrossRef]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991–52002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demory Beckler, M.; Higginbotham, J.N.; Franklin, J.L.; Ham, A.J.; Halvey, P.J.; Imasuen, I.E.; Whitwell, C.; Li, M.; Liebler, D.C.; Coffey, R.J. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell Proteom. 2013, 12, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, D.J.; Franklin, J.L.; Dou, Y.; Liu, Q.; Higginbotham, J.N.; Demory Beckler, M.; Weaver, A.M.; Vickers, K.; Prasad, N.; Levy, S.; et al. KRAS-dependent sorting of miRNA to exosomes. eLife 2015, 4, e07197. [Google Scholar] [CrossRef]
- Zhang, Q.; Jeppesen, D.K.; Higginbotham, J.N.; Demory Beckler, M.; Poulin, E.J.; Walsh, A.J.; Skala, M.C.; McKinley, E.T.; Manning, H.C.; Hight, M.R.; et al. Mutant KRAS Exosomes Alter the Metabolic State of Recipient Colonic Epithelial Cells. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 627.e6–629.e6. [Google Scholar] [CrossRef] [Green Version]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Makrodouli, E.; Oikonomou, E.; Koc, M.; Andera, L.; Sasazuki, T.; Shirasawa, S.; Pintzas, A. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: A comparative study. Mol. Cancer 2011, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Ekström, E.J.; Bergenfelz, C.; von Bülow, V.; Serifler, F.; Carlemalm, E.; Jönsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of exosomal miRNA on cancer biology and clinical applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Zavyalova, M.V.; Denisov, E.V.; Tashireva, L.A.; Savelieva, O.E.; Kaigorodova, E.V.; Krakhmal, N.V.; Perelmuter, V.M. Intravasation as a Key Step in Cancer Metastasis. Biochem 2019, 84, 762–772. [Google Scholar] [CrossRef]
- Knights, A.J.; Funnell, A.P.; Crossley, M.; Pearson, R.C. Holding Tight: Cell Junctions and Cancer Spread. Trends Cancer Res. 2012, 8, 61–69. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Nam, R.K.; Benatar, T.; Wallis, C.J.; Amemiya, Y.; Yang, W.; Garbens, A.; Naeim, M.; Sherman, C.; Sugar, L.; Seth, A. MiR-301a regulates E-cadherin expression and is predictive of prostate cancer recurrence. Prostate 2016, 76, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shan, Z.; Hong, J.; Yang, L. MicroRNA-92a promotes epithelial-mesenchymal transition through activation of PTEN/PI3K/AKT signaling pathway in non-small cell lung cancer metastasis. Int. J. Oncol. 2017, 51, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Wei, K.; Hu, L.Q.; Zhou, C.R.; Lu, Z.B.; Zhan, G.S.; Pan, X.L.; Pan, C.F.; Wang, J.; Wen, W.; et al. Exosome-mediated transfer of miR-1260b promotes cell invasion through Wnt/β-catenin signaling pathway in lung adenocarcinoma. J. Cell Physiol. 2020, 235, 6843–6853. [Google Scholar] [CrossRef]
- Bigagli, E.; Luceri, C.; Guasti, D.; Cinci, L. Exosomes secreted from human colon cancer cells influence the adhesion of neighboring metastatic cells: Role of microRNA-210. Cancer Biol. Ther. 2016, 17, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Ma, C.; Zhou, T.; Dong, X.; Luo, Q.; Geng, L.; Ding, L.; Zhang, Y.; Zhang, L.; Li, N.; et al. Exosomes derived from gemcitabine-resistant cells transfer malignant phenotypic traits via delivery of miRNA-222-3p. Mol. Cancer 2017, 16, 132. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell. Biochem. 2008, 105, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Shan, Y.; Chen, J.; Yue, H.; You, B.; Shi, S.; Li, X.; Cao, X. Matrix metalloproteinase 13-containing exosomes promote nasopharyngeal carcinoma metastasis. Cancer Sci. 2015, 106, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.Y.; Dugas-Ford, J.; Seiki, M.; Chang, J.H.; Azar, D.T. Evidence for the Involvement of MMP14 in MMP2 Processing and Recruitment in Exosomes of Corneal Fibroblasts. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5323–5329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, A.; Yoshioka, Y.; Yamamoto, Y.; Ishikawa, M.; Ikeda, S.I.; Kato, T.; Kiyono, T.; Takeshita, F.; Kajiyama, H.; Kikkawa, F.; et al. Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 2017, 8, 14470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.; Liu, R.; Shi, Y.J.; Yin, L.H.; Pu, Y.P. Exosome-shuttling microRNA-21 promotes cell migration and invasion-targeting PDCD4 in esophageal cancer. Int. J. Oncol. 2016, 48, 2567–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J. Considering Exosomal miR-21 as a Biomarker for Cancer. J. Clin. Med. 2016, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.J.; Wang, Z.Y.; Chen, R.; Xiong, J.; Yao, Y.L.; Wu, J.H.; Li, G.X. Macrophage-secreted Exosomes Delivering miRNA-21 Inhibitor can Regulate BGC-823 Cell Proliferation. Asian Pac. J. Cancer Prev. 2015, 16, 4203–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.H.; Tian, D.; Yang, Z.C.; Li, J.L. Exosomal miR-21 promotes proliferation, invasion and therapy resistance of colon adenocarcinoma cells through its target PDCD4. Sci. Rep. 2020, 10, 8271. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.H.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lu, J.; Chen, L.; Bian, H.; Hu, J.; Li, D.; Xia, C.; Xu, H. Tumor-Derived EV-Encapsulated miR-181b-5p Induces Angiogenesis to Foster Tumorigenesis and Metastasis of ESCC. Mol. Ther. Nucleic. Acids 2020, 20, 421–437. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, N.; Caetano, M.S.; Cumpian, A.M.; Unver, N.; De la Garza Ramos, C.; Noble, O.; Daliri, S.; Hernandez, B.J.; Gutierrez, B.A.; Evans, S.E.; et al. IL22 Promotes Kras-Mutant Lung Cancer by Induction of a Protumor Immune Response and Protection of Stemness Properties. Cancer Immunol. Res. 2018, 6, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Todoric, J.; Karin, M. The Fire within: Cell-Autonomous Mechanisms in Inflammation-Driven Cancer. Cancer Cell 2019, 35, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of Exosomes in Crosstalk Between Cancer-Associated Fibroblasts and Cancer Cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Kugeratski, F.G.; Kalluri, R. Exosomes as mediators of immune regulation and immunotherapy in cancer. Febs. j. 2021, 288, 10–35. [Google Scholar] [CrossRef]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef]
- Duan, B.; Shi, S.; Yue, H.; You, B.; Shan, Y.; Zhu, Z.; Bao, L.; You, Y. Exosomal miR-17-5p promotes angiogenesis in nasopharyngeal carcinoma via targeting BAMBI. J. Cancer 2019, 10, 6681–6692. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.S.; Kanugula, S.S.; Sudhir, S.; Pereira, M.P.; Jain, S.; Aghi, M.K. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012, 40, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef]
- Gu, J.; Qian, H.; Shen, L.; Zhang, X.; Zhu, W.; Huang, L.; Yan, Y.; Mao, F.; Zhao, C.; Shi, Y.; et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-β/Smad pathway. PLoS ONE 2012, 7, e52465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’Ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M.G.; et al. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015, 106, 1362–1369. [Google Scholar] [CrossRef]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther Nucleic Acids 2020, 19, 654–667. [Google Scholar] [CrossRef]
- Yue, X.; Lan, F.; Xia, T. Hypoxic Glioma Cell-Secreted Exosomal miR-301a Activates Wnt/β-catenin Signaling and Promotes Radiation Resistance by Targeting TCEAL7. Mol. Ther. 2019, 27, 1939–1949. [Google Scholar] [CrossRef]
- Cao, M.; Seike, M.; Soeno, C.; Mizutani, H.; Kitamura, K.; Minegishi, Y.; Noro, R.; Yoshimura, A.; Cai, L.; Gemma, A. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. Int. J. Oncol. 2012, 41, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhu, W.; Chen, Q.; Yuan, Y.; Wang, Y.; Wang, J.; Wu, X. Ovarian cancer cell-secreted exosomal miR-205 promotes metastasis by inducing angiogenesis. Theranostics 2019, 9, 8206–8220. [Google Scholar] [CrossRef]
- Sun, L.P.; Xu, K.; Cui, J.; Yuan, D.Y.; Zou, B.; Li, J.; Liu, J.L.; Li, K.Y.; Meng, Z.; Zhang, B. Cancer-associated fibroblast-derived exosomal miR-382-5p promotes the migration and invasion of oral squamous cell carcinoma. Oncol. Rep. 2019, 42, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.B.; Li, Z.L.; Luo, D.H.; Huang, B.J.; Chen, Y.S.; Zhang, X.S.; Cui, J.; Zeng, Y.X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yang, Y.; Wang, W.; Zhang, Y.; Chen, Z.; Hao, C.; Zhang, J. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4(+) T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Gao, C.; Huang, X.Y.; Lu, J.C.; Guo, X.J.; Shi, G.M.; Cai, J.B.; Ke, A.W. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol. Cancer 2020, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briand, J.; Garnier, D.; Nadaradjane, A.; Clément-Colmou, K.; Potiron, V.; Supiot, S.; Bougras-Cartron, G.; Frenel, J.S.; Heymann, D.; Vallette, F.M.; et al. Radiotherapy-induced overexpression of exosomal miRNA-378a-3p in cancer cells limits natural killer cells cytotoxicity. Epigenomics 2020, 12, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Zhou, L.; Qian, Y.; Fu, M.; Chen, J.; Chen, J.; Xiang, J.; Wu, Z.; Jiang, G.; Cao, L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 2015, 6, 29877–29888. [Google Scholar] [CrossRef] [Green Version]
- Tung, S.L.; Boardman, D.A.; Sen, M.; Letizia, M.; Peng, Q.; Cianci, N.; Dioni, L.; Carlin, L.M.; Lechler, R.; Bollati, V.; et al. Regulatory T cell-derived extracellular vesicles modify dendritic cell function. Sci. Rep. 2018, 8, 6065. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yu, J.; Chen, L.; Tao, T.; Yi, S.; Hanley, S.J.B.; Yue, J.; Watari, H.; Sakuragi, N. Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene 2018, 37, 5257–5268. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef]
- Karimi, N.; Ali Hosseinpour Feizi, M.; Safaralizadeh, R.; Hashemzadeh, S.; Baradaran, B.; Shokouhi, B.; Teimourian, S. Serum overexpression of miR-301a and miR-23a in patients with colorectal cancer. J. Chin. Med. Assoc. 2019, 82, 215–220. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef]
- Park, J.E.; Dutta, B.; Tse, S.W.; Gupta, N.; Tan, C.F.; Low, J.K.; Yeoh, K.W.; Kon, O.L.; Tam, J.P.; Sze, S.K. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene 2019, 38, 5158–5173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerloff, D.; Lützkendorf, J.; Moritz, R.K.C.; Wersig, T.; Mäder, K.; Müller, L.P.; Sunderkötter, C. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J. Cell Mol. Med. 2020, 24, 9560–9573. [Google Scholar] [CrossRef]
- Di Modica, M.; Regondi, V.; Sandri, M.; Iorio, M.V.; Zanetti, A.; Tagliabue, E.; Casalini, P.; Triulzi, T. Breast cancer-secreted miR-939 downregulates VE-cadherin and destroys the barrier function of endothelial monolayers. Cancer Lett. 2017, 384, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat. Commun. 2015, 6, 6716. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhang, H.; Deng, T.; Ning, T.; Liu, R.; Liu, D.; Bai, M.; Ying, G.; Ba, Y. Exosomes Carrying MicroRNA-155 Target Forkhead Box O3 of Endothelial Cells and Promote Angiogenesis in Gastric Cancer. Mol. Ther. Oncolytics 2019, 15, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Wang, Y.; Dong, L.; Zhang, Y.; Zhang, Y.; Wang, C.; Zhang, Q.; Yang, S.; Cao, L.; Zhang, X.; et al. Hypoxic exosomes facilitate angiogenesis and metastasis in esophageal squamous cell carcinoma through altering the phenotype and transcriptome of endothelial cells. J. Exp. Clin. Cancer Res. 2019, 38, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Dang, W.; Zhang, S.; Yue, W.; Yang, L.; Zhai, X.; Yan, Q.; Lu, J. The role of exosomal noncoding RNAs in cancer. Mol. Cancer 2019, 18, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Lin, Y.S.; Pan, Y.C.; Tsai, P.H.; Wu, C.Y.; Kuo, P.L. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene 2017, 36, 4929–4942. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.J.; Lin, X.J.; Tang, X.Y.; Zheng, T.T.; Lin, Y.Y.; Hua, K.Q. Exosomal Metastasis-Associated Lung Adenocarcinoma Transcript 1 Promotes Angiogenesis and Predicts Poor Prognosis in Epithelial Ovarian Cancer. Int. J. Biol. Sci. 2018, 14, 1960–1973. [Google Scholar] [CrossRef] [Green Version]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar]
- Zhou, C.F.; Ma, J.; Huang, L.; Yi, H.Y.; Zhang, Y.M.; Wu, X.G.; Yan, R.M.; Liang, L.; Zhong, M.; Yu, Y.H.; et al. Cervical squamous cell carcinoma-secreted exosomal miR-221-3p promotes lymphangiogenesis and lymphatic metastasis by targeting VASH1. Oncogene 2019, 38, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Luo, Y.; He, W.; Zhao, Y.; Kong, Y.; Liu, H.; Zhong, G.; Li, Y.; Li, J.; Huang, J.; et al. Exosomal long noncoding RNA LNMAT2 promotes lymphatic metastasis in bladder cancer. J. Clin. Invest. 2020, 130, 404–421. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazaei, S.; Nouraee, N.; Moradi, A.; Mowla, S.J. A novel signaling role for miR-451 in esophageal tumor microenvironment and its contribution to tumor progression. Clin. Transl. Oncol. 2017, 19, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [Green Version]
- Egen, J.G.; Ouyang, W.; Wu, L.C. Human Anti-tumor Immunity: Insights from Immunotherapy Clinical Trials. Immunity 2020, 52, 36–54. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Capello, M.; Vykoukal, J.V.; Katayama, H.; Bantis, L.E.; Wang, H.; Kundnani, D.L.; Aguilar-Bonavides, C.; Aguilar, M.; Tripathi, S.C.; Dhillon, D.S.; et al. Exosomes harbor B cell targets in pancreatic adenocarcinoma and exert decoy function against complement-mediated cytotoxicity. Nat. Commun. 2019, 10, 254. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [Green Version]
- Batista, I.A.; Melo, S.A. Exosomes and the Future of Immunotherapy in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 567. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The effect of tumor-derived exosomes on immune regulation and cancer immunotherapy. Future Oncol. 2017, 13, 2583–2592. [Google Scholar] [CrossRef] [PubMed]
- O′Brien, D.I.; Nally, K.; Kelly, R.G.; O’Connor, T.M.; Shanahan, F.; O’Connell, J. Targeting the Fas/Fas ligand pathway in cancer. Expert Opin. Ther. Targets 2005, 9, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yang, F.; Yu, L.; Yu, Z.; Jiang, L.; Wang, Q.; Yang, Y.; Wang, L.; Cao, X.; Wang, J. Activated T cell exosomes promote tumor invasion via Fas signaling pathway. J. Immunol. 2012, 188, 5954–5961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Sharma, P.; Theodoraki, M.N.; Pietrowska, M.; Yerneni, S.S.; Lang, S.; Ferrone, S.; Whiteside, T.L. Molecular and Functional Profiles of Exosomes From HPV(+) and HPV(-) Head and Neck Cancer Cell Lines. Front. Oncol. 2018, 8, 445. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Sung, Y.H.; Pack, C.G.; Jung, M.K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fan, Y.; Che, X.; Hou, K.; Zhang, C.; Li, C.; Wen, T.; Wang, S.; Cheng, Y.; Liu, Y.; et al. 5-FU-Induced Upregulation of Exosomal PD-L1 Causes Immunosuppression in Advanced Gastric Cancer Patients. Front. Oncol. 2020, 10, 492. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, D.; Gnatta, E.; Padoan, A.; Fogar, P.; Furlanello, S.; Aita, A.; Bozzato, D.; Zambon, C.F.; Arrigoni, G.; Frasson, C.; et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget 2017, 8, 84928–84944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotelli, M.R.; Reinhart-King, C.A. Mechanical Forces in Tumor Angiogenesis. Adv. Exp. Med. Biol. 2018, 1092, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Jászai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1989–2000. [Google Scholar] [CrossRef]
- Sruthi, T.V.; Edatt, L.; Raji, G.R.; Kunhiraman, H.; Shankar, S.S.; Shankar, V.; Ramachandran, V.; Poyyakkara, A.; Kumar, S.V.B. Horizontal transfer of miR-23a from hypoxic tumor cell colonies can induce angiogenesis. J. Cell Physiol. 2018, 233, 3498–3514. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Langheinrich, M.C.; Schellerer, V.; Perrakis, A.; Lohmüller, C.; Schildberg, C.; Naschberger, E.; Stürzl, M.; Hohenberger, W.; Croner, R.S. Molecular mechanisms of lymphatic metastasis in solid tumors of the gastrointestinal tract. Int. J. Clin. Exp. Pathol. 2012, 5, 614–623. [Google Scholar] [PubMed]
- Rahman, M.; Mohammed, S. Breast cancer metastasis and the lymphatic system. Oncol. Lett. 2015, 10, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Pak, K.H.; Park, K.C.; Cheong, J.H. VEGF-C induced by TGF- β1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis. BMC Cancer 2019, 19, 799. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Ji, H.; Feng, N.; Zhang, Y.; Yang, X.; Andersson, P.; Sun, Y.; Tritsaris, K.; Hansen, A.J.; Dissing, S.; et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 15894–15899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.; Shaker, H.; Morris, K.; Tugwood, J.D.; Kirwan, C.C. The significance of circulating tumour cells in breast cancer: A review. Breast 2014, 23, 552–560. [Google Scholar] [CrossRef]
- Hou, J.M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.J.; Greystoke, A.; Zhou, C.; Morris, K.; et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef]
- Brandt, B.; Junker, R.; Griwatz, C.; Heidl, S.; Brinkmann, O.; Semjonow, A.; Assmann, G.; Zänker, K.S. Isolation of prostate-derived single cells and cell clusters from human peripheral blood. Cancer Res. 1996, 56, 4556–4561. [Google Scholar] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Zhang, Q.; Lou, Y.; Yang, J.; Nie, G.; Chen, Q.; Chen, Y.; Zhang, J.; Wang, J.; Wei, T.; et al. Primary tumor-derived exosomes facilitate metastasis by regulating adhesion of circulating tumor cells via SMAD3 in liver cancer. Oncogene 2018, 37, 6105–6118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labernadie, A.; Kato, T.; Brugués, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; González-Tarragó, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial-mesenchymal plasticity. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.t.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell-Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, P.; Martínez-Pena, I.; Piñeiro, R. Dangerous Liaisons: Circulating Tumor Cells (CTCs) and Cancer-Associated Fibroblasts (CAFs). Cancers 2020, 12, 2861. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Karpatkin, S.; Pearlstein, E.; Ambrogio, C.; Coller, B.S. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J. Clin. Investig. 1988, 81, 1012–1019. [Google Scholar] [CrossRef]
- Gil-Bernabé, A.M.; Lucotti, S.; Muschel, R.J. Coagulation and metastasis: What does the experimental literature tell us? Br. J. Haematol. 2013, 162, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Hisada, Y.; Mackman, N. Cancer-associated pathways and biomarkers of venous thrombosis. Blood 2017, 130, 1499–1506. [Google Scholar] [CrossRef]
- Khorana, A.A. Venous thromboembolism and prognosis in cancer. Thromb. Res. 2010, 125, 490–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, D.; Magnus, N.; Lee, T.H.; Bentley, V.; Meehan, B.; Milsom, C.; Montermini, L.; Kislinger, T.; Rak, J. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J. Biol. Chem. 2012, 287, 43565–43572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Dean, W.L.; Lee, M.J.; Cummins, T.D.; Schultz, D.J.; Powell, D.W. Proteomic and functional characterisation of platelet microparticle size classes. Thromb. Haemost. 2009, 102, 711–718. [Google Scholar] [CrossRef]
- Dovizio, M.; Alberti, S.; Sacco, A.; Guillem-Llobat, P.; Schiavone, S.; Maier, T.J.; Steinhilber, D.; Patrignani, P. Novel insights into the regulation of cyclooxygenase-2 expression by platelet-cancer cell cross-talk. Biochem. Soc. Trans. 2015, 43, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Reymond, N.; d’Água, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Köhler, S.; Ullrich, S.; Richter, U.; Schumacher, U. E-/P-selectins and colon carcinoma metastasis: First in vivo evidence for their crucial role in a clinically relevant model of spontaneous metastasis formation in the lung. Br. J. Cancer 2010, 102, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, A.; Honma, K.; Saji, S.; Moriwaki, H.; Ochiya, T.; Hoffman, R.M. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv. Drug Deliv. Rev. 2013, 65, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Chan, Y.K.; Zhang, H.; Liu, P.; Tsao, S.W.; Lung, M.L.; Mak, N.K.; Ngok-Shun Wong, R.; Ying-Kit Yue, P. Proteomic analysis of exosomes from nasopharyngeal carcinoma cell identifies intercellular transfer of angiogenic proteins. Int. J. Cancer 2015, 137, 1830–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, T.; Castellana, D.; Klingbeil, P.; Cuesta Hernández, I.; Vitacolonna, M.; Orlicky, D.J.; Roffler, S.R.; Brodt, P.; Zöller, M. CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 2009, 11, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; von Au, A.; Schnölzer, M.; Hackert, T.; Zöller, M. CD44v6-competent tumor exosomes promote motility, invasion and cancer-initiating cell marker expression in pancreatic and colorectal cancer cells. Oncotarget 2016, 7, 55409–55436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, S.; Pucci, M.; Giallombardo, M.; Di Bella, M.A.; Santarpia, M.; Reclusa, P.; Gil-Bazo, I.; Rolfo, C.; Alessandro, R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep. 2017, 7, 3170. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Donoso-Quezada, J.; Ayala-Mar, S.; González-Valdez, J. The role of lipids in exosome biology and intercellular communication: Function, analytics and applications. Traffic 2021, 22, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhou, J.; Yuan, C.; Zhang, L.; Li, D.; Si, D.; Xiu, D.; Zhong, L. Metabolomics identifies serum and exosomes metabolite markers of pancreatic cancer. Metab. Off. J. Metab. Soc. 2019, 15, 86. [Google Scholar] [CrossRef]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-Y.; Sung, C.W.-H.; Chen, C.; Cheng, C.-M.; Lin, D.P.-C.; Huang, C.-T.; Hsu, M.-Y. Advances in exosomes technology. Clin. Chim. Acta 2019, 493, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef]
- Buscail, E.; Chauvet, A.; Quincy, P.; Degrandi, O.; Buscail, C.; Lamrissi, I.; Moranvillier, I.; Caumont, C.; Verdon, S.; Brisson, A.; et al. CD63-GPC1-Positive Exosomes Coupled with CA19-9 Offer Good Diagnostic Potential for Resectable Pancreatic Ductal Adenocarcinoma. Transl. Oncol. 2019, 12, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Frampton, A.E.; Prado, M.M.; Lopez-Jimenez, E.; Fajardo-Puerta, A.B.; Jawad, Z.A.R.; Lawton, P.; Giovannetti, E.; Habib, N.A.; Castellano, L.; Stebbing, J.; et al. Glypican-1 is enriched in circulating-exosomes in pancreatic cancer and correlates with tumor burden. Oncotarget 2018, 9, 19006–19013. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, S.; Iinuma, H.; Wada, K.; Takahashi, K.; Minezaki, S.; Kainuma, M.; Shibuya, M.; Miura, F.; Sano, K. Exosome-encapsulated microRNA-4525, microRNA-451a and microRNA-21 in portal vein blood is a high-sensitive liquid biomarker for the selection of high-risk pancreatic ductal adenocarcinoma patients. J. Hepatobiliary Pancreat. Sci. 2019, 26, 63–72. [Google Scholar] [CrossRef]
- Yuan, X.; Qian, N.; Ling, S.; Li, Y.; Sun, W.; Li, J.; Du, R.; Zhong, G.; Liu, C.; Yu, G.; et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics 2021, 11, 1429–1445. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Wang, P.Y.; Li, X.Y.; Chen, J.X.; Li, Y.; Zhang, X.Z.; Zhang, C.G.; Jiang, T.; Li, W.B.; Ding, W.; et al. Exosomal levels of miRNA-21 from cerebrospinal fluids associated with poor prognosis and tumor recurrence of glioma patients. Oncotarget 2015, 6, 26971–26981. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, Y.; Lu, J.; Sun, Y.; Xiao, H.; Liu, M.; Tian, L. Combined detection of serum exosomal miR-21 and HOTAIR as diagnostic and prognostic biomarkers for laryngeal squamous cell carcinoma. Med. Oncol. 2014, 31, 148. [Google Scholar] [CrossRef] [PubMed]
- Ohzawa, H.; Kumagai, Y.; Yamaguchi, H.; Miyato, H.; Sakuma, Y.; Horie, H.; Hosoya, Y.; Kawarai Lefor, A.; Sata, N.; Kitayama, J. Exosomal microRNA in peritoneal fluid as a biomarker of peritoneal metastases from gastric cancer. Ann. Gastroenterol. Surg. 2020, 4, 84–93. [Google Scholar] [CrossRef]
- Shin, S.; Park, Y.H.; Jung, S.H.; Jang, S.H.; Kim, M.Y.; Lee, J.Y.; Chung, Y.J. Urinary exosome microRNA signatures as a noninvasive prognostic biomarker for prostate cancer. NPJ Genom. Med. 2021, 6, 45. [Google Scholar] [CrossRef]
- De Miguel Perez, D.; Rodriguez Martinez, A.; Ortigosa Palomo, A.; Delgado Urena, M.; Garcia Puche, J.L.; Robles Remacho, A.; Exposito Hernandez, J.; Lorente Acosta, J.A.; Ortega Sanchez, F.G.; Serrano, M.J. Extracellular vesicle-miRNAs as liquid biopsy biomarkers for disease identification and prognosis in metastatic colorectal cancer patients. Sci. Rep. 2020, 10, 3974. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer*. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möhrmann, L.; Huang, H.J.; Hong, D.S.; Tsimberidou, A.M.; Fu, S.; Piha-Paul, S.A.; Subbiah, V.; Karp, D.D.; Naing, A.; Krug, A.; et al. Liquid Biopsies Using Plasma Exosomal Nucleic Acids and Plasma Cell-Free DNA Compared with Clinical Outcomes of Patients with Advanced Cancers. Clin. Cancer Res. 2018, 24, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.S.; Hoff, D.V.; Cridebring, D.; Goel, A. Extracellular Vesicles in Diagnosis and Treatment of Pancreatic Cancer: Current State and Future Perspectives. Cancers 2020, 12, 1530. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Bernard, V.; San Lucas, F.A.; Allenson, K.; Capello, M.; Kim, D.U.; Gascoyne, P.; Mulu, F.C.; Stephens, B.M.; Huang, J.; et al. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann. Oncol. 2018, 29, 223–229. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Wang, F.; Li, L.; Piontek, K.; Sakaguchi, M.; Selaru, F.M. Exosome miR-335 as a novel therapeutic strategy in hepatocellular carcinoma. Hepatology 2018, 67, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Raimondi, L.; Costa, V.; De Luca, A.; Carina, V.; Maglio, M.; Fini, M.; Alessandro, R.; Giavaresi, G. Engineered exosomes: A new promise for the management of musculoskeletal diseases. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Gonzalo, D.H.; Feely, M.; Rinaldi, C.; Belsare, S.; Zhai, H.; Kalra, K.; Gerber, M.H.; Forsmark, C.E.; Hughes, S.J. Stroma-derived extracellular vesicles deliver tumor-suppressive miRNAs to pancreatic cancer cells. Oncotarget 2018, 9, 5764–5777. [Google Scholar] [CrossRef] [Green Version]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.L.; Yao, J.L.; Wang, K.; Ai, H. Human Umbilical Cord Mesenchymal Stem Cell-Derived Extracellular Vesicles Inhibit Endometrial Cancer Cell Proliferation and Migration through Delivery of Exogenous miR-302a. Stem. Cells Int. 2019, 2019, 8108576. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D. Exosomal miRNA Cargo as Mediator of Immune Escape Mechanisms in Neuroblastoma. Cancer Res. 2019, 79, 1293–1294. [Google Scholar] [CrossRef] [Green Version]
- Lunavat, T.R.; Jang, S.C.; Nilsson, L.; Park, H.T.; Repiska, G.; Lässer, C.; Nilsson, J.A.; Gho, Y.S.; Lötvall, J. RNAi delivery by exosome-mimetic nanovesicles-Implications for targeting c-Myc in cancer. Biomaterials 2016, 102, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, S.; Bjornetro, T.; Lyckander, L.G.; Flatmark, K.; Dueland, S.; Samiappan, R.; Johansen, C.; Kalanxhi, E.; Ree, A.H.; Redalen, K.R. Circulating Exosomal miR-141-3p and miR-375 in Metastatic Progression of Rectal Cancer. Transl. Oncol. 2019, 12, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Chen, T.; Zheng, X.; Yang, S.; Xu, K.; Chen, X.; Xu, F.; Wang, L.; Shen, Y.; Wang, T.; et al. Colorectal cancer-derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis 2018, 39, 1368–1379. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, X.; Gao, S.; Jing, F.; Yang, Y.; Du, L.; Zheng, G.; Li, P.; Li, C.; Wang, C. Exosomal long noncoding RNA CRNDE-h as a novel serum-based biomarker for diagnosis and prognosis of colorectal cancer. Oncotarget 2016, 7, 85551–85563. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.Y.; Gu, R.H.; Yan, B. Downregulation of exosome-encapsulated miR-548c-5p is associated with poor prognosis in colorectal cancer. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microRNA levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Ma, Y.Y.; Wang, J.; Zeng, X.F.; Li, R.; Kang, W.; Hao, X.K. Exosomal microRNA-141 is upregulated in the serum of prostate cancer patients. Onco Targets Ther. 2016, 9, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagirath, D.; Yang, T.L.; Bucay, N.; Sekhon, K.; Majid, S.; Shahryari, V.; Dahiya, R.; Tanaka, Y.; Saini, S. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res. 2018, 78, 1833–1844. [Google Scholar] [CrossRef] [Green Version]
- Tokuhisa, M.; Ichikawa, Y.; Kosaka, N.; Ochiya, T.; Yashiro, M.; Hirakawa, K.; Kosaka, T.; Makino, H.; Akiyama, H.; Kunisaki, C.; et al. Exosomal miRNAs from Peritoneum Lavage Fluid as Potential Prognostic Biomarkers of Peritoneal Metastasis in Gastric Cancer. PLoS ONE 2015, 10, e0130472. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fu, H.; Wang, B.; Zhang, X.; Mao, J.; Li, X.; Wang, M.; Sun, Z.; Qian, H.; Xu, W. Exosomal miR-423-5p targets SUFU to promote cancer growth and metastasis and serves as a novel marker for gastric cancer. Mol. Carcinog. 2018, 57, 1223–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, T.; Feng, D.; Li, J.; Wu, M.; Peng, X.; Wang, B.; Zhan, X.; Fu, P. Screening of non-invasive miRNA biomarker candidates for metastasis of gastric cancer by small RNA sequencing of plasma exosomes. Carcinogenesis 2020, 41, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Muller, V.; Milde-Langosch, K.; Trillsch, F.; Pantel, K.; Schwarzenbach, H. Diagnostic and prognostic relevance of circulating exosomal miR-373, miR-200a, miR-200b and miR-200c in patients with epithelial ovarian cancer. Oncotarget 2016, 7, 16923–16935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Kamohara, H.; Kinoshita, K.; Kurashige, J.; Ishimoto, T.; Iwatsuki, M.; Watanabe, M.; Baba, H. Clinical impact of serum exosomal microRNA-21 as a clinical biomarker in human esophageal squamous cell carcinoma. Cancer 2013, 119, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Wu, J.; Wu, J.; Ji, A.; Qiang, G.; Jiang, Y.; Jiang, C.; Ding, Y. Exosomal miR-665 as a novel minimally invasive biomarker for hepatocellular carcinoma diagnosis and prognosis. Oncotarget 2017, 8, 80666–80678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Nan, A.; Chen, L.; Li, X.; Jia, Y.; Qiu, M.; Dai, X.; Zhou, H.; Zhu, J.; Zhang, H.; et al. Circular RNA circSATB2 promotes progression of non-small cell lung cancer cells. Mol. Cancer 2020, 19, 101. [Google Scholar] [CrossRef]
- Que, R.; Ding, G.; Chen, J.; Cao, L. Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J. Surg. Oncol. 2013, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, Z.; Jiang, P.; Peng, M.; Zhang, X.; Chen, K.; Liu, H.; Bi, H.; Liu, X.; Li, X. Circular RNA IARS (circ-IARS) secreted by pancreatic cancer cells and located within exosomes regulates endothelial monolayer permeability to promote tumor metastasis. J. Exp. Clin. Cancer Res. 2018, 37, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.R.; Grossmann, K.F.; Cassidy, P.B.; Yang, C.H.; Fan, M.; Kopelovich, L.; Leachman, S.A.; Pfeffer, L.M. Detection of Exosomal miRNAs in the Plasma of Melanoma Patients. J. Clin. Med. 2015, 4, 2012–2027. [Google Scholar] [CrossRef]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release Off. Control. Release Soc. 2015, 220, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Delivery of Exogenous miR-124 to Glioblastoma Multiform Cells by Wharton’s Jelly Mesenchymal Stem Cells Decreases Cell Proliferation and Migration, and Confers Chemosensitivity. Stem Cell Rev. Rep. 2018, 14, 236–246. [Google Scholar] [CrossRef]
- De la Fuente, A.; Alonso-Alconada, L.; Costa, C.; Cueva, J.; Garcia-Caballero, T.; Lopez-Lopez, R.; Abal, M. M-Trap: Exosome-Based Capture of Tumor Cells as a New Technology in Peritoneal Metastasis. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Mechanisms of sEV biogenesis and uptake.

Figure 2.
Functions of sEVs and ncRNA cargos in the metastatic cascade.


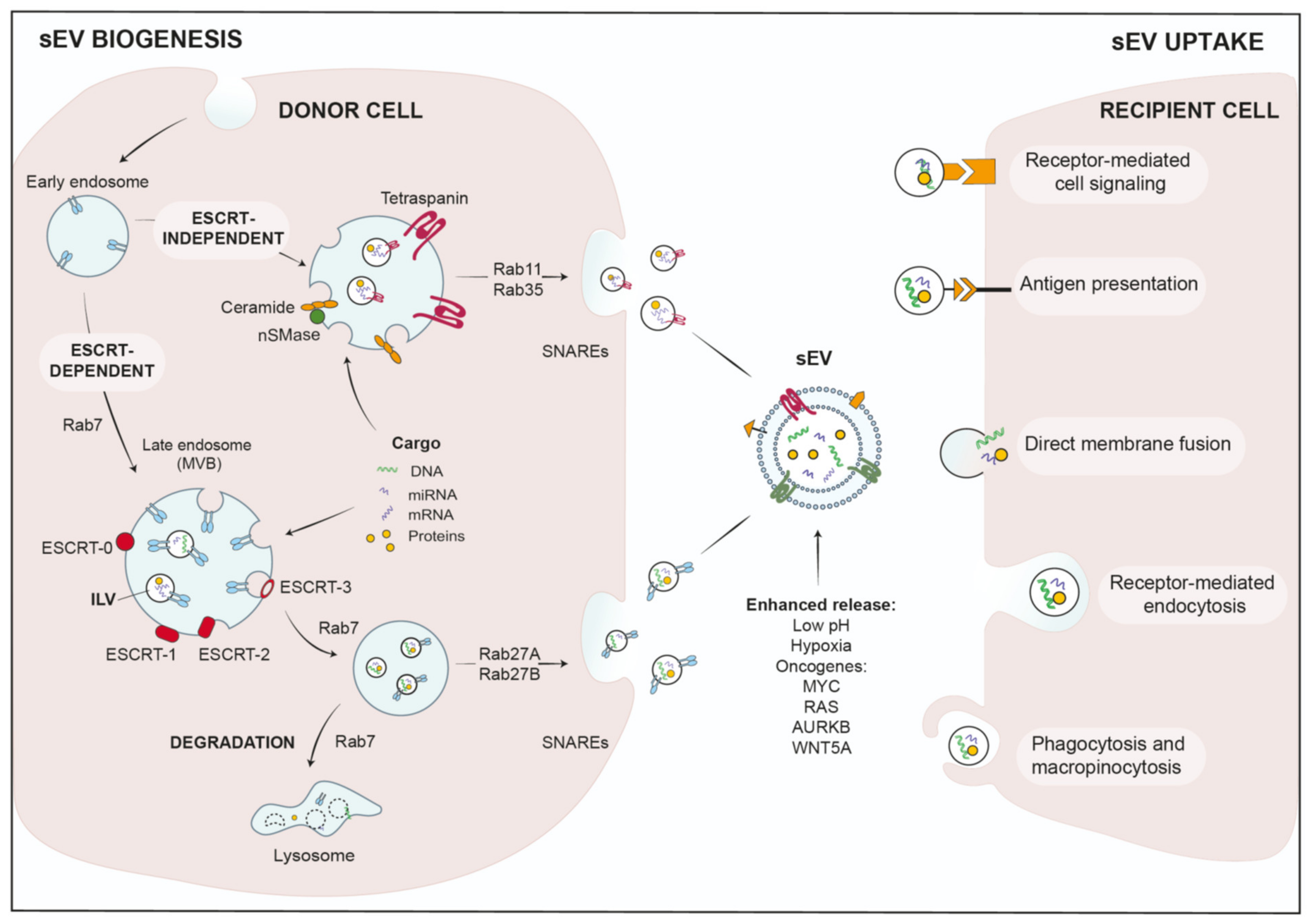{kind=link}
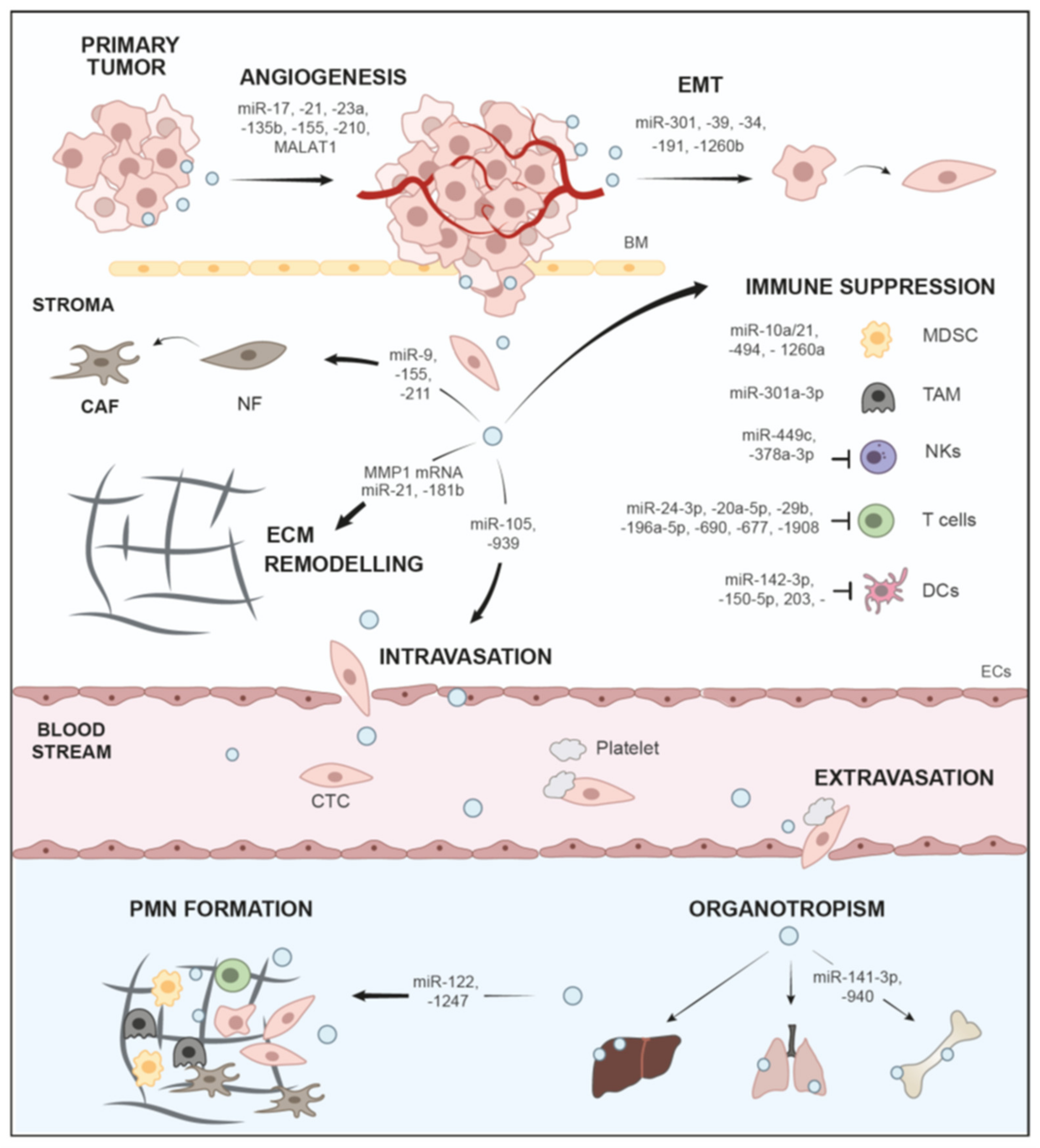{kind=link}
Table 1.
Origin and biological effects of sEV-derived ncRNA.
| Step | Molecule | Cell of Origin | Effect | Ref. |
|---|---|---|---|---|
| EMT | miR-301a | GBM, PC | Activation of Wnt/β-catenin signaling, suppression of TCEAL7, p63 and E-cadherin | [89,124] |
| miR-92a | NSCLC | Activation of PTEN/PI3K/AKT | [90] | |
| miR-191 | Melanoma | Activation of MAPK signaling | [14] | |
| miR-1260b | LC | Activation of Wnt/β-catenin signaling via inhibition of sFRP1 and Smad4 | [91] | |
| miR-181d-5p | CAFs in BC | Suppression of CDC2 and HOXA5 | ||
| miR-23a | NSCLC | Induction of TGFβ signaling and inhibition of E-cadherin | [125] | |
| miR-21 miR-143 miR-378e | CAFs in BC | Upregulation of NANOG, SOX2, SNAIL and ZEB | [126] | |
| miR-499a-5p | LC | Activation of mTOR signaling | [127] | |
| miR-19b-3p | CCRCC | Downregulation of PTEN | [127] | |
| miR-382-5p | CAFs in OSCC | Upregulation of ß-catenin and N-cadherin | [128] | |
| ECM degradation | miR-21 | CRC, EC | MMP activation via inhibition of PDCD4 and TIMP-3 | [101,102,104] |
| miR-181b | HCC, ESCC | MMP activation via inhibition of TIMP-3 | [105,106] | |
| MMP1 mRNA | OC | Enhanced MMP1 expression in recipient cells | [100] | |
| miR-382-5p | CAFs in OSCC | Upregulation of MMP-9 and MMP-3 | [128] | |
| Anoikis resistance | miR-210 | CRC | [92] | |
| miR-222-3p | NSCLC | Inhibition of SOCS3 | [93] | |
| Stroma | miR-9 | BC | CAF formation | [121] |
| miR-211 | Melanoma | CAF formation via MAPK activation | [12] | |
| miR-155 | PDAC | CAF formation via downregulation of TP53INP1 | [122] | |
| Immune modulation | miR-24-3p, miR-891a, miR-106a-5p, miR-20a-5p, miR-1908 | NPC | T-cell exhaustion via downregulation of the MARK1 signaling pathway | [129] |
| miR-690, miR-677, miR-29b | Melanoma | CD4+ T-cell apoptosis, increase of caspase-3, caspase-7 and caspase-9, downregulation of BCL-2 | [130] | |
| circUHRF1 | HCC | NK dysfunction via inhibition of IFN-γ and TNF-α secretion | [131] | |
| miR-301a-3p | PDAC | M2 macrophage polarization via activation of PTEN/PI3Kγ signaling | [132] | |
| miR-378a-3p | Various cell lines | Decreased NK cytotoxicity via inhibition of granzyme B | [133] | |
| miR-212-3p | PDAC | DC dysfunction via PFXAP inhibition and compromised MHCII expression | [134] | |
| miR-150-5p, miR-142-3p | Tregs | DC exhaustion | [135] | |
| miR-203 | PDAC | DC dysfunction via TLR4 inhibition | [136] | |
| miR-18a | CeC | T-cell exhaustion, suppression of PTEN, WNK2 and SOX6, and enhanced PD-L1 levels | [137] | |
| miR-23a | HCC | T-cell exhaustion, suppression of PTEN and enhanced PD-L1 levels | [138,139] | |
| miR-10a, miR-21 | Glioma | MDSC expansion by targeting RAR and PTEN | [140] | |
| miR-1246 | Mutant p53 cancer cell lines | M2 macrophage polarization via targeting TERF2IP | [141,142] | |
| Let7 | Hypoxic tumor cells | M2 polarization and metabolic reprogramming in macrophages via suppression of AKT-mTOR signaling | [143] | |
| miR-29a-3p and miR-21-5p | TAMs in OC | Suppression of STAT3 in CD4 T-cells and corresponding Treg induction | [144] | |
| miR-125b-5p | Melanoma | M2 macrophage polarization via downregulation of LIPA | [145] | |
| miR-27a-3p | BC | PD-L1-mediated immune evasion via targeting MAGI2/PTEN/PI3K signaling | [146] | |
| Intra- vasation | miR-939 | BC | Downregulation of VE-Cadherin in ECs | [147] |
| miR-105 | BC | Downregulation of ZO-1 in ECs | [148] | |
| miR-181c | BC | Destruction of blood–brain barrier and promotion of brain metastasis via downregulation of PDPK1 | [149] | |
| Angio- genesis | miR-17-5p | NPC | Promoting angiogenic activity in ECs via AKT/VEGF-A expression | [115] |
| Mir-155 | GC | Suppression of FOXO3a and c-MYC to enhance the expression of VEGF in ECs | [150] | |
| miR-27a | PDAC | Proliferation of ECs via inhibition of BTG2 | [13] | |
| miR-135b, miR-210, miR-21, miR-23a | Hypoxic tumor cells | Induction of blood vessel formation | [151,152,153] | |
| MALAT1 | OC | Pro-angiogenic gene expression in HUVECs | [154] | |
| miR-205 | OC | Promotes angiogenesis in ECs via PTEN-AKT signaling | [127] | |
| lncRNA-Ccat2 | Glioma | Proliferation of ECs via upregulation of VEGF-A and TGFβ. Inhibition of apoptosis by targeting Bax and caspase-3 | [155] | |
| lncRNA-Pouf3 | Glioma | Pro-angiogenic gene expression in ECs | [155] | |
| Lymphangiogenesis | miR-221-3p | CeC | Expansion of lymphatic vessels via downregulation of VASH | [156] |
| LNMAT2 | Bladder cancer | Growth of lymphatic vessels via upregulation of PROX1 | [157] | |
| Organo- tropism | miR-141-3p | PC | Bone metastasis via increased osteoblast activity | [15] |
| miR-940 | PC, BC | Bone metastasis via increased osteoblast activity | [158] | |
| PMN | miR-122 | BC | Suppression of glycolytic enzymes in non-tumor cells | [116] |
| miR-1247-3p | HCC | CAF formation | [159] | |
| miR-451 | CAFs in ESCC | Pro-tumor PMN formation | [160] | |
| miR-25-3p | CRC | Enhanced vascular permeability and angiogenesis in PMN via targeting KLF2 and KLF4 | [161] |
Abbreviations: BC: breast cancer; CCRCC: clear cell renal cell carcinoma; CeC: cervical cancer; CRC: colorectal cancer; ESCC: esophageal squamous cell carcinoma; GBM: glioblastoma; GC: gastric cancer; HCC: hepatocellular carcinoma; LC: lung cancer; NPC: nasopharyngeal carcinoma; NSCLC: non-small-cell lung cancer; OC: ovarian cancer; OSCC: oral squamous cell carcinoma; PC: prostate cancer; PDAC: pancreatic ductal adenocarcinoma; TAM: tumor-associated macrophage; CAF: cancer-associated fibroblast; PMN: pre-metastatic niche.
Table 2.
sEV-ncRNA cargo used as diagnostic and prognostic biomarkers in cancer.
| Cancer | sEV Cargo | Source | Diagnostic/Prognostic Value | Reference |
|---|---|---|---|---|
| CRC | miR-92a-3p | Serum | Liver and lung metastasis | [262] |
| miR-193a | Plasma | Liver metastasis | [47] | |
| miR-25-3p | Serum | Metastasis (Liver and lung metastasis in mice; involved PMN formation) | [161] | |
| miR-141-3p miR-375 | Plasma | Liver metastasis | [263] | |
| miR-21-5p | Plasma | Liver metastasis | [264] | |
| lncRNA CRNDE-h | Serum | Regional lymph node and distant metastasis | [265] | |
| miR-19b, miR-21, miR-222, miR-92a | Serum | Metastasis | [246] | |
| Low miR-548c-5p | Serum | Liver metastasis | [266] | |
| PC | miR-141 miR-375 | Serum | Metastasis | [267,268] |
| Low miR-636 High miR-21 High miR-16 High miR-142-3p High miR-451 | Urine | Metastasis | [245] | |
| miR-1246 | Serum | Metastasis | [269] | |
| GC | miR-21 miR-1225-5p | PLF | Peritoneal metastasis | [270] |
| miR-21-5p miR-92a-3p miR-223-3p miR-342-3p | PLF | Peritoneal metastasis | [244] | |
| miR-423-5p | Serum | Lymph node metastasis | [271] | |
| miR-10b-5p | Plasma | Lymph node metastasis | [272] | |
| miR-101-3p | Plasma | Ovarian metastasis | [272] | |
| miR-143-5p | Plasma | Liver metastasis | [272] | |
| OC | miR-200b miR-200c | Serum | Lymph node metastasis | [273] |
| ESCC | miR-21 | Serum | Metastasis | [274] |
| HCC | miR-665 | Serum | Metastasis | [275] |
| miR-1247-3p | Serum | Lung metastasis | [159] | |
| LSCC | miR-21 | Serum | Lymph node metastasis | [243] |
| Glioma | miR-21 | CSF | Tumor spinal/ventricle metastasis | [242] |
| BC | miR-105 | serum | Distant metastasis | [148] |
| miR-21 | Serum | Bone metastasis | [241] | |
| NSCLC | circSATB2 | Serum | Lymphatic metastasis | [276] |
| PDAC | miR-17-5p | Serum | Metastasis | [277] |
| Circ-IARS | Plasma | Tumor-node metastasis and liver metastasis | [278] | |
| miR-21 | Plasma | Lymph node metastasis | [240] | |
| Melanoma | miR-17 miR-19a miR-21 miR-126 miR-149 | Plasma | Metastasis | [279] |
Abbreviations: BC: breast cancer; CRC: colorectal cancer; ESCC: esophageal squamous cell carcinoma; GC: gastric cancer; HCC: hepatocellular carcinoma; LSCC: Laryngeal squamous cell carcinoma; NSCLC: non-small cell lung cancer; OC: ovarian cancer; PC: prostate cancer; PDAC: pancreatic ductal adenocarcinoma; CSF: cerebrospinal fluid; PLF: peritoneal lavage fluid.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seibold, T.; Waldenmaier, M.; Seufferlein, T.; Eiseler, T. Small Extracellular Vesicles and Metastasis—Blame the Messenger. Cancers 2021, 13, 4380. https://doi.org/10.3390/cancers13174380
AMA Style
Seibold T, Waldenmaier M, Seufferlein T, Eiseler T. Small Extracellular Vesicles and Metastasis—Blame the Messenger. Cancers. 2021; 13(17):4380. https://doi.org/10.3390/cancers13174380
Chicago/Turabian StyleSeibold, Tanja, Mareike Waldenmaier, Thomas Seufferlein, and Tim Eiseler. 2021. "Small Extracellular Vesicles and Metastasis—Blame the Messenger" Cancers 13, no. 17: 4380. https://doi.org/10.3390/cancers13174380
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.