
Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma



Department of Oncological Sciences, Department of Pathology, and Huntsman Cancer Institute, University of Utah, Salt Lake City, UT 84112, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(16), 4202; https://doi.org/10.3390/cancers13164202
Submission received: 26 June 2021
/
Revised: 12 August 2021
/
Accepted: 15 August 2021
/
Published: 20 August 2021
Abstract
:Simple Summary
Liver cancer is one of the deadliest human cancers. Two of the most common molecular aberrations in liver cancer are: (1) activating mutations in the gene encoding β-catenin (CTNNB1); and (2) promoter mutations in telomerase reverse transcriptase (TERT). Here, we review recent findings regarding the interplay between TERT and β-catenin in order to better understand their role in liver cancer.
Abstract
Hepatocellular carcinoma (HCC) is one of the deadliest human cancers. Activating mutations in the telomerase reverse transcriptase (TERT) promoter (TERTp) and CTNNB1 gene encoding β-catenin are widespread in HCC (~50% and ~30%, respectively). TERTp mutations are predicted to increase TERT transcription and telomerase activity. This review focuses on exploring the role of TERT and β-catenin in HCC and the current findings regarding their interplay. TERT can have contradictory effects on tumorigenesis via both its canonical and non-canonical functions. As a critical regulator of proliferation and differentiation in progenitor and stem cells, activated β-catenin drives HCC; however, inhibiting endogenous β-catenin can also have pro-tumor effects. Clinical studies revealed a significant concordance between TERTp and CTNNB1 mutations in HCC. In stem cells, TERT acts as a co-factor in β-catenin transcriptional complexes driving the expression of WNT/β-catenin target genes, and β-catenin can bind to the TERTp to drive its transcription. A few studies have examined potential interactions between TERT and β-catenin in HCC in vivo, and their results suggest that the coexpression of these two genes promotes hepatocarcinogenesis. Further studies are required with vertebrate models to better understand how TERT and β-catenin influence hepatocarcinogenesis.
1. Introduction
Hepatocellular carcinoma (HCC) forms 80% of all primary liver cancers and is the fourth leading cause of cancer-related mortality globally. Effective treatments for this disease are limited due to its molecularly heterogeneous nature [1]. Telomerase reverse transcriptase (TERT), TP53 (tumor protein p53), and CTNNB1 encoding β-catenin are the top three most frequently mutated genes in HCC, altered in 47.1%, 29.2%, and 27.4% of cases from large-scale sequencing projects [2].
Both TERT and β-catenin play essential roles in liver development, tumorigenesis, and stemness pathways. In embryonic stem and progenitor cells, the TERT and WNT/β-catenin signaling axes crosstalk such that TERT enhances the transcription of β-catenin target genes, while β-catenin enhances the transcription of TERT by binding to its promoter region [3,4]. In HCC clinical specimens, activating β-catenin and TERT promoter (TERTp) mutations show significant concordance such that 74% of CTNNB1-mutated HCC also have TERTp mutations [5].
This CTNNB1-TERT association is special because CTNNB1 mutations are mutually exclusive to TP53 mutations [6] and to other perturbations in WNT pathway genes, such as AXIN1 and CCND1 [7]. The chromatin remodelers ARID2 (AT rich interactive domain 2) and ARID1A (AT rich interactive domain 1A) are associated with mutations in CTNNB1 (p-value = 0.005) and AXIN1 (p-value = 0.002), respectively [6]. Potential mechanisms underlying the ARID2-CTNNB1 and ARID1A-AXIN1 associations in liver cancer have not been explored in depth, perhaps because these genes are less commonly altered in HCC (~10%) than TERT or CTNNB1 [2].
As TERTp and activating β-catenin mutations are concordant in HCC and the two signaling axes directly interact in stem cells, it is crucial to define how these pathways cooperate in HCC. This review focuses on current advances in delineating the role of TERT and CTNNB1 in HCC and the relationship between these two genes during carcinogenesis.
2. Telomeres, Telomerase, and TERT
Telomeres are ribonucleoprotein caps composed of short repetitive non-coding DNA sequences and associated proteins that protect the ends of eukaryotic chromosomes [8]. Telomere length varies among species and serves to mitigate two biological complications. First, together with the associated Shelterin complex, telomeres help protect DNA near chromosome ends from the DNA damage response (DDR), which is induced by double-stranded breaks in the DNA and can lead to cell cycle arrest, apoptosis, or repair. Second, telomeres provide a cushion of non-coding DNA to postpone the end replication crisis, in which genome instability results from gradual shortening of the lagging strand in semi-conservative DNA replication. Due to the presence of telomeres, chromosomes can withstand 50–90 replication cycles while losing around 50–150 nucleotides per mitosis, even in the absence of telomere maintenance mechanisms [9].
Telomerase, a multi-subunit reverse transcriptase enzyme, plays a significant role in telomere maintenance. Telomerase levels are undetectable in most somatic cells; however, this enzyme is active in certain proliferating cells, germ cells, and the majority of cancers (~90%) [10]. The telomerase holoenzyme is composed of a catalytic protein subunit encoded by TERT, an RNA template component (TERC), and auxiliary proteins. TERT consists of four main domains: the TERT N-terminal domain (TEN), the TERT RNA binding domain (TRBD), a reverse transcriptase domain (RT), and a C-terminal extension domain. The TEN domain binds to the ssDNA of telomere overhangs at the 3′ end, the TRBD domain interacts with TERC, and the RT and C-terminal extension domains are involved in catalyzing the addition of telomere repeats to the 3′ end and binding to the resultant DNA/RNA hybrid [8,10]. In somatic cells, TERT is transcriptionally repressed via epigenetic mechanisms, mainly in the TERTp region [9].
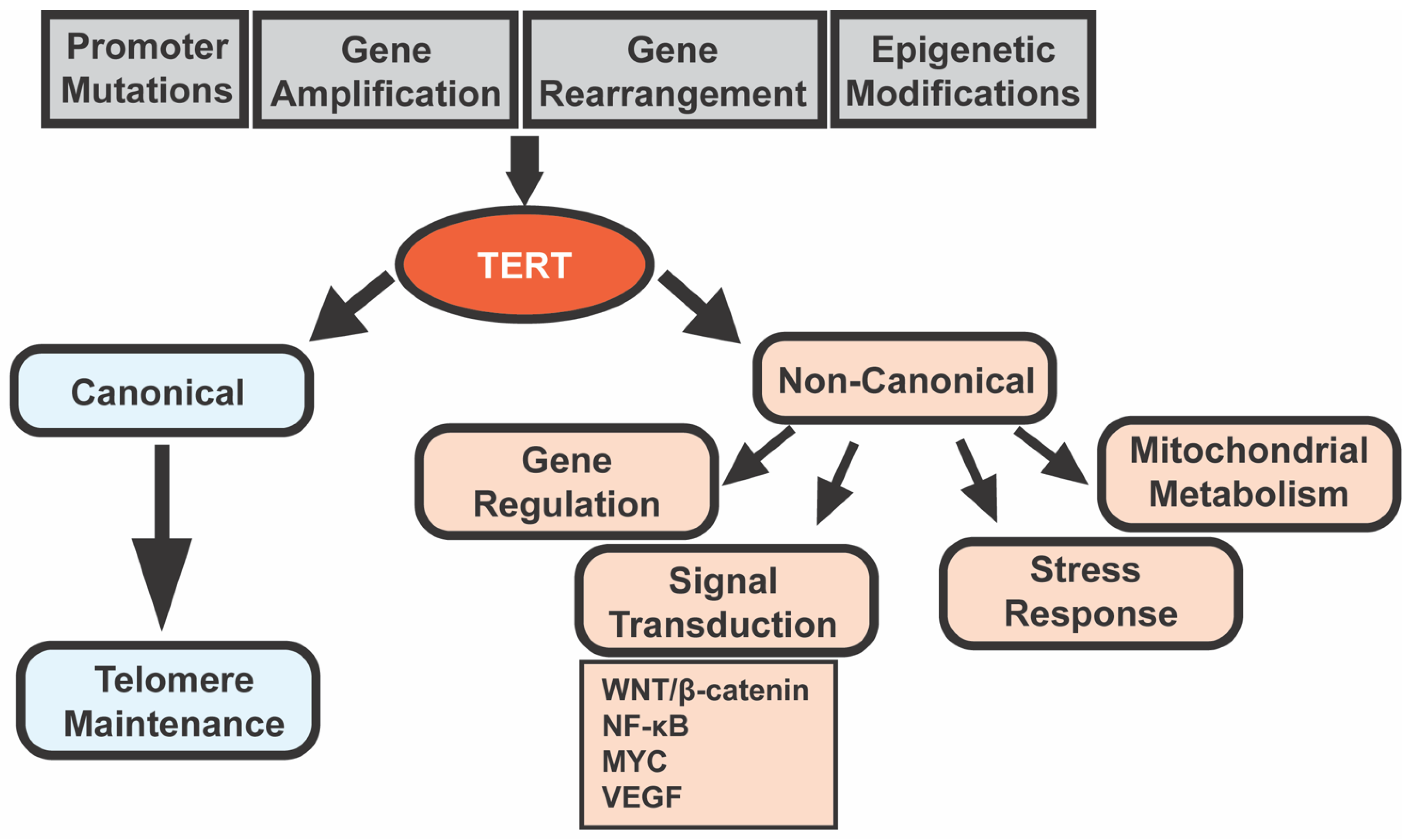
TERT is also involved in many roles independent of its telomere lengthening activity [11]. These non-canonical functions of TERT include regulating gene expression, signal transduction, mitochondrial metabolism, and resistance to ionizing radiation [12]. Some of the most well-studied pathways regulated by TERT include the WNT/β-catenin pathway and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, along with MYC, vascular endothelial growth factor (VEGF), and others [11]. TERT is found not only in the nucleus but also in the cytoplasm and mitochondria and is involved in tumorigenesis and cancer therapy resistance independent of telomere lengthening [12]. For example, the overexpression of mitochondrial TERT by HCC cells increases chemotherapeutic resistance (in vitro and in vivo) by increasing the mitochondrial membrane potential and inhibiting the mitochondrial apoptotic pathway [13]. The diverse roles of TERT are highlighted in Figure 1.
3. Telomerase and Cancer
Without a telomere maintenance mechanism, cells undergoing oncogenesis and rapid divisions gradually suffer from telomere attrition, which eventually triggers replicative senescence or apoptosis [14]. To overcome this replication barrier caused by critical telomere length, cancer cells usually increase the TERT levels and/or TERT activity via (1) TERTp mutations, which increase TERT mRNA levels by creating new ETS binding motifs [15]; (2) amplification of TERT and/or TERC; (3) TERT rearrangement; and/or (4) epigenetic regulation of TERT and its promoter [8]. As cells in high-turnover tissues have higher telomerase activity, they may be able to stably upregulate TERT without acquiring activating mutations like TERTp mutations [14]. Supporting this possibility, cancers originating from tissues with high turnover rates tend to have a lower frequency of TERTp mutations than those from low-turnover tissues [14].
Thus, telomerase promotes telomere maintenance, and telomere maintenance is a cancer hallmark that allows malignant cells to overcome replicative senescence. TERT also promotes cellular transformation and proliferation independent of its telomerase activity [11]. Therefore, one would predict that loss-of-function mutations in TERT or TERC would inhibit tumorigenesis, while overexpression of TERT would promote tumor formation. Table 1 summarizes recent findings from cancer models with manipulation of Tert or Terc in mice or zebrafish. Supporting the aforementioned prediction, Tert overexpression in mice leads to an increased incidence of T-cell lymphomas [16], skin papillomas [17], and mammary tumors [18], depending on the promoter used to drive its expression (Table 1). Tert loss of function mutants display a delayed onset of mammary tumors [19], lymphomas [20], and early HCCs [21] (Table 1).
On the other hand, Terc knockdown in mice results in the converse effect: enhancement of tumor formation (Table 1). Terc−/− mice treated with carbon tetrachloride (CCl4) or diethylnitrosamine (DEN) display increased HCC “initiation foci”—microscopic lesions with atypical cytologic or architectural features that suggest malignancy but are not sufficiently well-developed to diagnose as HCC—although HCC number and size were decreased [22]. Similarly, loss-of-function mutation of zebrafish tert or terc resulted in an earlier onset of spontaneous tumors, including intestinal adenocarcinomas, hematopoietic neoplasms, hepatocellular adenomas, and germ cell tumors [23].
Why does telomerase seem to promote tumorigenesis in some animal models yet inhibit it in others? One possible explanation is that long-term inhibition of Tert or Terc, as in these genetic animal models, results in compensation by other telomere maintenance programs, such as alternative lengthening of telomeres (ALT). ALT is a telomerase-independent mechanism for telomere maintenance. ALT-mediated telomere synthesis occurs via homologous recombination between non-allelic telomeres, sister chromatids at telomeric sites, and/or linear or circular extrachromosomal telomeric repeats [24]. This compensatory mechanism might not be relevant to human hepatocarcinogenesis, during which TERT could potentially be inhibited more acutely using drugs. Inducible and/or conditional animal models in which Tert and/or Terc can be turned on at different time points during tumorigenesis would be helpful to dissect the importance of acute versus chronic telomerase inhibition.
Another possible reason why telomerase might inhibit tumorigenesis in some animal models while promoting it in others is related to the balance between canonical and non-canonical effects of Tert. Laboratory mouse models have long telomeres (25–40 kb vs. 10–15 kb in humans) that are likely sufficient to permit malignant transformation even in the absence of telomerase enzyme [12]. Therefore, Tert overexpression and mutant phenotypes in mouse tumor models may be at least partially independent of Tert telomere lengthening activity [16,17,18]. As discussed in the section above, non-canonical functions of TERT tend to be tumorigenic and anti-apoptotic. Thus, Tert deficiency in mice may delay tumorigenesis due to the resultant reduction in its non-canonical activity [19,21].
Zebrafish have telomere lengths similar to humans (~5–15 kb) [25] and, like humans, exhibit drastic shortening of telomeres with age [23]; therefore, the manipulation of tert levels is more likely to impact telomere length than in mice. Thus, the canonical effects of tert may be relatively more important to its role in zebrafish carcinogenesis. Zebrafish tert deficiency promotes the initiation of tumorigenesis through shortened telomeres that induce DNA damage responses, inflammation, and genetic instability [23]. A similar phenomenon occurs in aged late-generation Terc−/− mice, wherein shortened telomeres are associated with ulcerated skin lesions, defective wound healing, and genomic instability [26].
These findings highlight that TERT influences tumorigenesis in both canonical and non-canonical ways. Whether the net effects of TERT are pro- or anti-tumor depends on the stage of disease, tissue type, telomere biology in different animal models, when during tumor formation TERT is manipulated, and other factors. Additional studies of vertebrate HCC models, including rigorous examination of telomere length during carcinogenesis, could help define the importance of TERT’s telomerase activity to HCC pathogenesis.

{kind=link}
{kind=link}
{kind=link}
Table 1.
TERT cancer models in mice and zebrafish.
| Gene (Animal) | Site Specificity | Expression * | Result ** | Ref. |
|---|---|---|---|---|
| Tert (mouse) | Thymocytes and peripheral T cells | + | ↑ T-Cell Lymphomas | [16] |
| Tert (mouse) | Basal keratinocytes | + | ↑ skin papillomas (DMBA + TPA induction) | [17] |
| Tert (mouse) | Whole body | + | ↑ mammary tumors in aged females | [18] |
| tert and terc (zebrafish) | Neural progenitor cells | + | ↓ aggressiveness of RAS-mediated brain tumors | [27] |
| Tert (mouse) | Whole body | − | Delayed onset of lymphomas in EμMYC mice | [20] |
| Tert (mouse) | Whole body | − | Delayed onset of mammary tumors in PyMT mice | [19] |
| Tert (mouse) | Whole body | − | ↓ HCC “initiation foci” (CCl4 induction) | [21] |
| Terc (mouse) | Whole body | − | ↑ HCC “initiation foci” but ↓ HCC progression (uPA, CCl4 or DEN induction) | [22] |
| Terc (mouse) | Whole body | − | ↑ tumors (lymphomas, teratocarcinomas, HCC, squamous cell carcinoma) | [26] |
| Terc (mouse) | Whole body | − | ↓ skin papillomas (DMBA + TPA induction) | [28] |
| Terc (mouse) | Whole body | − | ↑ epithelial cancers in TP53−/− mice | [29] |
| Terc (mouse) | Whole body | − | ↑ adenoma initiation but ↓ progression in ApcMin mice | [30] |
| terthu3430 (zebrafish) | Whole body | − | Earlier onset of tumors (germ cell tumors, hematopoietic neoplasms, HCA, etc.) | [23] |
| terthu3430 (zebrafish) | Whole body | − | ↑ tumor incidence and aggressiveness of melanoma model *** | [31] |
*: +, gene overexpression; −, gene knockout or knockdown. **: ↑, increased; ↓, decreased. *** mitfa: HRAS (gives rise to melanomas) blastula cells transplanted into tert−/− casper embryos. Abbreviations: DMBA, 7,12-dimethylbenz[a]anthracene; TPA, 12-o-tetradecanoylphorbol 13-acetate; PyMT, polyomavirus middle T oncogene; CCl4, carbon tetrachloride; uPA, urokinase plasminogen activator; DEN, diethylnitrosamine; HCA, hepatocellular adenoma; Apc, adenomatous polyposis coli; ApcMin, multiple intestinal neoplasia (mutant) allele of Apc gene; and terthu3430, tert mutant line (allele hu3430) with a non-sense mutation resulting in a premature stop codon in exon 2 of tert gene.
4. TERT Promoter Mutations in HCC
HCC is an extremely heterogeneous disease with its prevalence, etiology, and epidemiology showing marked variance across cohorts separated by geography, ancestry, and sex. Eighty percent of HCCs arise in a cirrhotic background caused by various insults, including hepatitis C virus (HCV) and hepatitis B virus (HBV) infections, alcohol intake, non-alcoholic steatohepatitis (NASH), and aflatoxin exposure [32]. Less commonly, HCC occurs via malignant transformation in the non-cirrhotic liver or hepatocellular adenoma (HCA) [32]. The variable etiological and epidemiological causes contribute to the molecularly heterogeneous nature of HCC [32].
TERT re-expression is observed in approximately 90% of HCC cases [33]. Genetic alterations in TERT can be due to TERTp mutations (54% of cases), TERT focal amplification (6.7%), or HBV genome integration into the TERT promoter (22% of HBV-related HCC) [7]. TERTp mutations are mutually exclusive with TERT focal amplifications and HBV genome integration in HBV positive cases [7].
TERTp mutations in HCC typically involve one of two mutational hotspots of cytosine to thymine transitions, 1,295,228 C > T (termed C228T) and 1,295,250 C > T (C250T) at −124 bp and −146 bp, respectively [34]. Each of these mutually exclusive mutations creates de novo binding sites for ETS transcription factors within the TERT promoter. These mutations are predicted to increase TERT expression levels, enzymatic activity, and telomere length; they are also associated with decreased survival in cancer patients [35,36,37,38].
For example, TERTp mutations significantly increased TERT mRNA levels in human malignant pleural mesothelioma samples (only C228T mutation detected; p-value = 0.02) [39] and were significantly correlated with thyroid cancer mortality (83% C228T, 17% C250T; p-value < 0.001) [40]. These mutations are linked to increased TERT expression (p-value < 0.0001 and p-value = 0.007) in HCC patients as well [5,41].
In accordance with HCC heterogeneity, the prevalence of TERTp mutations also varies based on etiological/epidemiological factors. We performed a PubMed search using the search terms “TERTp”, “mutations”, and “hepatocellular carcinoma”. We reviewed all studies using archived tumor genomic DNA or formalin fixed tumor tissue samples with matching non-tumor controls. Table 2 summarizes these studies, delineating the prevalence of TERTp mutations across diverse geographies and etiologies. This table focuses on the two common hotspot mutations mentioned in the above paragraph and excludes other rare TERTp mutations, if detected. Overall, TERTp mutations are enriched in HCV-related HCC compared to HBV or non-viral HCC cases [7,35,42,43,44,45,46] and may be associated with the male sex [5,35,44]. C228T mutations are the dominant TERTp mutation regardless of etiology (Table 2).
An analysis of 1061 HCC genomes from four different geographical locations showed a significant link between TERTp mutations and more aggressive tumors (p-value = 1.02 × 10−5), relapse (p-value = 2.33 × 10−12), and decreased overall survival (p-value = 2.81 × 10−5) [47].
Table 2.
Landscape of TERTp mutations in HCC.
| Region | Etiology (N) | C228T Incidence % (N) | C250T Incidence % (N) | Association * | Ref. |
|---|---|---|---|---|---|
| (1) Asia (2) Africa (3) Europe | 52.3% HBV, rest unknown (regions 1–3) (44) | (1) 16% (3) (2) 33% (5) (3) 20% (2) | (1) 5% (1) (2) 20% (3) (3) 10% (1) | ↑ frequency in Africans vs. non-Africans (p = 0.056) and in HBV− vs. HBV+ (p = 0.295) (regions 1–3) | [48] |
| Southern Italy | (67) | 41.8% (28) | 0% (0) | ↑ TERT expression in mutated tumors vs. control tissue (p < 0.0001) | [41] |
| Southern Italy | 7.9% HBV+, 86.6% HCV+ (127) | 48.8% (62) | 1.6% (2) | ↑ frequency in HCV+ vs. HBV+ (p < 0.001) | [43] |
| Germany | (78) | 47.4% (37) | [49] | ||
| France, Italy and Spain | 14% HBV+, 26% HCV+ (243) | 54.3% (132) | 2% (5) | [6] | |
| France | 22% HBV+, 26% HCV+ (305) | 55% (168) | 3.6% (11) | ↑ frequency in males vs. females (p = 0.001) and in HBV− vs. HBV+ (p < 0.0001); ↑ TERT expression in mutated HCC vs. normal liver, cirrhosis, and HCA (p = 0.0007) | [5] |
| USA | 24.6% HBV+, 26% HCV+ (61) | 42.6% (26) | 1.6% (1) | no correlation with etiology, sex, age, or ethnicity | [50] |
| (1) USA (2) Japan | (1) 14.6% HBV+, 57% HCV+ (89) (2) 28.6% HBV+, 37% HCV+ (374) | (1) 34.8% (31) (2) 55.6% (208) | (1) 2.2% (2) (2) 2.4% (9) | ↑ frequency in HCV+ vs. HCV− (p = 0.0016) | [7,42] |
| China | 94% HBV+ (276) | 30.5% (84) | 0.36% (1) | ↑ frequency in older vs. younger age (p = 0.04); no correlation with sex or etiology | [51] |
| China | (35) | 25.7% (9) | 5.7% (2) | [52] | |
| China | 83% HBV+ (190) | 26.3% (50) | 3.7% (7) | no correlation with age, sex, etiology, or tumor status | [53] |
| Japan | (11) | 81.8% (9) | [54] | ||
| Japan | 23% HBV+, 61.6% HCV+ (125) | 66.4% (83) | 1.6% (2) | ↑ frequency in HCV+ vs. HCV− (p = 0.0007) and in viral vs. non-viral (p = 0.0282) | [45] |
| South Korea | 36% HBV+, 3% HCV+ (160) | 20% (32) | 8.75% (14) | ↑ frequency in males vs. females (p = 0.027) and in HCV+ vs. HCV− (p = 0.285); no association with telomere length or HCC prognosis | [35] |
| South Korea | 74% HBV+, 5.7% HCV+ (105) | 37% (39) | 1.9% (2) | ↑ frequency in HCV+ vs. HCV− (p = 0.001) | [46] |
| Taiwan | 63% HBV+, 40.2% HCV+ (195) | 27.7% (54) | 1.5% (3) | ↑ frequency in HCV+ vs. HCV− (p = 0.0048), older vs. younger age (p = 0.0122), and HBV− vs. HBV+ (p = 0.0007) | [34] |
| USA, Canada, South Korea, Vietnam, and Russia | 22.4% HBV+, 17.8% HCV+ (196) | 40.8% (80) | 3.6% (7) | ↑ frequency in older vs. younger age (p = 0.0006), males vs. females (p = 0.006), HCV+ vs. HCV− (p = 0.04), and HBV− vs. HBV+ (p = 0.02); no association with TERT expression | [44] |
*: ↑, increased or higher.
Apart from the two hotspot mutations (C228T and C250T), the TERT promoter SNP rs2853669 is also common in several cancers and is predicted to decrease TERT expression [55]. However, little is known of its role in HCC. Ko et al. analyzed Korean HCC samples and observed that the combination of rs2853669 with TERTp mutation decreased patient survival and increased cancer recurrence risk. The SNP alone had no effect. This combination led to increased TERT expression and telomere length compared to rs2853669 only [56].
A study from Southern Italy analyzed 84 HCC cases and did not find a correlation between TERTp mutations and survival with or without the SNP [43]. However, the frequency of SNP genotypes was not in Hardy–Weinberg equilibrium in cases with TERTp mutations, indicating the presence of selective pressure for the combination of TERTp mutation and the rs2853669 SNP [43]. Other studies have noted a high prevalence of this SNP in HCC patients with no link to increased TERT expression or TERTp mutational frequency [41,57]. To better understand the prognostic value of this SNP’s association with TERTp mutations, further clinical studies with larger and diverse (in terms of etiology, sex, driver mutations, etc.) cohorts are needed as well as more mechanistic studies.
5. WNT/β-Catenin Signaling in Normal and Cancer Cells
The WNT/β-catenin signaling pathway, depicted in Figure 2A, is a critical regulator of the proliferation and differentiation of stem and progenitor cells [58]. In the absence of WNT ligands, a destruction complex comprising adenomatous polyposis coli (APC), AXIN, glycogen synthase kinase 3β (GSK-3β), and casein kinase 1α (CK1α) phosphorylates cytoplasmic β-catenin at its N-terminal serine/threonine residues. This phosphorylation triggers its ubiquitination followed by proteasomal degradation [58,59]. As cytoplasmic β-catenin gets degraded, it cannot accumulate and translocate into the nucleus to drive the expression of WNT target genes.
In the presence of WNT ligands, binding of the receptor complex destabilizes the destruction complex and leads to the stabilization and cytoplasmic accumulation of β-catenin [58,59]. β-catenin then undergoes nuclear translocation where it binds to T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors and replaces the transcriptional repressor Groucho. This complex formation drives the expression of WNT target genes, such as C-MYC, EPCAM, CYCLIN D1, and CD44, which promote cell cycle progression, proliferation, metabolism, cell survival, immune tolerance, and angiogenesis [58,59].
As a central regulator of the WNT cascade, β-catenin levels can be increased in cancer via stabilizing mutations, which increase the nuclear accumulation of β-catenin and upregulate WNT/β-catenin signaling [60,61]. Exon 3 is a mutational hotspot for such stabilizing missense mutations that target the serine/threonine sites that render β-catenin resistant to GSK-3β mediated phosphorylation and, thereby, to subsequent proteasomal degradation. Some of the most common mutational sites in this hotspot are S33, S37, S45, T41, D32, and G34 [60]. Activating mutations in CTNNB1 have been reported in several cancers, including colorectal cancer, HCC, endometrial cancer, melanoma, ovarian cancer, and thyroid cancer [61].
Figure 2.
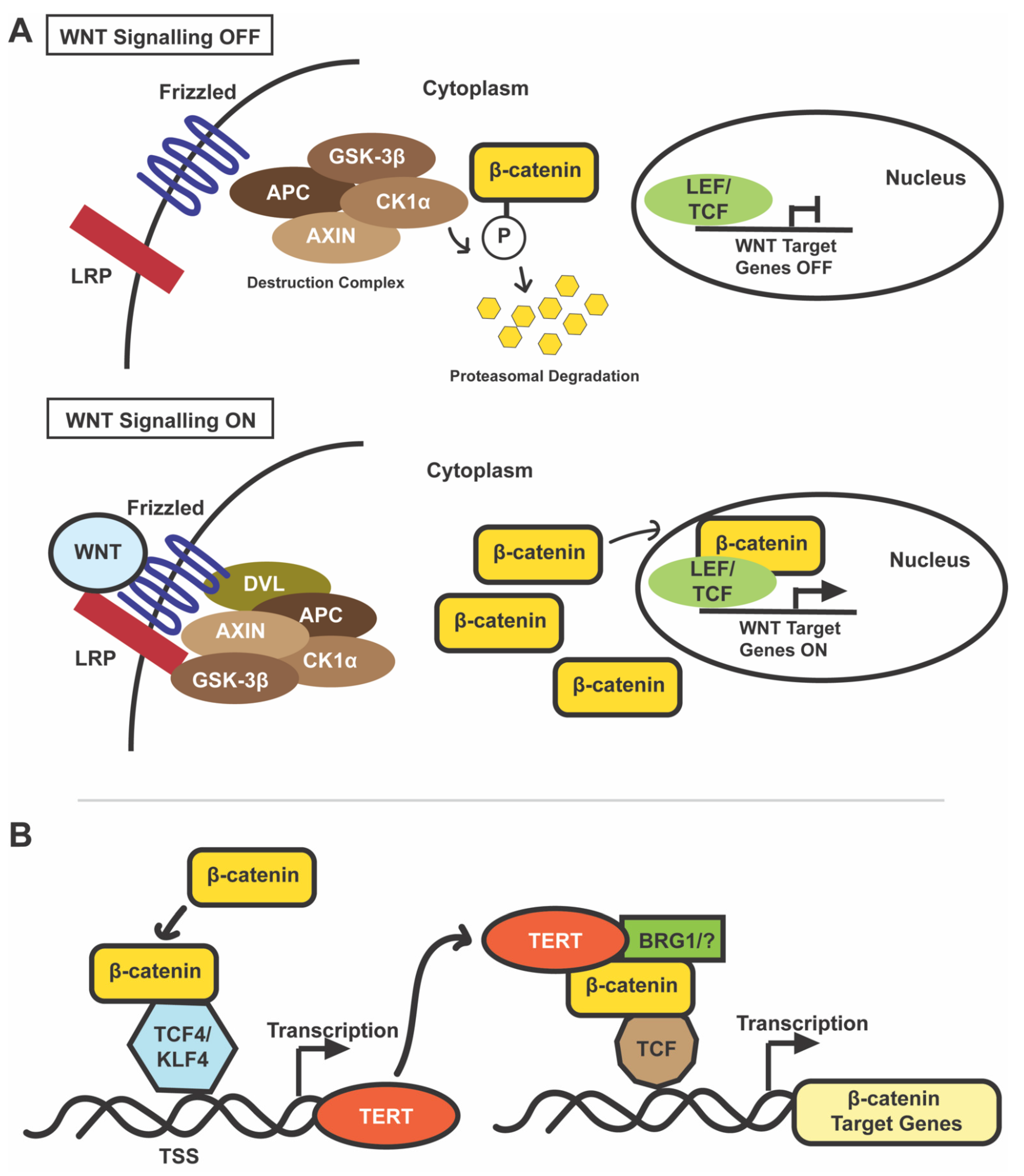
(A) Canonical WNT/β-catenin pathway. When WNT signaling is OFF (top), cytoplasmic β-catenin is phosphorylated by a destruction complex comprising of APC, AXIN, GSK-3β, and CK1α [58,59]. This phosphorylation marks it for proteasomal degradation, and consequently it is unavailable to bind to LEF/TCF elements in the nucleus to drive transcription of target genes [58,59]. When WNT signaling is ON, WNT ligands bind to LRP and Frizzled receptors leading to the destabilization of the destruction complex by a mechanism that may involve DVL protein [58,59]. The degradation complex is, thus, unable to phosphorylate and degrade cytoplasmic β-catenin [58,59]. β-catenin accumulates in the cytoplasm and then enters the nucleus to bind to TCF/LEF elements and drive transcription of WNT target genes [58,59]. (B) Direct interplay of TERT and β-catenin signaling. β-catenin enhances transcription of TERT by binding to its promoter region [4]. In mouse ES cells, this β-catenin-TERT interaction is facilitated by Klf4 [4]; another study showed that β-catenin induced an increase in TERT mRNA and telomerase activity is dependent on TCF4 binding to TERTp [62]. TERT modulates WNT signaling by interacting with BRG1 (a chromatin-remodeling protein; “?” indicates uncertainty as some groups did not observe this association), binding to the promoter of WNT responsive genes, and acting as a co-factor [3]. This transcriptional upregulation of WNT target genes by TERT is independent of its catalytic activity [3]. Abbreviations: APC, adenomatous polyposis coli; CK1α, casein kinase 1α; GSK-3β, glycogen synthase kinase 3β; and DVL, disheveled.
Figure 2.
(A) Canonical WNT/β-catenin pathway. When WNT signaling is OFF (top), cytoplasmic β-catenin is phosphorylated by a destruction complex comprising of APC, AXIN, GSK-3β, and CK1α [58,59]. This phosphorylation marks it for proteasomal degradation, and consequently it is unavailable to bind to LEF/TCF elements in the nucleus to drive transcription of target genes [58,59]. When WNT signaling is ON, WNT ligands bind to LRP and Frizzled receptors leading to the destabilization of the destruction complex by a mechanism that may involve DVL protein [58,59]. The degradation complex is, thus, unable to phosphorylate and degrade cytoplasmic β-catenin [58,59]. β-catenin accumulates in the cytoplasm and then enters the nucleus to bind to TCF/LEF elements and drive transcription of WNT target genes [58,59]. (B) Direct interplay of TERT and β-catenin signaling. β-catenin enhances transcription of TERT by binding to its promoter region [4]. In mouse ES cells, this β-catenin-TERT interaction is facilitated by Klf4 [4]; another study showed that β-catenin induced an increase in TERT mRNA and telomerase activity is dependent on TCF4 binding to TERTp [62]. TERT modulates WNT signaling by interacting with BRG1 (a chromatin-remodeling protein; “?” indicates uncertainty as some groups did not observe this association), binding to the promoter of WNT responsive genes, and acting as a co-factor [3]. This transcriptional upregulation of WNT target genes by TERT is independent of its catalytic activity [3]. Abbreviations: APC, adenomatous polyposis coli; CK1α, casein kinase 1α; GSK-3β, glycogen synthase kinase 3β; and DVL, disheveled.

6. WNT/β-Catenin in HCC
The WNT/β-catenin pathway is integral for liver development and homeostasis [59]. Its dysregulation is linked to various hepatic diseases, including HCC. Aberrant WNT/β-catenin signaling has been detected in up to 66% of HCC cases (mutually exclusive mutations in CTNNB1, AXIN1, and APC, and CCND1-FGF19 amplification) [7]. Deletions or missense mutations in the CTNNB1 gene are the most frequent WNT/β-catenin dysregulating event in HCC [7]. In terms of etiology, CTNNB1 mutations are strikingly more prevalent in HCV-related HCCs (~26.7%) than in HBV-related HCC (11.6%) or non-viral HCC (21.2%) (p-value < 0.0001) [63]. These percentages may vary between studies depending on the cohorts, but CTNNB1 mutations are unanimously higher in HCV-related HCCs than HBV- or non-viral-related HCCs [64].
The results are conflicting concerning the prognostic value of CTNNB1 mutations. Khalaf et al. summarized clinical studies linked to β-catenin and HCC and their outcomes [64]. Certain studies indicate that the presence of CTNNB1 mutations is associated with decreased aggressiveness, invasiveness, serum α-fetoprotein levels, and more differentiated HCC [64,65,66,67,68,69], while others linked it to poorer prognosis (small vessel invasion and tumor capsule invasion) or no effect on survival [64,70].
Gain and loss of function studies in vertebrate models have helped to elucidate the role of CTNNB1 mutations in HCC initiation, progression, and maintenance. Table 3 summarizes how overexpressing (+) or knocking down (−) wild-type or mutant CTNNB1 in hepatocytes affects HCC. Studies led by Satdarshan Monga, Makoto Taketo, and Christine Perret have indicated that the expression of wild-type or activated forms of β-catenin in mouse hepatocytes under control of the Alb (albumin) or Fabp (fatty acid-binding protein) promoter is not enough to drive HCC [71,72,73,74] (Table 3). Similarly, targeting mouse hepatocytes with activated β-catenin by hydrodynamic tail vein injection (HDT) is not sufficient to induce HCC [75,76,77] (Table 3).
On the other hand, activated β-catenin can collaborate with other oncogenes or chemical insults to drive HCC in mice [72,75,76,77,78] (Table 3). In addition, expression of activated β-catenin in mouse liver progenitor cells or zebrafish hepatocytes is sufficient to induce HCC [79,80,81]. Together these studies indicate that activating β-catenin mutations promote HCC, although additional genetic or epigenetic alterations may be required for HCC initiation or progression in some contexts, particularly if activated β-catenin is turned on after early development.
While activated β-catenin drives HCC, endogenous β-catenin plays a restraining role in hepatocarcinogenesis in at least some contexts. Knockout of endogenous β-catenin is pro-tumorigenic in mouse DEN-induced hepatocarcinogenesis, an effect that is abrogated by treatment with the antioxidant N-acetyl-L-cysteine (NAC) [82]. Knockout of endogenous β-catenin also promotes mouse HCC induced by hydrodynamic tail vein injection (HDT) with activated β-catenin and MET [83]. Gene expression analysis of HCC lacking endogenous β-catenin showed an enrichment of inflammatory and DNA damage response genes [83].
Together, these studies suggest that inhibiting endogenous β-catenin may promote HCC by creating a pro-inflammatory, pro-oxidant microenvironment. As therapeutic targeting of β-catenin becomes more widespread the clinic, it will be critical to keep the anti-oncogenic effects of endogenous β-catenin in mind, as they may influence recurrence after targeting the primary tumor.
Table 3.
HCC animal models with CTNNB1 manipulation.
| Animal | Method * | Expression ** | CTNNB1 Status *** | Results | Ref. |
|---|---|---|---|---|---|
| Mice | Alb promoter | + | Wildtype | Transgenic mice develop hepatomegaly but no tumors | [71] |
| Alb promoter | + | Ser45 mutated | Transgenic mice develop hepatomegaly (only in younger mice) but no tumors; Increased HCC (DEN induction) | [72] | |
| Fabp: Cre | + | Exon 3 deleted | Transgenic mice develop hepatomegaly but no tumors | [73] | |
| CaBP9K promoter | + | N131 deleted | Transgenic mice develop hepatomegaly but no tumors | [74] | |
| Cited1: CreER | + | Exon 3 deleted | Transgenic mice develop HCCs, hepatoblastomas, and lung metastases | [79] | |
| AdCMV-Cre | + | Exon 3 deleted (and H-RAS) | Double transgenic mice develop HCC, but no HCC develops with expression of mutated CTNNB1 alone | [78] | |
| SB-HDT | + | S45 or S33 mutated (and MET) | Mice expressing MET and either form of mutated CTNNB1, but not mutated CTNNB1 alone, develop HCC | [75] | |
| SB-HDT | + | N90 deleted (and activated YAP) | Mice expressing activated YAP and mutated CTNNB1, but not mutated CTNNB1 alone, develop HCC | [76] | |
| SB-HDT | + | S45 or S33 mutated (and K-RAS) | Mice expressing K-RAS and either form of mutated CTNNB1, but not mutated CTNNB1 alone, develop HCC | [77] | |
| siRNA | − | S45 mutated (HDT induction) | Decreased HCC (K-RAS plus S45-mutated CTNNB1 HDT induction) | [77] | |
| Alb:Cre | − | Wildtype (endogenous) | Increased HCC (DEN induction) | [82] | |
| Alb:Cre | − | Wildtype (endogenous) | Increased HCC (MET and N90-deleted CTNNB1 induction) | [83] | |
| Zebrafish | fabp10a promoter | + | S33A, S37A, T41A, and S45A mutated | Transgenic zebrafish develop HCC as adults (78% by 6 months) | [81] |
| fabp: CreERT2 | + | Transgenic zebrafish develop HCC as adults (13% by 6 months) | [80] |
*: Method indicates promoter or method used to express or target CTNNB1. **: +, CTNNB1 overexpression or expression of activated form of CTNNB1; -, CTNNB1 knockout or knockdown. ***: For gain-of-function (+) studies, CTNNB1 status indicates the wildtype or activated form of the gene that is expressed in each study. Other genes targeted in the model are shown in parentheses. For loss-of-function (−) studies, the form of the targeted CTNNB1 allele is shown, with its origin indicated in parentheses. Abbreviations: Alb, albumin; Fabp, fatty acid-binding protein; Fabp: Cre, Fabp promoter driving expression of Cre recombinase; CaBP9K, calbindin-D9K; Cited1: CreER, Cited1 driving expression of tamoxifen-inducible Cre (CreER); AdCMV-Cre, recombinant adenovirus with cytomegalovirus promoter driving Cre recombinase; SB-HDT, sleeping beauty transposase-mediated hydrodynamic transfection; Alb: Cre, albumin promoter driving Cre recombinase; and DEN, diethylnitrosamine.
In patient HCC samples with activating mutations in β-catenin, immunostaining for cytoplasmic and nuclear β-catenin—indicative of active WNT/β-catenin signaling—is typically patchy or focal, confined to a small percentage of tumor cells [70]. This observation seems to result from actual differences in WNT/β-catenin signaling activity among tumor cells rather than from a lack of antibody sensitivity. Our lab has shown that WNT reporter activity and expression levels of WNT/β-catenin target genes are highly variable from cell to cell in zebrafish β-catenin-driven HCC [80,81].
Furthermore, expression of activated β-catenin in even a small subset of larval hepatocytes is sufficient to drive HCC in zebrafish [80]. Similar results were reported by Mokapatti et al., who found that expression of activated β-catenin in hepatic progenitor cells (4% of fetal liver cells) initiated HCC in mice [79]. These results highlight the importance of β-catenin heterogeneity in the initiation and progression of HCC.
7. Interactions between TERT and β-Catenin in Cultured Cells and Animal Models
Manipulations in TERT or WNT/β-catenin signaling result in strikingly similar phenotypes in mouse skin, suggesting these two pathways may cooperate in this system. Sarin et al. investigated the role of TERT in multipotent stem cells/progenitor cells by conditional TERT expression in mouse skin [84]. The hair follicle region consists of multipotent stem cells and progenitor cells, which cycle from telogen phase (resting phase) to anagen phase (active phase) [84]. TERT modulates this transition by increasing the proliferation of the stem cell population [84].
This anagen-promoting effect is independent of the telomerase activity of TERT, as these effects are retained even in the absence of TERC [84] and mirror the phenotype of β-catenin overexpression in hair follicles [85,86,87,88]. These results highlight the similarities in function between non-canonical TERT activity and the WNT/β-catenin pathway in stem cells. Choi et al. further underlined that TERT regulates stem cell activity independently of its telomerase activity by overexpressing a catalytically inactive form of TERT (TERTci) [89]. TERTci had the same effect as TERT in promoting anagen in hair follicles and keratinocyte proliferation in the skin. They found strong associations between TERT and WNT/β-catenin pathways through genome-wide analysis of transcriptional changes wherein TERT specifically regulated WNT target genes with TCF/LEF elements [89].
Studies predominantly performed in mouse stem cells delineate a feedback loop wherein Wnt/β-catenin signaling and TERT upregulate each other (Figure 2B). A key paper in this field by Park et al. illustrated that TERT regulates WNT/β-catenin signaling by acting as a co-factor in the β-catenin transcription complex [3]. They found that, in cultured mouse ES cells, endogenous TERT associated with the bromodomain of BRG1 to regulate transcription of β-catenin target genes [3]. TERT overexpression hyperactivated WNT/β-catenin signaling independent of TERT catalytic activity [3].
In mouse stomach cells, endogenous TERT occupied TCF binding elements together with β-catenin, and TERTci overexpression increased promoter activity and/or expression of β-catenin target genes, such as AXIN2 and CD44 [3]. TERT knockout in mouse ES cells decreased both the basal and WNT3A-induced expression of WNT/β-catenin target gene AXIN2 [3]. Corroborating Park et al., Hrdlickova et al. showed that the overexpression of TERT/TERTci splice variants increased proliferation and activated WNT signaling in TERT-deficient and TERT-expressing human cell lines [90].
Hoffmeyer et al. described the role of WNT/β-catenin signaling in regulating TERT in stem cells and cancer cells [4]. Using ES cells, they showed that β-catenin knockdown resulted in reduced TERT (but not TERC) mRNA levels as well as decreased telomerase activity while its overexpression increased TERT mRNA, protein, and telomerase activity [4]. Telomere length increased upon β-catenin overexpression and decreased in β-catenin-deficient cells relative to wildtype. In terms of this upregulation mechanism, β-catenin binds at the transcriptional start site (TSS) of TERT, possibly by forming a complex that requires Klf4 [4]. These results indicate that β-catenin promotes TERT transcription in ES cells [4]. Similarly, β-catenin was found at the TSS of TERT in adult mouse stem cells, primary mouse neurospheres, and human cancer cell lines, correlating with TERT expression [4].
Similar results were observed by Zhang et al. in 293T (human embryonic kidney), HCT116 (human colon cancer), and MCF7 (human breast cancer) cell lines wherein WNT/β-catenin upregulates TERT expression levels and telomerase activity [62]. Mechanistically, they found that TCF4 (and not TCF1/3 or LEF1) was involved as a binding partner of β-catenin, responsible for increasing TERT promoter activity [62]. The WNT/β-catenin target MYC also binds at the TSS of TERT to increase its expression [91,92]. Another study using human colorectal Caco2 and SW620 cells showed that β-catenin directly regulated TERT via TCF4 binding, and this effect was independent of MYC [93].
Some groups have failed to discern interactions between TERT and WNT/β-catenin during transcriptional regulation. Listerman et al. overexpressed TERT in breast cancer cell lines with varying endogenous levels of TERT expression and saw WNT/β-catenin signaling activation only in those cell lines that had low endogenous telomerase activity [94]. They did not observe an interaction between FLAG-TERT and β-catenin or BRG1 and proposed that the results seen by Park et al. were due to their anti-FLAG antibody binding to a molecule other than β-catenin but with similar electrophoretic mobility [94].
Additionally, Strong et al. did not find abnormalities in WNT/β-catenin-signaling-related developmental processes in Tert−/− mice [95]. Their TOPFLASH assay did not reveal any difference in WNT activation levels in Tert−/− mice vs. wildtype [95]. Similarly, another study in Tert−/− and Terc−/− G1 mouse embryonic fibroblasts reported no transcriptional profile changes, including WNT/β-catenin target genes, compared to wildtype [96]. Possible reasons for these contradictions could be germline compensation in WNT/β-catenin signaling in these mutants [11], different mouse backgrounds, different epitope tags or antibodies used, and different assay sensitivities.
WNT and TERT are also linked by their relationship to telomere capping [97,98]. Late generation Terc−/− mice show telomere shortening leading to uncapping. These capping defects can downregulate WNT/β-catenin signaling in high turnover cells, and WNT agonists can rescue the effects [98]. This rescue is not mediated by telomere lengthening but by increased expression of Shelterin complex proteins (Trf2 and Pot1a) [98]. This group proposed that the phenotype observed by Park et al. may be due to the presence of a shortened telomere in the Tert+/− parent that led to the downregulation of WNT/β-catenin signaling at some critical phase of development of the G1 Tert−/− offspring [98]. These differences highlight that the impact of TERT overexpression or deletion on WNT/β-catenin signaling is not a universal effect and may depend on telomere length, developmental stage, cell type, or other factors.
8. TERT and β-Catenin in Human Cancer
Associations between TERT and β-catenin have been reported in diverse cancer types, including gastrointestinal cancers, medulloblastoma, breast cancer, and osteosarcoma. The expression of CTNNB1, its target genes, and TERT have been reported at the invasive edges of colorectal cancer [93]. Jaitner et al. analyzed 24 human colorectal cancer samples with invasive fronts and found that TERT expression was higher in tumor cells with nuclear β-catenin, while tumor cells without nuclear β-catenin did not express TERT [93]. Similarly, significant protein–protein interactions between β-catenin and TERT were detected in 104 esophageal cancer patient samples [99].
In gastric cancer cell lines, TERT overexpression enhances WNT/β-catenin signaling and increases the expression of WNT/β-catenin target genes, such as c-MYC and Cyclin D1 [100]. This TERT-induced c-MYC upregulation increases the expression of heparanase, a metastasis-promoting protein, leading to increased aggressiveness and poor prognosis [100]. TERT mutations are recurrent in the WNT subgroup of medulloblastoma although they do not impact prognosis [101]. In breast cancer, TERT can upregulate WNT signaling but only in a cell line dependent manner: out of four cell lines tested, only one showed WNT reporter activity in response to TERT overexpression [94]. TERT-induced miR-500a upregulation increased tumor aggressiveness in osteosarcoma cell lines, and WNT/β-catenin is predicted to be one of the affected downstream signaling pathways [102].
9. TERT and β-Catenin in HCC Patients
Several small- and large-scale genome sequencing efforts in HCC samples with diverse etiologies, summarized in Table 4, provide clues regarding the association between the two genes. A seminal study by Nault et al. illustrated the concordance between TERT and β-catenin while also indicating their interplay in HCC initiation and progression [5]. They screened 305 HCC cases from two French hospitals and found that 59% of cases had TERTp mutations, significantly correlated with CTNNB1 mutations (p-value < 0.0001, χ2-test) [5]. The same group further confirmed this association in a study with 720 patients (p-value = 0.0000001) [103].
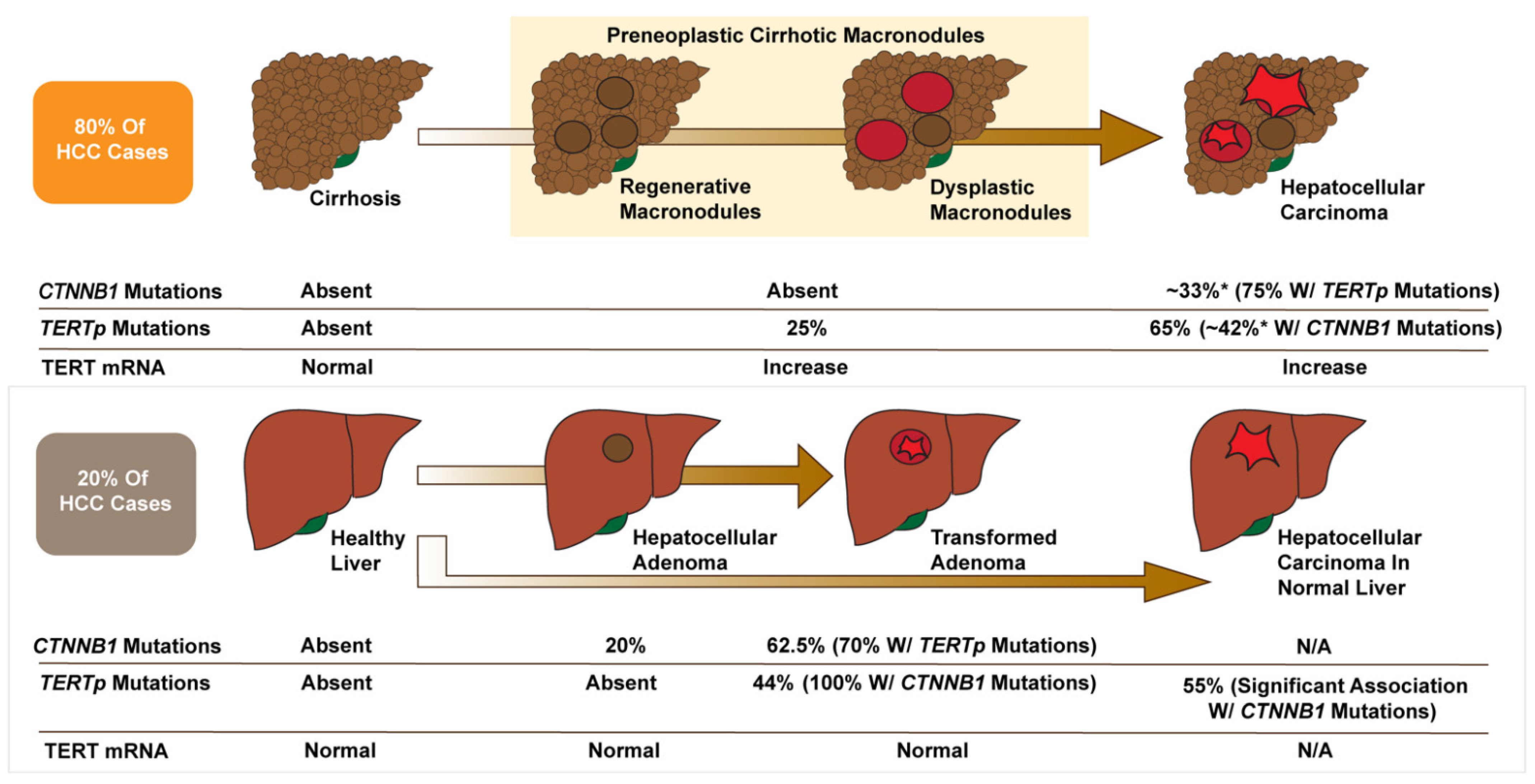
For HCC arising in cirrhotic livers, TERTp mutations seemed to be the first genetic alteration leading to malignancy, being found in 25% of preneoplastic lesions compared to 65% of malignant tumors (42% of these with concurrent β-catenin mutations) (Figure 3) [5]. TERTp mutations were absent in cirrhotic livers without preneoplastic lesions but enriched as the cirrhotic tissue progressed into dysplastic nodules and HCC, correlating with increased TERT expression (Figure 3) [5]. These results were corroborated in another study by Torrecilla et al. that identified TERT as the gatekeeper mutation and CTNNB1 as a driver mutation in cirrhotic hepatocarcinogenesis in patient samples from the USA, Italy, and Spain [104].
For HCC arising from HCA, CTNNB1 mutations appear to be the initiating event, being found in 20% of HCA without malignant transformation (0% with TERT mutations) (Figure 3) [105]. β-catenin activated HCAs, which are characterized by activating CTNNB1 mutations, nuclear β-catenin staining, and strong diffuse staining for the WNT/β-catenin target gene GLUL, are significantly more likely to transform to HCC than other HCA subtypes [106,107]. In transformed adenomas, 44% have TERTp mutations, all associated with β-catenin mutations (Figure 3) [5,105]. These studies suggest a gatekeeping role of TERT in HCA: β-catenin mutations are an early event driving proliferation and a significant risk factor for malignancy; however, TERTp mutations are critical for malignant transformation [108,109,110].
A study done on a European cohort with a high prevalence of HCV-related HCC also showed the coexistence of TERTp and CTNNB1 mutations, as 57.6% of CTNNB1-mutated HCC had TERTp mutations (p-value = 0.4192) [43]. Another study with Asian (Japanese) and American (mixed ancestry) populations observed significant concordance between TERTp mutations and WNT pathway alterations (CTNNB1, AXIN1, or APC) in HCV and non-virus related HCCs (p-value < 0.001) [7].
In HCC cases arising in patients with non-alcoholic fatty liver disease (NAFLD), TERTp mutations are very high and more frequent than in HCC associated with HBV or even HCV [42,54]. In a Japanese study, all NAFLD-HCC with CTNNB1 mutations were mutated for TERTp; however, the association was statistically insignificant (p-value = 0.4545), likely due to the small sample size of 11 [54].
The aforementioned studies all demonstrated a high concordance between TERTp and β-catenin mutations; however, none of them found evidence to support the direct regulation of one gene by the other [5,7,43,54,103]. In HCC with TERTp mutations and not CTNNB1 mutations, β-catenin target genes (GLUL and LGR5) were not upregulated compared to HCC without mutations in TERT or β-catenin [5]. In HCA with CTNNB1 mutations, TERT transcription was not increased [5].
Not all clinical studies have identified an association between TERTp and β-catenin mutations. Studies done on Taiwanese, Korean, and Chinese HCC cohorts did not reveal any concurrence between the two mutations [34,46,53]. Another study that mined HCC data from TCGA and American HCC samples indicated a very low prevalence of TERTp mutations in NAFLD-HCC patients [111]. Clinical studies that found a significant association between TERTp and CTNNB1 mutations tended to have a larger sample size than studies that did not identify a significant correlation (Table 4). Other potential reasons why the strength of the TERTp-CTNNB1 association has varied between studies include racial or ethnic differences among the patients, different etiologies, and/or diverse disease course in HBV-endemic and non-endemic areas.
10. TERT and β-Catenin in Vertebrate HCC Models
The strong correlation between TERTp and CTNNB1 mutations reported in large clinical studies of HCC have prompted a few groups to examine the TERT-CTNNB1 cooperation using animal liver tumor models. Forced β-catenin overexpression in human fetal hepatocytes immortalized with hTERT (FH-hTERT) led to a malignant phenotype of loss of contact inhibition, increased anchorage-independent growth, and improved ability to form tumors in athymic nude mice [112]. These FH-hTERT pbcatS33Y cells have persistent clonal chromosomal translocations, which are not seen in FH-hTERT cells without β-catenin overexpression [112]. This finding indicates that the coexpression of TERT and β-catenin leads to tolerance for chromosomal structural instability, which may be important for the transformed phenotype. Additionally, using RNAi to knockdown TERT and TERC, this study showed that telomerase activity is not required for the short-term expansion of transformed cells [112].
Molina-Sánchez et al. showed cooperation between TERT and β-catenin in HCC with an elegant in vivo screen [113]. They used hydrodynamic tail-vein injections to deliver 23 different combinations of genetic alterations into mouse livers and examined the tumor formation six months later. Three out of nine mice overexpressing Tert and CTNNB1 developed HCC, compared to zero of six mice overexpressing CTNNB1 alone [113]. However, in a follow-up experiment (n = 9), no Tert; CTNNB1 mice developed liver tumors [113]. The authors suggested that cooperation between Tert and β-catenin may be insufficient for HCC, and additional spontaneous genetic alterations may be required [113]. Future experiments in vertebrate models, including those combining both gain- and loss-of-function alterations in TERT and CTNNB1, will be critical for confirming and extending the results of Molina-Sánchez et al. Catalytically inactive TERT (TERTci) constructs can be particularly useful in determining whether interactions between TERT and β-catenin require the telomerase activity of TERT or act through non-canonical mechanisms.
11. Conclusions and Future Directions
The key points from the above studies are: (1) TERTp and CTNNB1 mutations are concordant in patient HCC; (2) in animal models, overexpression of TERT or CTNNB1 promotes HCC, although the effect is modest in most models and often requires another oncogene or insult; (3) in cultured stem cells, TERT and CTNNB1 cooperate via direct or indirect interactions during the transcription of target genes; and (4) both CTNNB1 and, to a lesser extent, TERT, have key roles in cellular homeostasis, making the potent inhibition of endogenous genes unrealistic from a therapeutic perspective.
Using cultured human liver cancer cells and vertebrate HCC models will be critical for confirming that the mechanism of TERT/β-catenin interaction identified in stem cells applies to HCC and establishing that non-canonical effects of TERT drive its interactions with β-catenin and the resulting HCC-promoting effects. Delineating exactly how TERT and β-catenin interplay would suggest promising treatment strategies for TERTp and CTNNB1-mutated HCC, including: (1) targeting non-canonical effects of TERT, critical for its tumor-promoting interactions with β-catenin; (2) targeting the interaction between TERT and β-catenin; and (3) combining relatively low doses of Wnt/β-catenin pathway and TERT inhibitors.
With respect to the latter approach, several drugs targeting either the WNT/β-catenin pathway or TERT are in early clinical trials or preclinical testing. The porcupine inhibitor CGX1321 and the Frizzled8 decoy receptor OMP-54F28 are currently in Phase I clinical trials [114,115]. The antibiotic and LRP5/6 inhibitor salinomycin and the Tankyrase inhibitor NVP-TNKS656 are in the preclinical stage for HCC [114,115]. All four of these drugs target WNT/β-catenin signaling upstream of β-catenin and, thus, may be ineffective in the setting of activating mutations in CTNNB1.
One option for inhibiting WNT/β-catenin signaling further downstream is to block β-catenin’s interactions with its transcriptional partners to prevent the expression of target genes. To that effect, molecules targeting β-catenin binding to CREB-binding protein (CBP) (PRI-724 (Phase 1 and 2)) or to TCF/LEF elements (LF3, iCRT3/5, ZINC02092166, and NLS-StAx-h; all in preclinical stages) are being tested [114,115]. CBP-inhibitors, such as ICG001 and Isoquercitrin, are also under preclinical evaluation [114].
Current strategies for the therapeutic targeting of TERT include: (1) small molecule inhibitors against TERT, like BIBR1532 [116]; (2) gene therapies against TERT, like telomelysin [117]; (3) gene silencing via microRNAs against TERT [117]; (4) G-quadruplex stabilizers that interfere with telomere structure and may inhibit telomerase activity, reviewed in detail by Alessandrini et al. [118]; (5) inhibitors of telomere- and telomerase-associated proteins, such as HSP90 [119]; (6) immunotherapies utilizing TERT as a tumor antigen [120]; and (7) oligonucleotides that mimic telomere overhangs (T-oligos) or inhibit telomerase (GRN163L) [117]. As with some of the WNT/β-catenin inhibitors, GRN163L shows promising anticancer activity, but its use as a single chemotherapeutic agent is limited by toxicity [117]. Combining a WNT/β-catenin inhibitor with a TERT inhibitor offers the potential to decrease serious side effects by using a lower dose of each drug.
The association of CTNNB1 and TERTp mutations in HCC presents attractive diagnostic and prognostic possibilities. Risk stratification could be done based on the clues we have regarding the temporal play of gatekeeper and driver oncogenes. TERTp mutations are the first changes associated with neoplasia in cirrhotic livers. Thus, they could potentially be used to predict HCC risk in cirrhotic patients, just as β-catenin activation is currently used to predict HCC risk in patients with HCA. Further studies examining the prevalence of TERTp mutations in cirrhotic patients without HCC would be required to determine the feasibility of this approach.
HCC poses a therapeutic challenge due to the late diagnosis and paucity of effective treatments for advanced disease. The molecular heterogeneity in HCC further exacerbates this challenge, highlighting the need for increased identification and understanding of biomarkers and their prognostic and therapeutic value. Current HCC treatments are not tailored to patients based on the molecular features of their tumors, as no genetic perturbations have been definitively shown to influence therapeutic response in patients. As the armamentarium of approved HCC therapeutics expands, it will be critical to correlate molecular subtypes, including those with TERTp and CTNNB1 mutations, with response to specific treatments to guide future patient treatment regimens.
Author Contributions
Conceptualization, S.K. and K.J.E.; writing—original draft preparation, S.K.; writing—review and editing, K.J.E.; funding acquisition, K.J.E. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the NIH/NCI, grant number R01CA222570, and the Damon Runyon Cancer Research Foundation, Damon Runyon-Rachleff Innovation Award DRR-61-20.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Lee, J.-S. The mutational landscape of hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 220–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/β-Catenin Signaling Regulates Telomerase in Stem Cells and Cancer Cells. Science 2012, 336, 1549. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.B.; Sá, A.; Lopes, J.M.; Sobrinho-Simões, M.; Soares, P.; Vinagre, J. Telomere Maintenance Mechanisms in Cancer. Genes 2018, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. Rev. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kim, N.-K.; Feigon, J. Architecture of human telomerase RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20325–20332. [Google Scholar] [CrossRef] [Green Version]
- Ségal-Bendirdjian, E.; Geli, V. Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019, 7, 332. [Google Scholar] [CrossRef]
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The non-canonical functions of telomerase: To turn off or not to turn off. Mol. Biol. Rep. 2019, 46, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Zhou, Y.; Chen, D.; Li, L.; Yang, X.; You, Y.; Ling, X. Impact of mitochondrial telomerase over-expression on drug resistance of hepatocellular carcinoma. Am. J. Transl. Res. 2015, 7, 88–99. [Google Scholar]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Dai, M.; Xu, D. TERT promoter mutations and GABP transcription factors in carcinogenesis: More foes than friends. Cancer Lett. 2020, 493, 1–9. [Google Scholar] [CrossRef]
- Canela, A.; Martín-Caballero, J.; Flores, J.M.; Blasco, M.A. Constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in Lck-Tert mice. Mol. Cell Biol. 2004, 24, 4275–4293. [Google Scholar] [CrossRef] [Green Version]
- González-Suárez, E.; Samper, E.; Ramírez, A.; Flores, J.M.; Martín-Caballero, J.; Jorcano, J.L.; Blasco, M.A. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001, 20, 2619–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artandi, S.E.; Alson, S.; Tietze, M.K.; Sharpless, N.E.; Ye, S.; Greenberg, R.A.; Castrillon, D.H.; Horner, J.W.; Weiler, S.R.; Carrasco, R.D.; et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8191–8196. [Google Scholar] [CrossRef] [Green Version]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Akıncılar, S.C.; Raju, A.; Lakshmanan, M.; Maury, J.J.P.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Investig. 2016, 126, 4045–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farazi, P.A.; Glickman, J.; Horner, J.; DePinho, R.A. Cooperative Interactions of p53 Mutation, Telomere Dysfunction, and Chronic Liver Damage in Hepatocellular Carcinoma Progression. Cancer Res. 2006, 66, 4766. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential Impact of Telomere Dysfunction on Initiation and Progression of Hepatocellular Carcinoma. Cancer Res. 2003, 63, 5021. [Google Scholar] [PubMed]
- Carneiro, M.C.; Henriques, C.M.; Nabais, J.; Ferreira, T.; Carvalho, T.; Ferreira, M.G. Short Telomeres in Key Tissues Initiate Local and Systemic Aging in Zebrafish. PLoS Genet. 2016, 12, e1005798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzec, P.; Armenise, C.; Pérot, G.; Roumelioti, F.-M.; Basyuk, E.; Gagos, S.; Chibon, F.; Déjardin, J. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 2015, 160, 913–927. [Google Scholar] [CrossRef] [Green Version]
- Cayuela, M.L.; Claes, K.B.M.; Ferreira, M.G.; Henriques, C.M.; van Eeden, F.; Varga, M.; Vierstraete, J.; Mione, M.C. The Zebrafish as an Emerging Model to Study DNA Damage in Aging, Cancer and Other Diseases. Front. Cell Dev. Biol. 2019, 6, 178. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, K.L.; Chang, S.; Lee, H.-W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Idilli, A.I.; Cusanelli, E.; Pagani, F.; Berardinelli, F.; Bernabé, M.; Cayuela, M.L.; Poliani, P.L.; Mione, M.C. Expression of tert Prevents ALT in Zebrafish Brain Tumors. Front. Cell Dev. Biol. 2020, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- González-Suárez, E.; Samper, E.; Flores, J.M.; Blasco, M.A. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat. Genet. 2000, 26, 114–117. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.-L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.L.; Millard, M.; Bosenberg, M.W.; DePinho, R.A. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat. Genet. 2001, 28, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lex, K.; Maia Gil, M.; Lopes-Bastos, B.; Figueira, M.; Marzullo, M.; Giannetti, K.; Carvalho, T.; Ferreira, M.G. Telomere shortening produces an inflammatory environment that increases tumor incidence in zebrafish. Proc. Natl. Acad. Sci. USA 2020, 117, 15066. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhu, Y.-J.; Wang, H.-Y.; Chen, L. Gender disparity in hepatocellular carcinoma (HCC): Multiple underlying mechanisms. Sci. China Life Sci. 2017, 60, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.-C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Jeng, Y.-M.; Chang, C.-N.; Lee, H.-J.; Hsu, H.-C.; Lai, P.-L.; Yuan, R.-H. TERT promoter mutation in resectable hepatocellular carcinomas: A strong association with hepatitis C infection and absence of hepatitis B infection. Int. J. Surg. 2014, 12, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Park, T.I.; Jang, S.Y.; Park, S.Y.; Park, W.-J.; Jung, S.-J.; Lee, J.-H. Clinicopathological characteristics of TERT promoter mutation and telomere length in hepatocellular carcinoma. Medicine 2017, 96, e5766. [Google Scholar] [CrossRef]
- Qu, Y.; Dang, S.; Wu, K.; Shao, Y.; Yang, Q.; Ji, M.; Shi, B.; Hou, P. TERT promoter mutations predict worse survival in laryngeal cancer patients. Int. J. Cancer 2014, 135, 1008–1010. [Google Scholar] [CrossRef]
- Bell, R.J.A.; Rube, H.T.; Xavier-Magalhães, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT Promoter Mutations: A Common Path to Immortality. Mol. Cancer Res. 2016, 14, 315. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Yuan, X.; Xu, D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes 2016, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Tallet, A.; Nault, J.-C.; Renier, A.; Hysi, I.; Galateau-Sallé, F.; Cazes, A.; Copin, M.-C.; Hofman, P.; Andujar, P.; Le Pimpec-Barthes, F.; et al. Overexpression and promoter mutation of the TERT gene in malignant pleural mesothelioma. Oncogene 2014, 33, 3748–3752. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.; Da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, C.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, D.; Saitta, C.; Giosa, D.; Di Tocco, F.C.; Musolino, C.; Caminiti, G.; Chines, V.; Franzè, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [PubMed]
- Pezzuto, F.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Frequency and geographic distribution of TERT promoter mutations in primary hepatocellular carcinoma. Infect. Agent Cancer 2017, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzuto, F.; Izzo, F.; Buonaguro, L.; Annunziata, C.; Tatangelo, F.; Botti, G.; Buonaguro, F.M.; Tornesello, M.L. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget 2016, 7, 54253–54262. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Electronic address: [email protected]; Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341 e23. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Nishimura, T.; Kaido, T.; Minaga, K.; Yamao, K.; Kamata, K.; Takenaka, M.; Ida, H.; Hagiwara, S.; Minami, Y.; et al. Molecular Scoring of Hepatocellular Carcinoma for Predicting Metastatic Recurrence and Requirements of Systemic Chemotherapy. Cancers 2018, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Chang, S.-H.; Kim, W.Y.; Lim, S.D.; Kim, W.S.; Hwang, T.S.; Han, H.S. Frequent somatic TERT promoter mutations and CTNNB1 mutations in hepatocellular carcinoma. Oncotarget 2016, 7, 69267–69275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Xu, W.; Kang, W.; Wong, S.H.; Wang, M.; Zhou, Y.; Fang, X.; Zhang, X.; Yang, H.; Wong, C.H.; et al. Genomic analysis of liver cancer unveils novel driver genes and distinct prognostic features. Theranostics 2018, 8, 1740–1751. [Google Scholar] [CrossRef] [Green Version]
- Cevik, D.; Yildiz, G.; Ozturk, M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J. Gastroenterol. 2015, 21, 311–317. [Google Scholar] [CrossRef]
- Quaas, A.; Oldopp, T.; Tharun, L.; Klingenfeld, C.; Krech, T.; Sauter, G.; Grob, T.J. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014, 465, 673–677. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Guo, X.; Chen, Y.; Chen, G.; Ma, Y.; Huang, K.; Zhang, Y.; Zhao, Q.; Winkler, C.A.; An, P.; et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget 2016, 7, 27838–27847. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.-S.; Wang, Z.; He, X.-J.; Diplas, B.H.; Yang, R.; Killela, P.J.; Meng, Q.; Ye, Z.-Y.; Wang, W.; Jiang, X.-T.; et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer 2015, 51, 969–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Cheng, G.; Yu, J.; Zheng, S.; Sun, C.; Sun, Q.; Li, K.; Lin, Z.; Liu, T.; Li, P.; et al. The TERT promoter mutation incidence is modified by germline TERT rs2736098 and rs2736100 polymorphisms in hepatocellular carcinoma. Oncotarget 2017, 8, 23120–23129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT promoter mutations and chromosome 8p loss are characteristic of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Int. J. Cancer 2016, 139, 2512–2518. [Google Scholar] [CrossRef]
- Park, C.-K.; Lee, S.-H.; Kim, J.Y.; Kim, J.E.; Kim, T.M.; Lee, S.-T.; Choi, S.H.; Park, S.-H.; Kim, I.H. Expression level of hTERT is regulated by somatic mutation and common single nucleotide polymorphism at promoter region in glioblastoma. Oncotarget 2014, 5, 3399–3407. [Google Scholar] [CrossRef] [Green Version]
- Ko, E.; Seo, H.-W.; Jung, E.S.; Kim, B.; Jung, G. The TERT promoter SNP rs2853669 decreases E2F1 transcription factor binding and increases mortality and recurrence risks in liver cancer. Oncotarget 2016, 7, 684–699. [Google Scholar] [CrossRef]
- Kim, Y.J.; Yoo, J.E.; Jeon, Y. Suppression of PROX1-mediated TERT expression in hepatitis B viral hepatocellular carcinoma. Int. J. Cancer 2018, 143, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Russell, J.O.; Monga, S.P. Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu. Rev. Pathol. 2018, 13, 351–378. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2017, 9, 5492–5508. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, M.K.; Shao, C.; Wang, J.; Wei, Q.; Wang, X.; Collier, Z.; Tang, S.; Liu, H.; Zhang, F.; Huang, J.; et al. Wnt/β-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis. 2016, 3, 11–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Toh, L.; Lau, P.; Wang, X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. J. Biol. Chem. 2012, 287, 32494–32511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalaf, A.M.; Fuentes, D.; Morshid, A.I.; Burke, M.R.; Kaseb, A.O.; Hassan, M.; Hazle, J.D.; Elsayes, K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma 2018, 5, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sheng, Y.-Y.; Gao, X.-M.; Wang, C.-Q.; Wang, X.-Y.; Lu, X.U.; Wei, J.-W.; Zhang, K.-L.; Dong, Q.-Z.; Qin, L.-X. β-catenin mutation is correlated with a favorable prognosis in patients with hepatocellular carcinoma. Mol. Clin. Oncol. 2015, 3, 936–940. [Google Scholar] [CrossRef] [Green Version]
- Mao, T.-L.; Chu, J.-S.; Jeng, Y.-M.; Lai, P.-L.; Hsu, H.-C. Expression of mutant nuclear β-catenin correlates with non-invasive hepatocellular carcinoma, absence of portal vein spread, and good prognosis. J. Pathol. 2001, 193, 95–101. [Google Scholar] [CrossRef]
- Yuan, R.-H.; Jeng, Y.-M.; Hu, R.-H.; Lai, P.-L.; Lee, P.-H.; Cheng, C.-C.; Hsu, H.-C. Role of p53 and β-catenin Mutations in Conjunction with CK19 Expression on Early Tumor Recurrence and Prognosis of Hepatocellular Carcinoma. J. Gastrointest. Surg. 2011, 15, 321–329. [Google Scholar] [CrossRef]
- Peng, S.-Y.; Chen, W.J.; Lai, P.-L.; Jeng, Y.-M.; Sheu, J.-C.; Hsu, H.-C. High α-fetoprotein level correlates with high stage, early recurrence and poor prognosis of hepatocellular carcinoma: Significance of hepatitis virus infection, age, p53 and β-catenin mutations. Int. J. Cancer 2004, 112, 44–50. [Google Scholar] [CrossRef]
- Kitao, A.; Matsui, O.; Yoneda, N.; Kozaka, K.; Kobayashi, S.; Sanada, J.; Koda, W.; Minami, T.; Inoue, D.; Yoshida, K.; et al. Hepatocellular Carcinoma with β-Catenin Mutation: Imaging and Pathologic Characteristics. Radiology 2015, 275, 708–717. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Apte, U.; Micsenyi, A.; Kotsagrelos, E.; Luo, J.-H.; Ranganathan, S.; Monga, D.K.; Bell, A.; Michalopoulos, G.K.; Monga, S.P.S. Epidermal growth factor receptor: A novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology 2005, 129, 285–302. [Google Scholar] [CrossRef] [Green Version]
- Nejak-Bowen, K.N.; Thompson, M.D.; Singh, S.; Bowen, W.C.; Dar, M.J.; Khillan, J.; Dai, C.; Monga, S.P.S. Accelerated Liver Regeneration And Hepatocarcinogenesis In Mice Overexpressing Serine-45 Mutant Beta-Catenin. Hepatology 2010, 51, 1603–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, N.; Miyoshi, H.; Murai, N.; Oshima, H.; Tamai, Y.; Oshima, M.; Taketo, M.M. Lack of Tumorigenesis in the Mouse Liver after Adenovirus-mediated Expression of a Dominant Stable Mutant of β-Catenin. Cancer Res. 2002, 62, 1971. [Google Scholar] [PubMed]
- Cadoret, A.; Ovejero, C.; Saadi-Kheddouci, S.; Souil, E.; Fabre, M.; Romagnolo, B.; Kahn, A.; Perret, C. Hepatomegaly in Transgenic Mice Expressing an Oncogenic Form of β-Catenin. Cancer Res. 2001, 61, 3245. [Google Scholar]
- Tao, J.; Xu, E.; Zhao, Y.; Singh, S.; Li, X.; Couchy, G.; Chen, X.; Zucman-Rossi, J.; Chikina, M.; Monga, S.P.S. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology 2016, 64, 1587–1605. [Google Scholar] [CrossRef]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu-Ebinger, S.; et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Zhang, R.; Singh, S.; Poddar, M.; Xu, E.; Oertel, M.; Chen, X.; Ganesh, S.; Abrams, M.; Monga, S.P. Targeting β-catenin in hepatocellular cancers induced by coexpression of mutant β-catenin and K-Ras in mice. Hepatology 2017, 65, 1581–1599. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Oshima, H.; Katoh, M.; Tamai, Y.; Oshima, M.; Taketo, M.M. Hepatocarcinogenesis in Mice with β-Catenin and Ha-Ras Gene Mutations. Cancer Res. 2004, 64, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokkapati, S.; Niopek, K.; Huang, L.; Cunniff, K.J.; Ruteshouser, E.C.; deCaestecker, M.; Finegold, M.J.; Huff, V. β-catenin activation in a novel liver progenitor cell type is sufficient to cause hepatocellular carcinoma and hepatoblastoma. Cancer Res. 2014, 74, 4515–4525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalasekar, S.M.; Kotiyal, S.; Conley, C.; Phan, C.; Young, A.; Evason, K.J. Heterogeneous beta-catenin activation is sufficient to cause hepatocellular carcinoma in zebrafish. Biol. Open 2019, 8, bio047829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evason, K.J.; Francisco, M.T.; Juric, V.; Balakrishnan, S.; Pazmino, M.d.P.L.; Gordan, J.D.; Kakar, S.; Spitsbergen, J.; Goga, A.; Stainier, D.Y.R. Identification of Chemical Inhibitors of β-Catenin-Driven Liver Tumorigenesis in Zebrafish. PLoS Genet. 2015, 11, e1005305. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Tan, X.; Zeng, G.; Misse, A.; Singh, S.; Kim, Y.; Klaunig, J.E.; Monga, S.P.S. Conditional beta-catenin loss in mice promotes chemical hepatocarcinogenesis: Role of oxidative stress and platelet-derived growth factor receptor alpha/phosphoinositide 3-kinase signaling. Hepatology 2010, 52, 954–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Feng, Y.; Zong, M.; Wei, X.-F.; Lee, J.; Feng, Y.; Li, H.; Yang, G.-S.; Wu, Z.-J.; Fu, X.-D.; et al. β-catenin deficiency in hepatocytes aggravates hepatocarcinogenesis driven by oncogenic β-catenin and MET. Hepatology 2018, 67, 1807–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarin, K.Y.; Cheung, P.; Gilison, D.; Lee, E.; Tennen, R.I.; Wang, E.; Artandi, M.K.; Oro, A.E.; Artandi, S.E. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 2005, 436, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Gat, U.; DasGupta, R.; Degenstein, L.; Fuchs, E. De Novo Hair Follicle Morphogenesis and Hair Tumors in Mice Expressing a Truncated β-Catenin in Skin. Cell 1998, 95, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Andl, T.; Reddy, S.T.; Gaddapara, T. Millar SE WNT signals are required for the initiation of hair follicle development. Dev. Cell. 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Lowry, W.E.; Blanpain, C.; Nowak, J.A.; Guasch, G.; Lewis, L.; Fuchs, E. Defining the impact of beta-catenin/Tcf transactivation on epithelial stem cells. Genes Dev. 2005, 19, 1596–1611. [Google Scholar] [CrossRef] [Green Version]
- Huelsken, J.; Vogel, R.; Erdmann, B.; Cotsarelis, G.; Birchmeier, W. Beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 2001, 105, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008, 4, e10. [Google Scholar] [CrossRef] [Green Version]
- Hrdličková, R.; Nehyba, J.; Bose, H.R. Alternatively Spliced Telomerase Reverse Transcriptase Variants Lacking Telomerase Activity Stimulate Cell Proliferation. Mol. Cell. Biol. 2012, 32, 4283. [Google Scholar] [CrossRef] [Green Version]
- Khattar, E.; Tergaonkar, V. Transcriptional Regulation of Telomerase Reverse Transcriptase (TERT) by MYC. Front. Cell Dev. Biol 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.-J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Jaitner, S.; Reiche, J.A.; Schäffauer, A.J.; Hiendlmeyer, E.; Herbst, H.; Brabletz, T.; Kirchner, T.; Jung, A. Human telomerase reverse transcriptase (hTERT) is a target gene of β-catenin in human colorectal tumors. Null 2012, 11, 3331–3338. [Google Scholar] [CrossRef] [Green Version]
- Listerman, I.; Gazzaniga, F.S.; Blackburn, E.H. An Investigation of the Effects of the Core Protein Telomerase Reverse Transcriptase on Wnt Signaling in Breast Cancer Cells. Mol. Cell. Biol. 2014, 34, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.A.; Vidal-Cardenas, S.L.; Karim, B.; Yu, H.; Guo, N.; Greider, C.W. Phenotypes in mTERT+/− and mTERT−/− mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol. Cell Biol. 2011, 31, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Cardenas, S.L.; Greider, C.W. Comparing effects of mTR and mTERT deletion on gene expression and DNA damage response: A critical examination of telomere length maintenance-independent roles of telomerase. Nucleic Acids Res. 2010, 38, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-L.B.; Chen, Q.; Deng, J.T.; Jagannathan, G.; Tobias, J.W.; Schultz, D.C.; Wang, S.; Lengner, C.J.; Rustgi, A.K.; Lynch, J.P.; et al. Mutual reinforcement between telomere capping and canonical Wnt signalling in the intestinal stem cell niche. Nat. Commun. 2017, 8, 14766. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, R.J., 3rd; Johnson, F.B. A regulatory loop connecting WNT signaling and telomere capping: Possible therapeutic implications for dyskeratosis congenita. Ann. N. Y. Acad. Sci. 2018, 1418, 56–68. [Google Scholar] [CrossRef]
- Singh, V.; Singh, A.P.; Sharma, I. Epigenetic deregulations of Wnt/β-catenin and transforming growth factor beta-Smad pathways in esophageal cancer: Outcome of DNA methylation. J. Cancer Res. Ther. 2019, 15, 192–203. [Google Scholar] [CrossRef]
- Tang, B.; Xie, R.; Qin, Y.; Xiao, Y.-F.; Yong, X.; Zheng, L.; Dong, H.; Yang, S.-M. Human telomerase reverse transcriptase (hTERT) promotes gastric cancer invasion through cooperating with c-Myc to upregulate heparanase expression. Oncotarget 2016, 7, 11364–11379. [Google Scholar] [CrossRef]
- Remke, M.; Ramaswamy, V.; Peacock, J.; Shih, D.J.H.; Koelsche, C.; Northcott, P.A.; Hill, N.; Cavalli, F.M.G.; Kool, M.; Wang, X.; et al. TERT promoter mutations are highly recurrent in SHH subgroup medulloblastoma. Acta Neuropathol. 2013, 126, 917–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernabé-García, M.; Martínez-Balsalobre, E.; García-Moreno, D.; García-Castillo, J.; Revilla-Nuin, B.; Blanco-Alcaina, E.; Mulero, V.; Alcaraz-Pérez, F.; Cayuela, M.L. TERT-mediated induction of MIR500A contributes to tumor invasiveness by targeting Hedgehog pathway. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.-C.; Martin, Y.; Caruso, S.; Hirsch, T.Z.; Bayard, Q.; Calderaro, J.; Charpy, C.; Copie-Bergman, C.; Ziol, M.; Bioulac-Sage, P.; et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology 2020, 71, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, S.; Sia, D.; Harrington, A.N.; Zhang, Z.; Cabellos, L.; Cornella, H.; Moeini, A.; Camprecios, G.; Leow, W.-Q.; Fiel, M.I.; et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. J. Hepatol. 2017, 67, 1222–1231. [Google Scholar] [CrossRef] [Green Version]
- Pinyol, R.; Tovar, V.; Llovet, J.M. TERT promoter mutations: Gatekeeper and driver of hepatocellular carcinoma. J. Hepatol. 2014, 61, 685–687. [Google Scholar] [CrossRef] [Green Version]
- Bioulac-Sage, P.; Rebouissou, S.; Thomas, C.; Blanc, J.-F.; Saric, J.; Sa Cunha, A.; Rullier, A.; Cubel, G.; Couchy, G.; Imbeaud, S.; et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology 2007, 46, 740–748. [Google Scholar] [CrossRef]
- Nault, J.-C.; Bioulac–Sage, P.; Zucman–Rossi, J. Hepatocellular Benign Tumors—From Molecular Classification to Personalized Clinical Care. Gastroenterology 2013, 144, 888–902. [Google Scholar] [CrossRef]
- Nault, J.-C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.-F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates With Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef] [Green Version]
- Pilati, C.; Letouzé, E.; Nault, J.-C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic Profiling of Hepatocellular Adenomas Reveals Recurrent FRK-Activating Mutations and the Mechanisms of Malignant Transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Sadot, E.; Vigidal, J.A.; Klimstra, D.S.; Balachandran, V.P.; Kingham, T.P.; Allen, P.J.; D’Angelica, M.I.; DeMatteo, R.P.; Jarnagin, W.R.; et al. Characterization of hepatocellular adenoma and carcinoma using microRNA profiling and targeted gene sequencing. PLoS ONE 2018, 13, e0200776. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Watt, G.P.; Stevenson, H.L.; Calderone, T.L.; Fisher-Hoch, S.P.; Ye, Y.; Wu, X.; Vierling, J.M.; Beretta, L. Telomerase reverse transcriptase mutations in plasma DNA in patients with hepatocellular carcinoma or cirrhosis: Prevalence and risk factors. Hepatol. Commun. 2018, 2, 718–731. [Google Scholar] [CrossRef]
- Wege, H.; Heim, D.; Lütgehetmann, M.; Dierlamm, J.; Lohse, A.W.; Brümmendorf, T.H. Forced Activation of β-Catenin Signaling Supports the Transformation of hTERT-Immortalized Human Fetal Hepatocytes. Mol. Cancer Res. 2011, 9, 1222. [Google Scholar] [CrossRef] [Green Version]
- Molina-Sánchez, P.; Ruiz de Galarreta, M.; Yao, M.A.; Lindblad, K.E.; Bresnahan, E.; Bitterman, E.; Martin, T.C.; Rubenstein, T.; Nie, K.; Golas, J.; et al. Cooperation Between Distinct Cancer Driver Genes Underlies Intertumor Heterogeneity in Hepatocellular Carcinoma. Gastroenterology 2020, 159, 2203–2220.e14. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Park, J.-I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Guterres, A.N.; Villanueva, J. Targeting telomerase for cancer therapy. Oncogene 2020, 39, 5811–5824. [Google Scholar] [CrossRef] [PubMed]
- Trybek, T.; Kowalik, A.; Góźdź, S.; Kowalska, A. Telomeres and telomerase in oncogenesis. Oncol. Lett. 2020, 20, 1015–1027. [Google Scholar] [CrossRef]
- Alessandrini, I.; Recagni, M.; Zaffaroni, N.; Folini, M. On the Road to Fight Cancer: The Potential of G-quadruplex Ligands as Novel Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 5947. [Google Scholar] [CrossRef] [PubMed]
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Telomerase reverse transcriptase (TERT) regulation and functions in cancer cells. Cancer cells may upregulate TERT by somatic TERTp mutations, TERT amplification and rearrangement, and/or epigenetic modifications in TERT and TERTp [8]. TERT performs both canonical (telomere lengthening) and non-canonical (gene regulation, signal transduction, stress response, and mitochondrial metabolism) functions [11,12]. Abbreviations: NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; and VEGF, vascular endothelial growth factor.
Figure 1.
Telomerase reverse transcriptase (TERT) regulation and functions in cancer cells. Cancer cells may upregulate TERT by somatic TERTp mutations, TERT amplification and rearrangement, and/or epigenetic modifications in TERT and TERTp [8]. TERT performs both canonical (telomere lengthening) and non-canonical (gene regulation, signal transduction, stress response, and mitochondrial metabolism) functions [11,12]. Abbreviations: NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; and VEGF, vascular endothelial growth factor.

Figure 3.
Multi-step hepatocarcinogenesis: Interplay of TERTp and CTNNB1 mutations. Adapted from Nault et al. and Pinyol et al. [5,105]. TERTp mutations (found in the majority of HCC cases) are the earliest somatic alteration found in preneoplastic cirrhotic lesions and are associated with increased TERT transcription. Activating CTNNB1 mutations and TERTp mutations show high concordance in HCC, and CTNNB1 mutations are enriched in later stages of malignancy, indicating their role as cancer drivers. In HCA, TERTp mutations might flip the switch from benign β-catenin-linked tumorigenesis to malignant HCC. *: Mutation incidence in total HCC samples irrespective of cirrhotic or non-cirrhotic background. N/A: Not applicable as not reported separately in large-scale sequencing studies.
Figure 3.
Multi-step hepatocarcinogenesis: Interplay of TERTp and CTNNB1 mutations. Adapted from Nault et al. and Pinyol et al. [5,105]. TERTp mutations (found in the majority of HCC cases) are the earliest somatic alteration found in preneoplastic cirrhotic lesions and are associated with increased TERT transcription. Activating CTNNB1 mutations and TERTp mutations show high concordance in HCC, and CTNNB1 mutations are enriched in later stages of malignancy, indicating their role as cancer drivers. In HCA, TERTp mutations might flip the switch from benign β-catenin-linked tumorigenesis to malignant HCC. *: Mutation incidence in total HCC samples irrespective of cirrhotic or non-cirrhotic background. N/A: Not applicable as not reported separately in large-scale sequencing studies.

Table 4.
Concordance between TERTp and CTNNB1 mutations in HCC: clues from clinical studies.
| Region | Etiology (N) | TERTp % Incidence (N) | CTNNB1 % Incidence (N) | Association | Ref. |
|---|---|---|---|---|---|
| France | 22% HBV+, 26% HCV+ (305) | 59% (179) | 33% (101) | Significant, p < 0.0001 | [5] |
| France | 20.7% HBV+, 26.5% HCV+ (801) | 58.1% (441) | 30.7% (229) | Significant, p = 0.0000001 | [103] |
| Southern Italy | 7.9% HBV+, 86.6% HCV+ (127) | 50.4% (64) | 26% (33) | Not significant, p = 0.4192 | [43] |
| USA and Japan | 25.6% HBV+, 42.6% HCV+ (469) | 54.1% (254) | 31.1% (146) | Significant, p < 0.0001 | [7] |
| Japan | NAFLD (11) | 82% (9) | 45% (5) | Not significant, p = 0.4545 | [54] |
| Taiwan | 63% HBV+, 40.2% HCV+ (195) | 29.2% (57) | 16.5% (31/188) | Not significant, p = 0.2055 | [34] |
| Korea | 74.3% HBV+, 5.7% HCV+; (105) | 39% (41) | 14.6% (15) | Not significant, p = 0.568 | [46] |
| China | 83% HBV+ (190) | 30% (57) | 24.3% (17/70) | Not significant, p = 0.535 | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kotiyal, S.; Evason, K.J. Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma. Cancers 2021, 13, 4202. https://doi.org/10.3390/cancers13164202
AMA Style
Kotiyal S, Evason KJ. Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma. Cancers. 2021; 13(16):4202. https://doi.org/10.3390/cancers13164202
Chicago/Turabian StyleKotiyal, Srishti, and Kimberley Jane Evason. 2021. "Exploring the Interplay of Telomerase Reverse Transcriptase and β-Catenin in Hepatocellular Carcinoma" Cancers 13, no. 16: 4202. https://doi.org/10.3390/cancers13164202
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.