
DUOX2, a New Biomarker for Disseminated Gastric Cancer’s Response to Low Dose Radiation in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Treatments
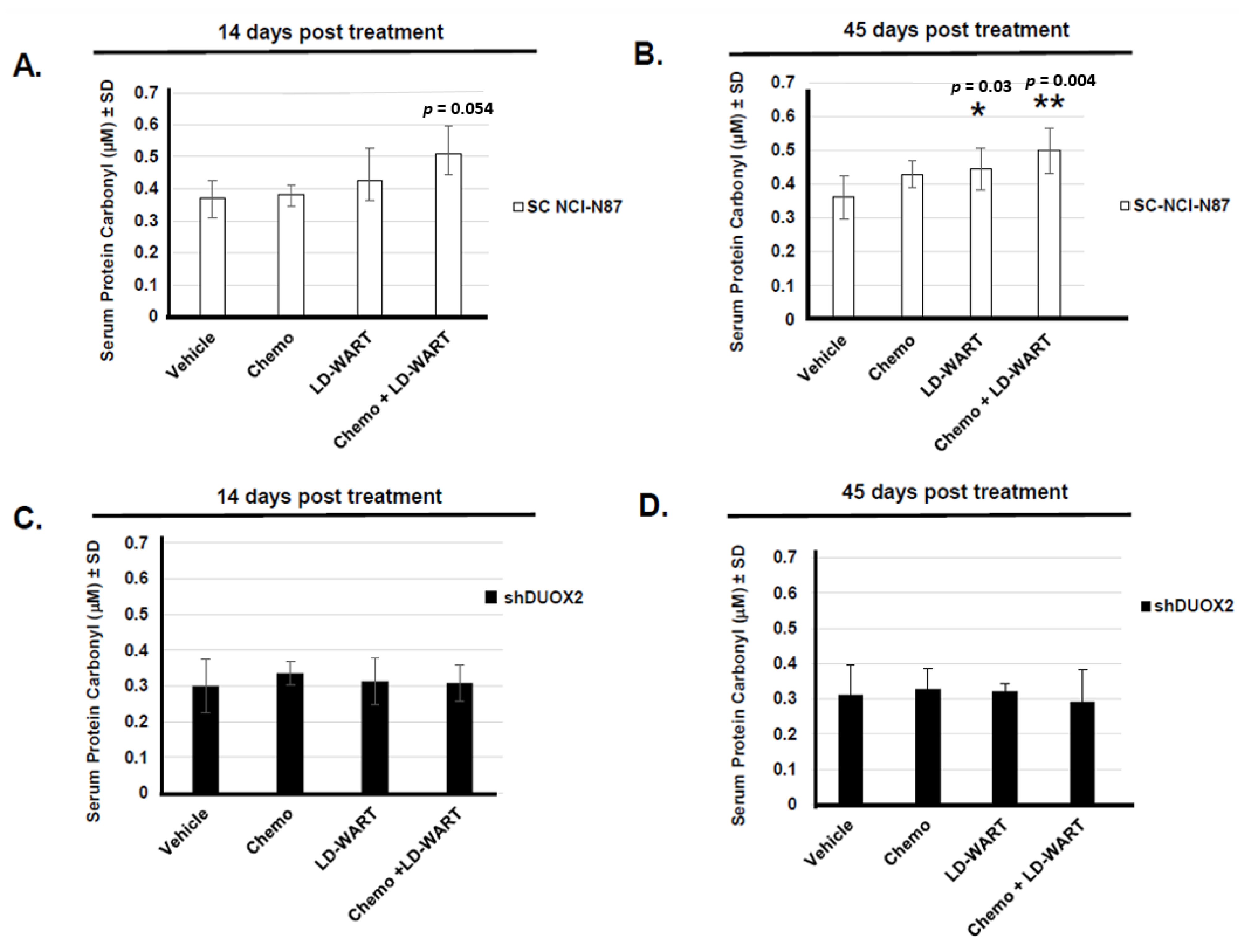
2.2. Protein Carbonyl
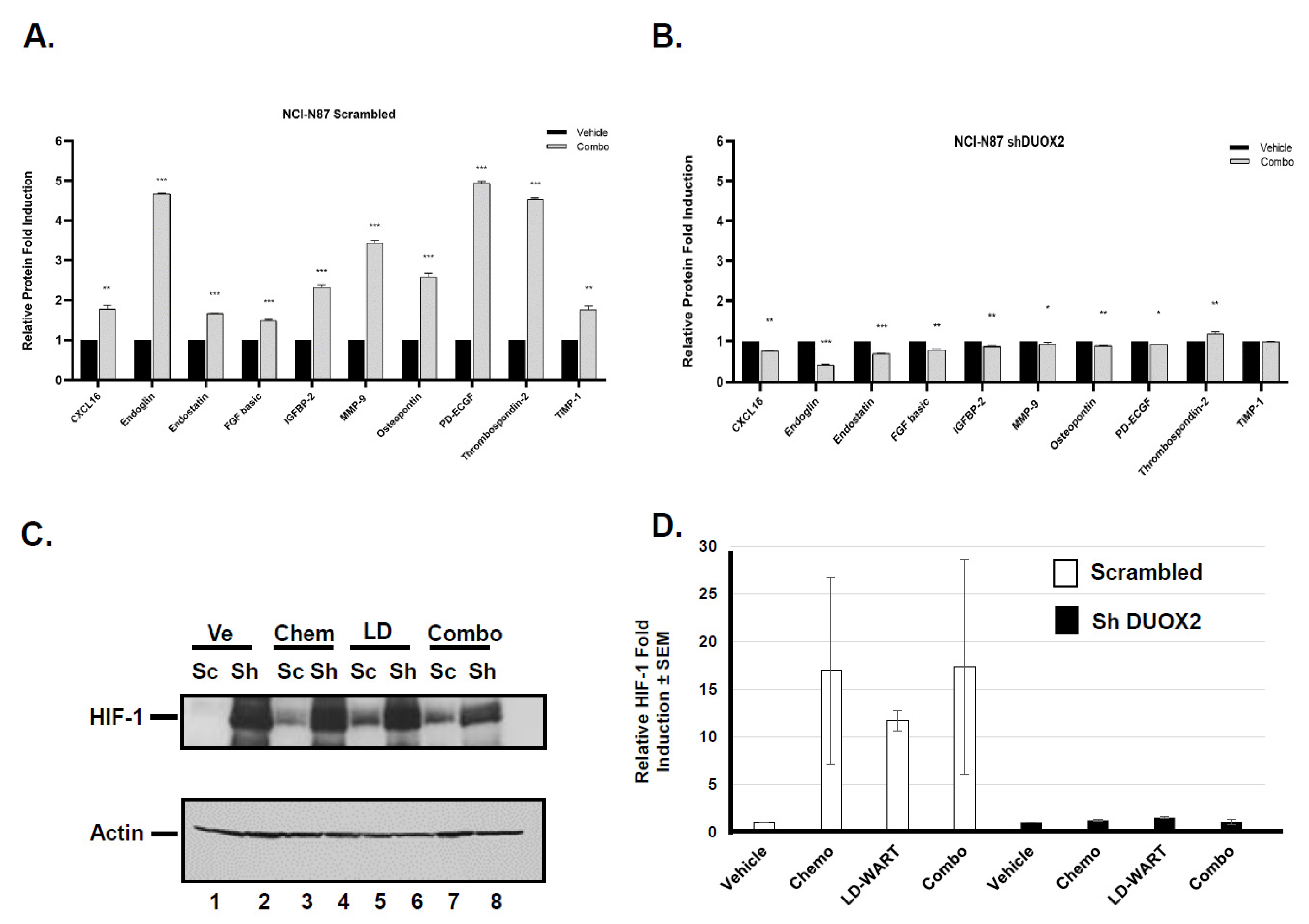
2.3. Mouse Angiogenesis Profiler Array
2.4. TCGA Stomach Adenocarcinoma
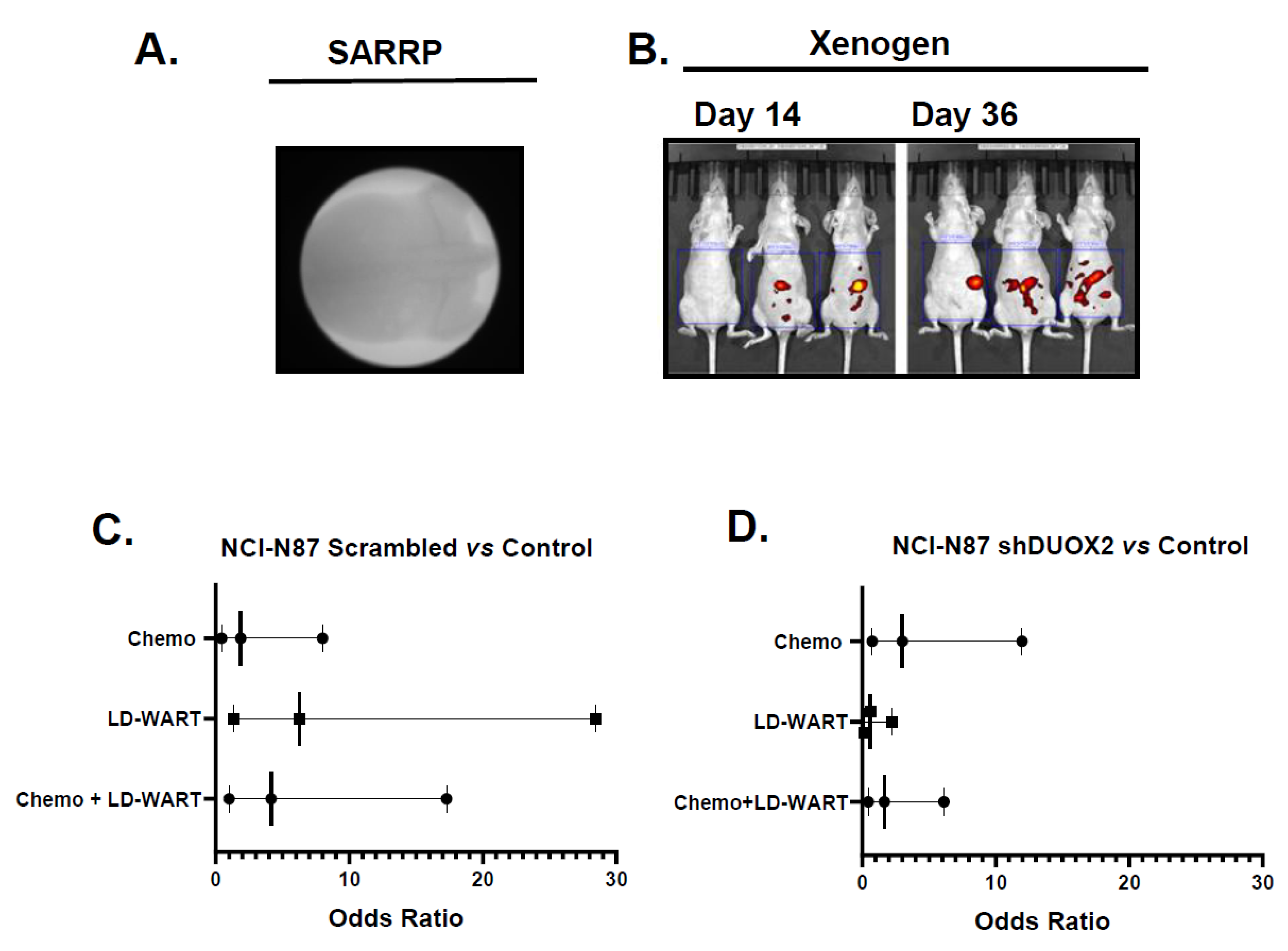
2.5. Animal Studies
2.6. Animal Irradiation
LD-WART
2.7. Electrophoretic Mobility Shift Assay (EMSA)
2.8. RT-PCR
2.9. Immuno Histo Chemistry
2.10. Statistics
3. Results
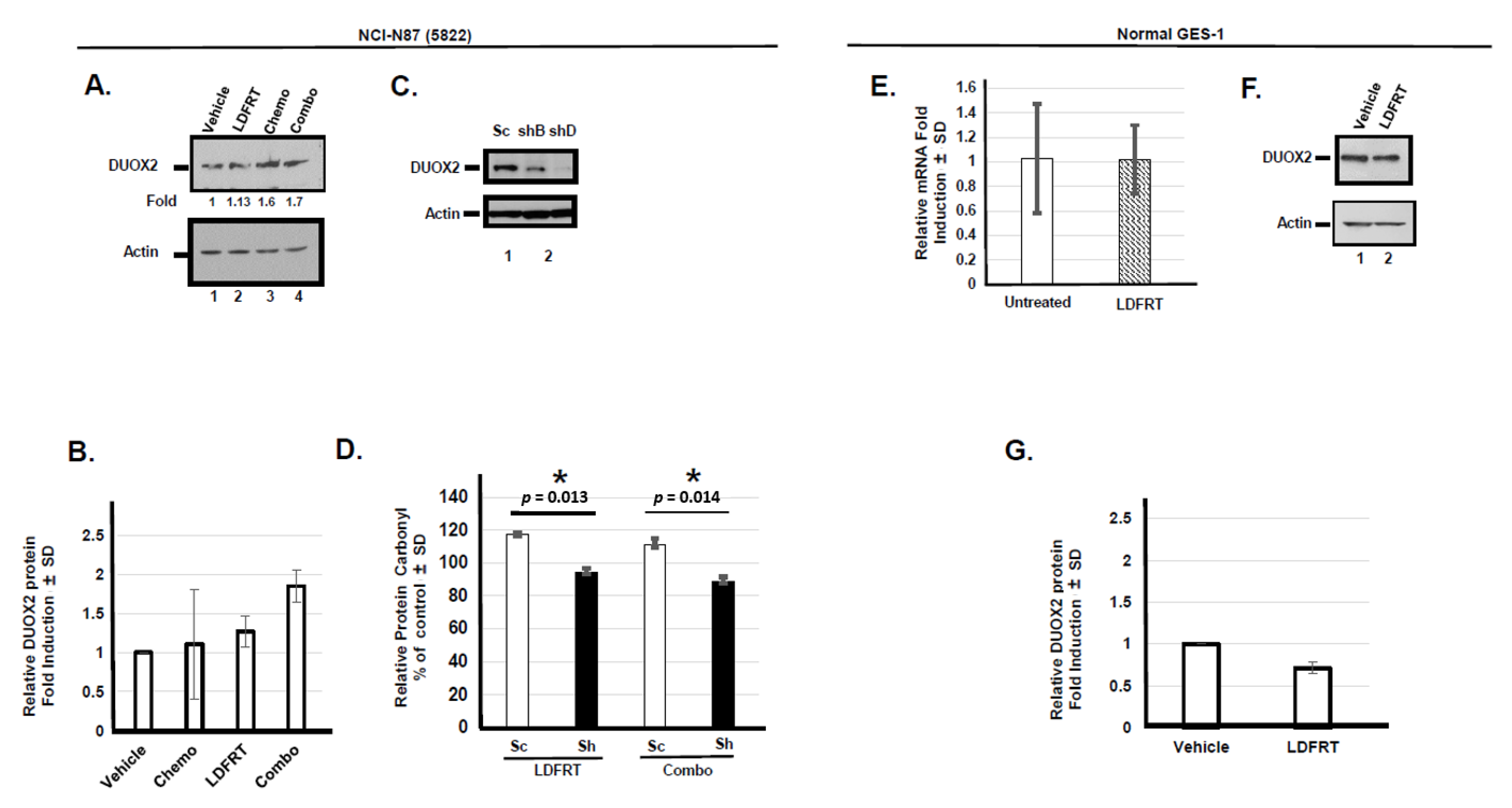
3.1. Upregulation of DUOX2 and Protein Oxidation In Vitro
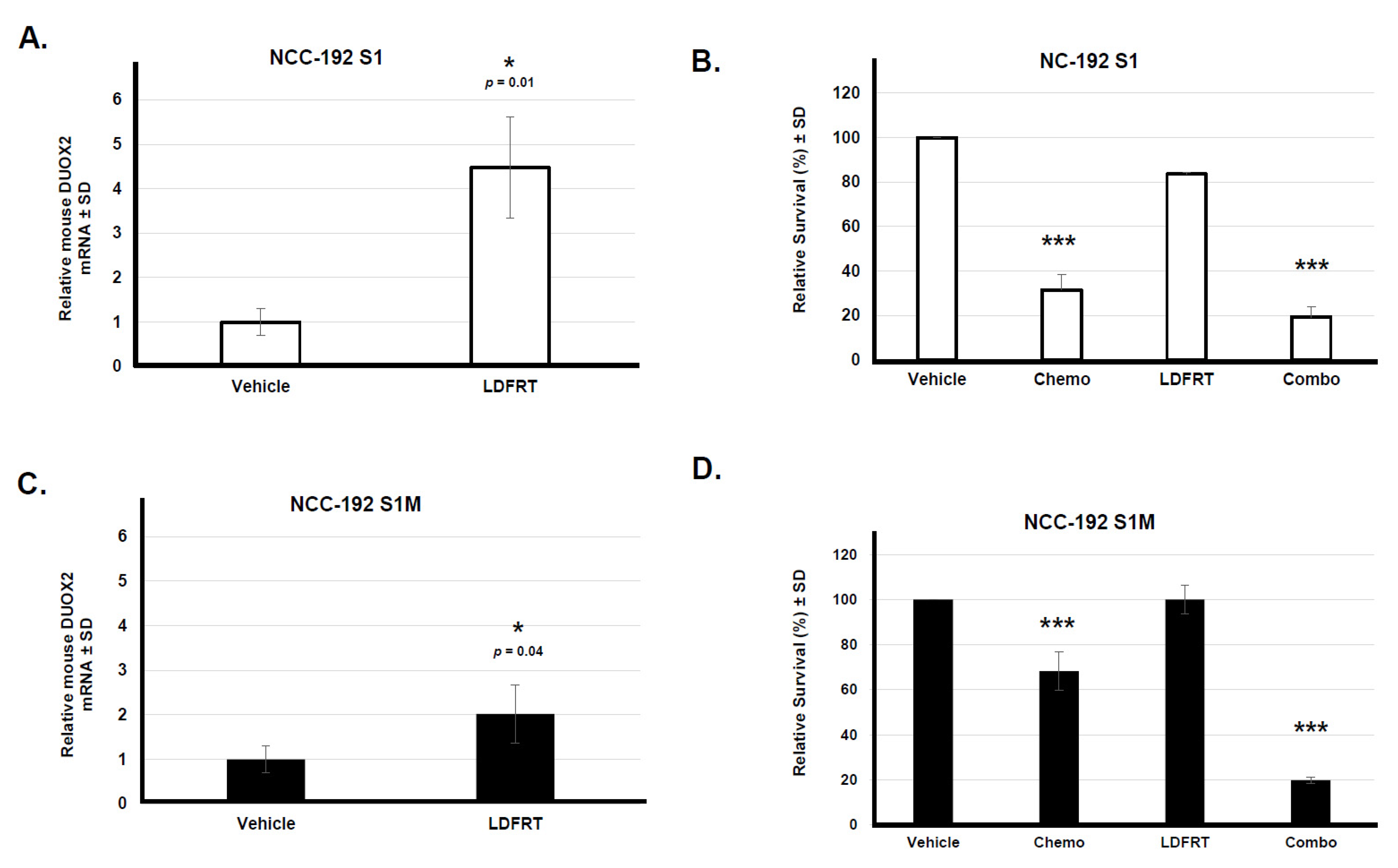
3.1.1. Role of DUOX2 in Disseminated Gastric Cancer Response to mDCF and LD-WART
3.1.2. Effect of LD-WART on Angiogenesis
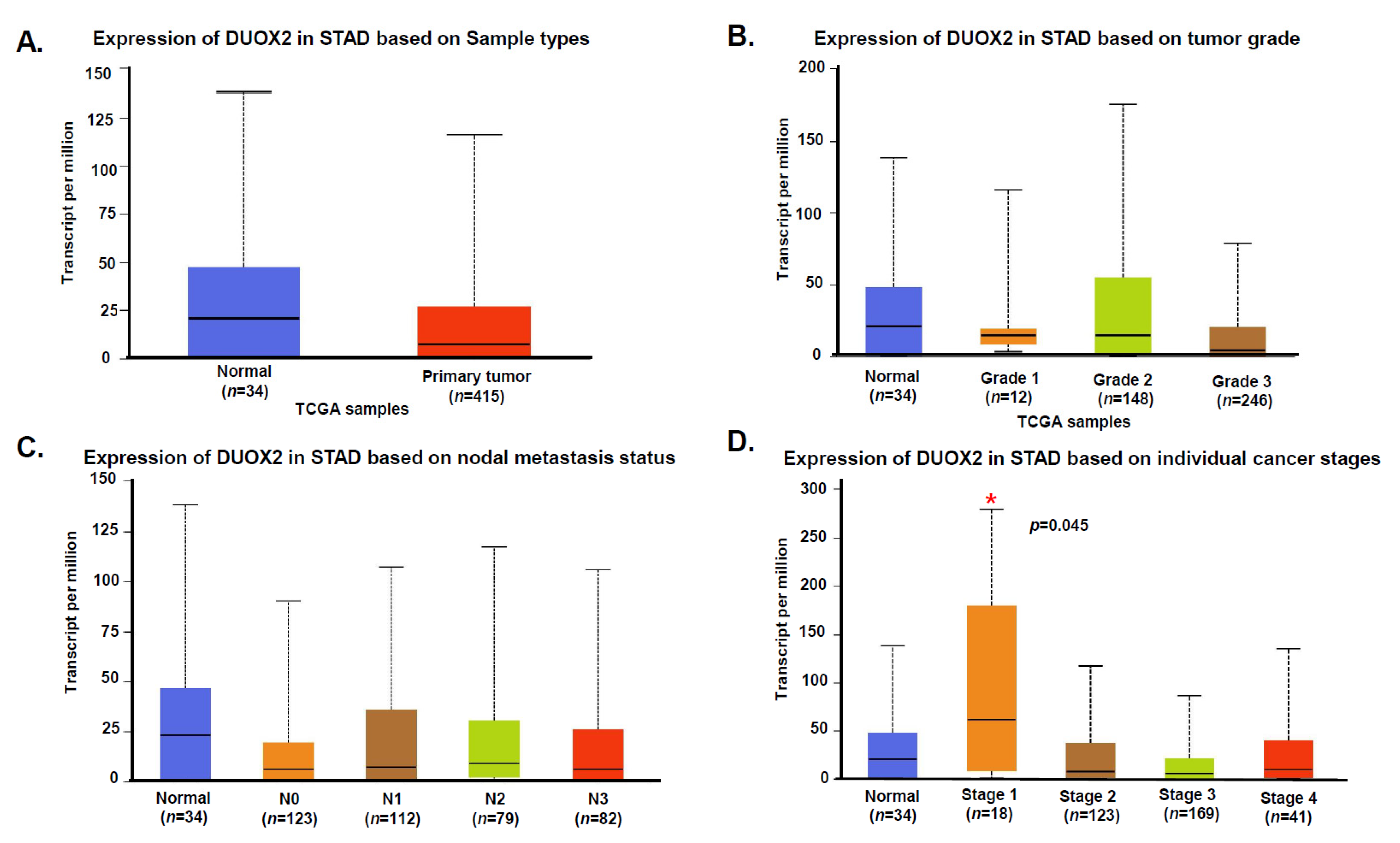
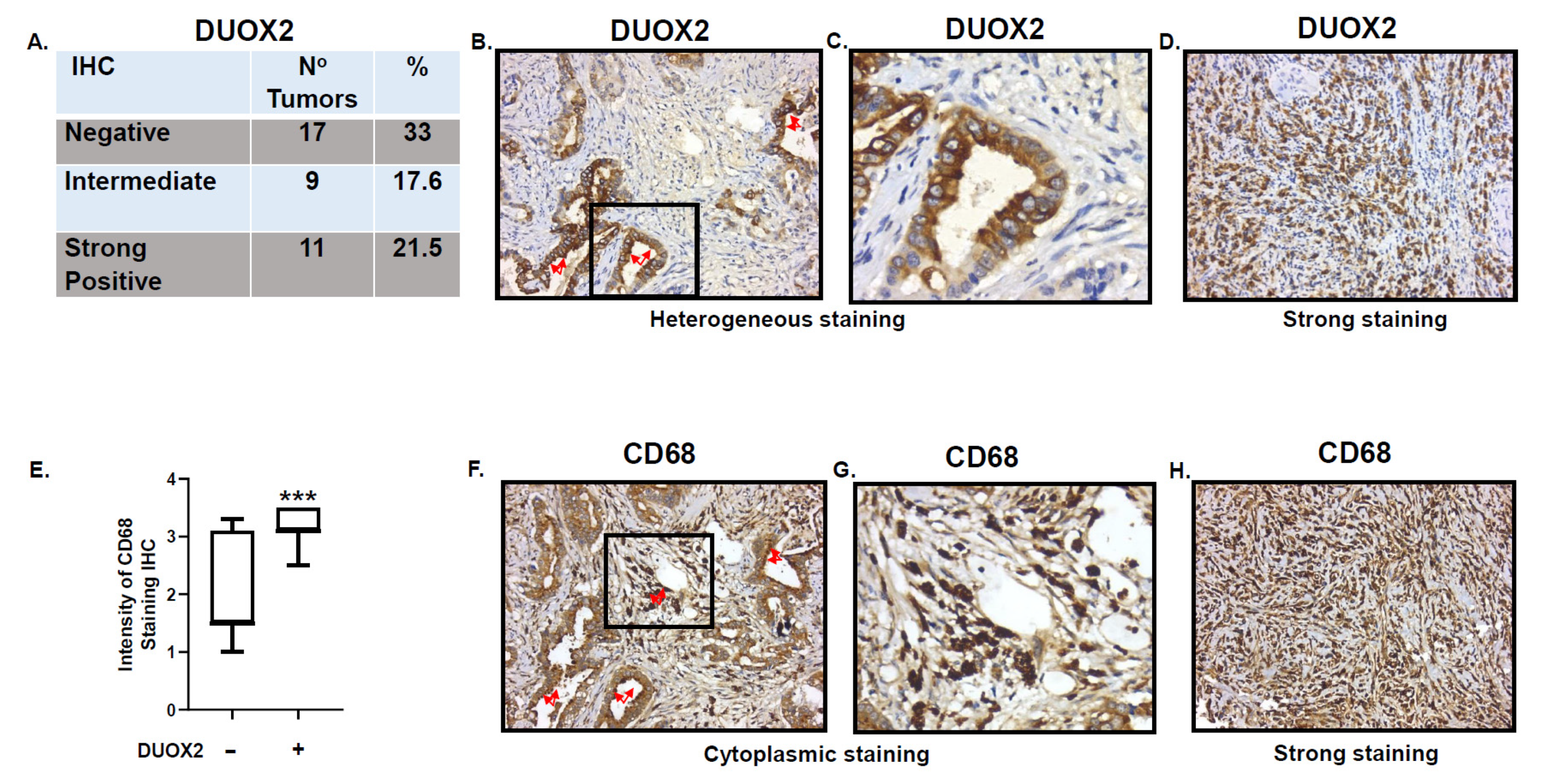
3.2. Expression of DUOX2 in Human Stomach Cancer
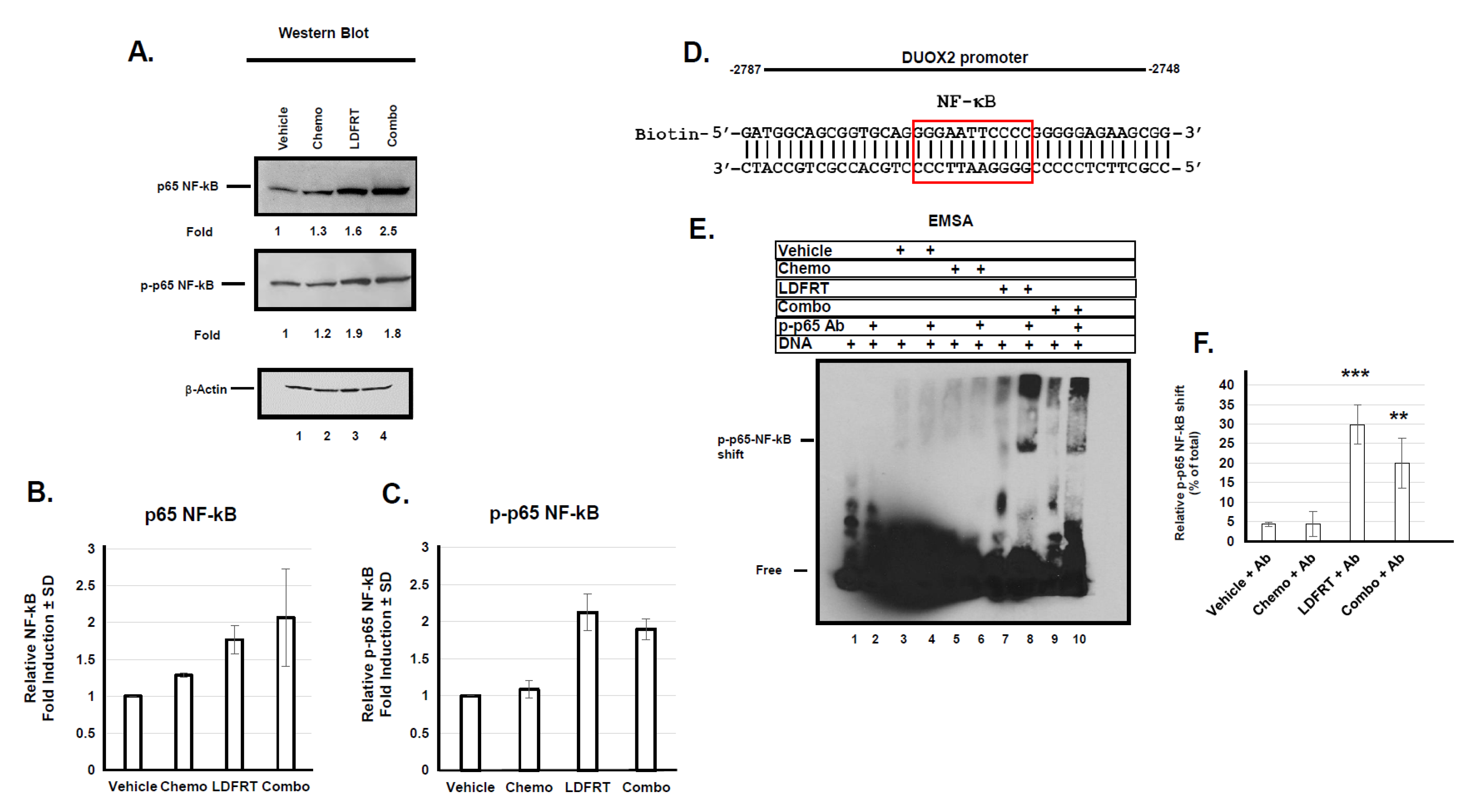
3.3. Regulation of DUOX2 Expression in Response to mDCF and LDFRT
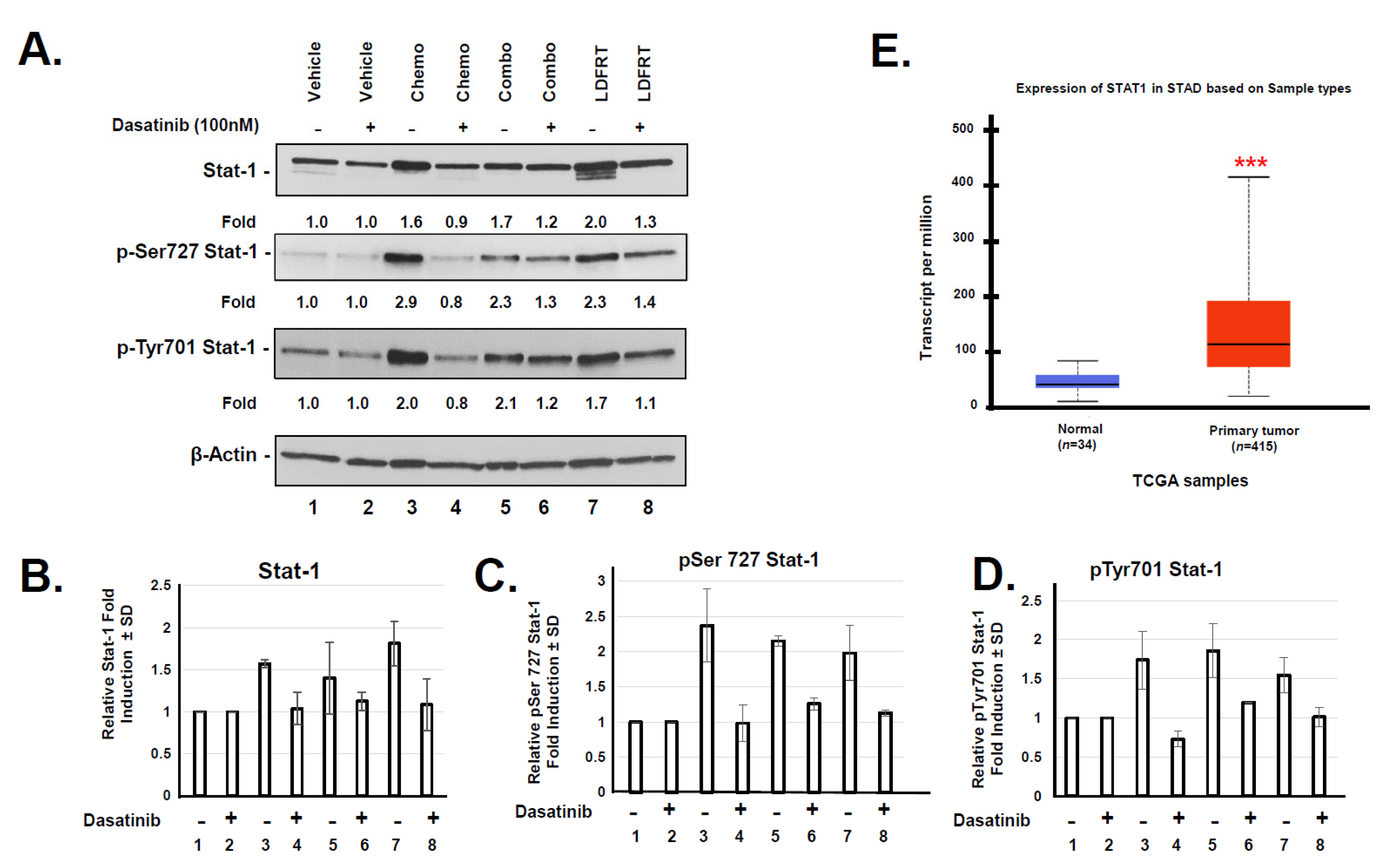
3.4. Potential Role of DUOX2 in Cancer Stem Cells Response to mDCF and LDFRT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Thomassen, I.; van Gestel, Y.R.; van Ramshorst, B.; Luyer, M.D.; Bosscha, K.; Nienhuijs, S.W.; Lemmens, V.E.; de Hingh, I.H. Peritoneal carcinomatosis of gastric origin: A population-based study on incidence, survival and risk factors. Int. J. Cancer 2014, 134, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Yarema, R.; Mielko, J.; Fetsych, T.; Ohorchak, M.; Skorzewska, M.; Rawicz-Pruszynski, K.; Mashukov, A.; Maksimovsky, V.; Jastrzebski, T.; Polkowski, W.; et al. Hyperthermic intraperitoneal chemotherapy (HIPEC) in combined treatment of locally advanced and intraperitonealy disseminated gastric cancer: A retrospective cooperative Central-Eastern European study. Cancer Med. 2019, 8, 2877–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanese, S.; Lordick, F. Targeted and immunotherapy in the era of personalised gastric cancer treatment. Best Pract. Res. Clin. Gastroenterol. 2021, 50–51, 101738. [Google Scholar] [CrossRef] [PubMed]
- Sugarbaker, P.H. Gastric cancer: Prevention and treatment of peritoneal metastases. J. Cancer Metastasis Treat. 2018, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Yao, X.; Ajani, J.A.; Song, S. Molecular biology and immunology of gastric cancer peritoneal metastasis. Transl. Gastroenterol. Hepatol. 2020, 5, 57. [Google Scholar] [CrossRef]
- Moehler, M.; Dvorkin, M.; Boku, N.; Ozguroglu, M.; Ryu, M.H.; Muntean, A.S.; Lonardi, S.; Nechaeva, M.; Bragagnoli, A.C.; Coskun, H.S.; et al. Phase III Trial of Avelumab Maintenance after First-Line Induction Chemotherapy Versus Continuation of Chemotherapy in Patients with Gastric Cancers: Results from JAVELIN Gastric 100. J. Clin. Oncol. 2021, 39, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.H.; Doi, T.; Moriwaki, T.; Kim, S.B.; Lee, S.H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Fabian, C.; Giri, S.; Estes, N.; Tangen, C.M.; Poplin, E.; Vogel, S.; Goodwin, W.; Rivkin, S.; Fleming, T.R.; Macdonald, J.S. Adjuvant continuous infusion 5-FU, whole-abdominal radiation, and tumor bed boost in high-risk stage III colon carcinoma: A Southwest Oncology Group Pilot study. Int. J. Radiat. Oncol. Biol. Phys. 1995, 32, 457–464. [Google Scholar] [CrossRef]
- Prasanna, A.; Ahmed, M.M.; Mohiuddin, M.; Coleman, C.N. Exploiting sensitization windows of opportunity in hyper and hypo-fractionated radiation therapy. J. Thorac. Dis. 2014, 6, 287–302. [Google Scholar] [CrossRef]
- Arnold, S.M.; Regine, W.F.; Ahmed, M.M.; Valentino, J.; Spring, P.; Kudrimoti, M.; Kenady, D.; Desimone, P.; Mohiuddin, M. Low-dose fractionated radiation as a chemopotentiator of neoadjuvant paclitaxel and carboplatin for locally advanced squamous cell carcinoma of the head and neck: Results of a new treatment paradigm. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Regine, W.F.; Hanna, N.; Garofalo, M.C.; Doyle, A.; Arnold, S.; Kataria, R.; Sims, J.; Tan, M.; Mohiuddin, M. Low-dose radiotherapy as a chemopotentiator of gemcitabine in tumors of the pancreas or small bowel: A phase I study exploring a new treatment paradigm. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Kunos, C.A.; Sill, M.W.; Buekers, T.E.; Walker, J.L.; Schilder, J.M.; Yamada, S.D.; Waggoner, S.E.; Mohiuddin, M.; Fracasso, P.M. Low-dose abdominal radiation as a docetaxel chemosensitizer for recurrent epithelial ovarian cancer: A phase I study of the Gynecologic Oncology Group. Gynecol. Oncol. 2011, 120, 224–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrenn, D.C.; Saigal, K.; Lucci, J.A., 3rd; Pearson, M.J.; Simpkins, F.; Schuman, S.; Twiggs, L.B.; Walker, G.R.; Wolfson, A.H. A Phase I Study using low-dose fractionated whole abdominal radiotherapy as a chemopotentiator to full-dose cisplatin for optimally debulked stage III/IV carcinoma of the endometrium. Gynecol. Oncol. 2011, 122, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.L.; Arnold, S.; Gleason, J.F., Jr.; Kudrimoti, M.; Brill, Y.; Van Meter, E.M.; Gal, T.J.; Valentino, J. p16 status predicts response to low dose fractionated radiation as a chemopotentiator of neoadjuvant chemotherapy for locally advanced squamous cell carcinoma of the head and neck. In Proceedings of the 8th International Conference on Head and Neck Cancer, Toronto, ON, Canada, 21–25 July 2012; p. 82. [Google Scholar]
- Galloway, T.J.; Zhang, Q.E.; Nguyen-Tan, P.F.; Rosenthal, D.I.; Soulieres, D.; Fortin, A.; Silverman, C.L.; Daly, M.E.; Ridge, J.A.; Hammond, J.A.; et al. Prognostic Value of p16 Status on the Development of a Complete Response in Involved Oropharynx Cancer Neck Nodes After Cisplatin-Based Chemoradiation: A Secondary Analysis of NRG Oncology RTOG 0129. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.M.; Parekh, P.R.; Chang, E.T.; Sharma, N.K.; Carrier, F. Contribution of Dual Oxidase 2 (DUOX2) to Hyper-Radiosensitivity in Human Gastric Cancer Cells. Radiat. Res. 2015, 184, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Murphy, D.L.; Sweasy, J.B. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis 2014, 3, e128. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Park, D.M.; Choi, B.K.; Kwon, B.S.; Seong, J.K.; Green, J.E.; Kim, D.Y.; Kim, H.K. Establishment and characterization of metastatic gastric cancer cell lines from murine gastric adenocarcinoma lacking Smad4, p53, and E-cadherin. Mol. Carcinog. 2015, 54, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Park, J.M.; Park, D.M.; Kim, D.Y.; Kim, H.K. Stem Cells Antigen-1 Enriches for a Cancer Stem Cell-Like Subpopulation in Mouse Gastric Cancer. Stem Cells 2016, 34, 1177–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Choyke, P.L.; Kobayashi, H. Photoimmunotherapy of gastric cancer peritoneal carcinomatosis in a mouse model. PLoS ONE 2014, 9, e113276. [Google Scholar] [CrossRef]
- Bozec, A.; Sudaka, A.; Etienne-Grimaldi, M.C.; Brunstein, M.C.; Fischel, J.L.; Milano, G. Antitumor activity of cetuximab associated with the taxotere-cisplatin-fluorouracil (TPF) combination on an orthotopic head and neck cancer model. Oral Oncol. 2011, 47, 940–945. [Google Scholar] [CrossRef]
- Ma, C.M.; Coffey, C.W.; DeWerd, L.A.; Liu, C.; Nath, R.; Seltzer, S.M.; Seuntjens, J.P. AAPM protocol for 40–300 kV X-ray beam dosimetry in radiotherapy and radiobiology. Med. Phys. 2001, 28, 868–893. [Google Scholar] [CrossRef]
- Iacovelli, R.; Pietrantonio, F.; Maggi, C.; de Braud, F.; Di Bartolomeo, M. Combination or single-agent chemotherapy as adjuvant treatment of gastric cancer: A systematic review and meta-analysis of published trials. Crit. Rev. Oncol. Hematol. 2016, 98, 24–28. [Google Scholar] [CrossRef]
- Lipinski, S.; Till, A.; Sina, C.; Arlt, A.; Grasberger, H.; Schreiber, S.; Rosenstiel, P. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci. 2009, 122, 3522–3530. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Shao, M.; Liu, M.; Peng, P.; Li, L.; Wu, W.; Wang, L.; Duan, F.; Zhang, M.; Song, S.; et al. PKCalpha promotes generation of reactive oxygen species via DUOX2 in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2015, 463, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Brouet, A.; Havaux, X.; Gregoire, V.; Dessy, C.; Balligand, J.L.; Feron, O. Irradiation-induced angiogenesis through the up-regulation of the nitric oxide pathway: Implications for tumor radiotherapy. Cancer Res. 2003, 63, 1012–1019. [Google Scholar]
- Heissig, B.; Rafii, S.; Akiyama, H.; Ohki, Y.; Sato, Y.; Rafael, T.; Zhu, Z.; Hicklin, D.J.; Okumura, K.; Ogawa, H.; et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J. Exp. Med. 2005, 202, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M. The hypoxic cell: A target for selective cancer therapy—Eighteenth Bruce F. Cain Memorial Award lecture. Cancer Res. 1999, 59, 5863–5870. [Google Scholar] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Schindl, A.; Heinze, G.; Schindl, M.; Pernerstorfer-Schon, H.; Schindl, L. Systemic effects of low-intensity laser irradiation on skin microcirculation in patients with diabetic microangiopathy. Microvasc. Res. 2002, 64, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Mohan, N.; Meltz, M.L. Induction of nuclear factor kappa B after low-dose ionizing radiation involves a reactive oxygen intermediate signaling pathway. Radiat. Res. 1994, 140, 97–104. [Google Scholar] [CrossRef]
- Yim, J.H.; Yun, J.M.; Kim, J.Y.; Nam, S.Y.; Kim, C.S. Estimation of low-dose radiation-responsive proteins in the absence of genomic instability in normal human fibroblast cells. Int. J. Radiat. Biol. 2017, 93, 1197–1206. [Google Scholar] [CrossRef]
- Lai, S.Y.; Johnson, F.M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: Implications for future therapeutic approaches. Drug Resist. Updates 2010, 13, 67–78. [Google Scholar] [CrossRef]
- Kharbanda, S.; Yuan, Z.M.; Rubin, E.; Weichselbaum, R.; Kufe, D. Activation of Src-like p56/p53lyn tyrosine kinase by ionizing radiation. J. Biol. Chem. 1994, 269, 20739–20743. [Google Scholar] [CrossRef]
- Andrianifahanana, M.; Singh, A.P.; Nemos, C.; Ponnusamy, M.P.; Moniaux, N.; Mehta, P.P.; Varshney, G.C.; Batra, S.K. IFN-gamma-induced expression of MUC4 in pancreatic cancer cells is mediated by STAT-1 upregulation: A novel mechanism for IFN-gamma response. Oncogene 2007, 26, 7251–7261. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Antony, S.; Juhasz, A.; Lu, J.; Ge, Y.; Jiang, G.; Roy, K.; Doroshow, J.H. Up-regulation and sustained activation of Stat1 are essential for interferon-gamma (IFN-gamma)-induced dual oxidase 2 (Duox2) and dual oxidase A2 (DuoxA2) expression in human pancreatic cancer cell lines. J. Biol. Chem. 2011, 286, 12245–12256. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Meyer, F.; Dubrovska, A.; Borgmann, K. Cancer Stem Cells and Radioresistance: DNA Repair and Beyond. Cancers 2019, 11, 862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Li, C.; Cheng, N.; Cui, X.; Xu, X.; Zhou, G. Redox Regulation in Cancer Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 750798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joiner, M.C.; Marples, B.; Lambin, P.; Short, S.C.; Turesson, I. Low-dose hypersensitivity: Current status and possible mechanisms. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 379–389. [Google Scholar] [CrossRef]
- Arnold, S.M.; Kudrimoti, M.; Dressler, E.V.; Gleason, J.F., Jr.; Silver, N.L.; Regine, W.F.; Valentino, J. Using low-dose radiation to potentiate the effect of induction chemotherapy in head and neck cancer: Results of a prospective phase 2 trial. Adv. Radiat. Oncol 2016, 1, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.S.; Smalley, S.R.; Benedetti, J.; Hundahl, S.A.; Estes, N.C.; Stemmermann, G.N.; Haller, D.G.; Ajani, J.A.; Gunderson, L.L.; Jessup, J.M.; et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N. Engl. J. Med. 2001, 345, 725–730. [Google Scholar] [CrossRef]
- Wegner, R.E.; Abel, S.; White, R.J.; Horne, Z.D.; Hasan, S.; Kirichenko, A.V. Trends in intensity-modulated radiation therapy use for rectal cancer in the neoadjuvant setting: A National Cancer Database analysis. Radiat. Oncol. J. 2018, 36, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Gu, X.; Ge, K.; Fu, G.; Chu, J.; Wei, W. The survival benefit of adjuvant radiotherapy for pathological T4N2M0 colon cancer in the Modern Chemotherapy Era: Evidence from the SEER database 2004–2015. Artif. Cells Nanomed. Biotechnol. 2020, 48, 834–840. [Google Scholar] [CrossRef]
- Marples, B.; Wouters, B.G.; Collis, S.J.; Chalmers, A.J.; Joiner, M.C. Low-dose hyper-radiosensitivity: A consequence of ineffective cell cycle arrest of radiation-damaged G2-phase cells. Radiat. Res. 2004, 161, 247–255. [Google Scholar] [CrossRef]
- Krueger, S.A.; Joiner, M.C.; Weinfeld, M.; Piasentin, E.; Marples, B. Role of apoptosis in low-dose hyper-radiosensitivity. Radiat. Res. 2007, 167, 260–267. [Google Scholar] [CrossRef]
- Gupta, S.; Koru-Sengul, T.; Arnold, S.M.; Devi, G.R.; Mohiuddin, M.; Ahmed, M.M. Low-dose fractionated radiation potentiates the effects of cisplatin independent of the hyper-radiation sensitivity in human lung cancer cells. Mol. Cancer Ther. 2011, 10, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Wykes, S.M.; Piasentin, E.; Joiner, M.C.; Wilson, G.D.; Marples, B. Low-dose hyper-radiosensitivity is not caused by a failure to recognize DNA double-strand breaks. Radiat. Res. 2006, 165, 516–524. [Google Scholar] [CrossRef]
- Rey, S.; Schito, L.; Koritzinsky, M.; Wouters, B.G. Molecular targeting of hypoxia in radiotherapy. Adv. Drug Deliv. Rev. 2017, 109, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tothill, P.; Klys, H.S.; Matheson, L.M.; McKay, K.; Smyth, J.F. The long-term retention of platinum in human tissues following the administration of cisplatin or carboplatin for cancer chemotherapy. Eur. J. Cancer 1992, 28A, 1358–1361. [Google Scholar] [CrossRef]
- Zhang, H.; Mi, J.Q.; Fang, H.; Wang, Z.; Wang, C.; Wu, L.; Zhang, B.; Minden, M.; Yang, W.T.; Wang, H.W.; et al. Preferential eradication of acute myelogenous leukemia stem cells by fenretinide. Proc. Natl. Acad. Sci. USA 2013, 110, 5606–5611. [Google Scholar] [CrossRef] [Green Version]
- Olivares-Urbano, M.A.; Grinan-Lison, C.; Marchal, J.A.; Nunez, M.I. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells 2020, 9, 1651. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parekh, P.R.; Solano-Gonzalez, E.; Martins, M.B.; Ma, X.; Tighe, K.; Casildo, A.; Zodda, A.; Johnstone, C.; Poirier, Y.; Mahmood, J.; et al. DUOX2, a New Biomarker for Disseminated Gastric Cancer’s Response to Low Dose Radiation in Mice. Cancers 2021, 13, 4186. https://doi.org/10.3390/cancers13164186
Parekh PR, Solano-Gonzalez E, Martins MB, Ma X, Tighe K, Casildo A, Zodda A, Johnstone C, Poirier Y, Mahmood J, et al. DUOX2, a New Biomarker for Disseminated Gastric Cancer’s Response to Low Dose Radiation in Mice. Cancers. 2021; 13(16):4186. https://doi.org/10.3390/cancers13164186
Chicago/Turabian StyleParekh, Palak R., Eduardo Solano-Gonzalez, Mariana B. Martins, Xinrong Ma, Kayla Tighe, Andrea Casildo, Andrew Zodda, Christopher Johnstone, Yannick Poirier, Javed Mahmood, and et al. 2021. "DUOX2, a New Biomarker for Disseminated Gastric Cancer’s Response to Low Dose Radiation in Mice" Cancers 13, no. 16: 4186. https://doi.org/10.3390/cancers13164186