
WIP1 Inhibition by GSK2830371 Potentiates HDM201 through Enhanced p53 Phosphorylation and Activation in Liver Adenocarcinoma Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
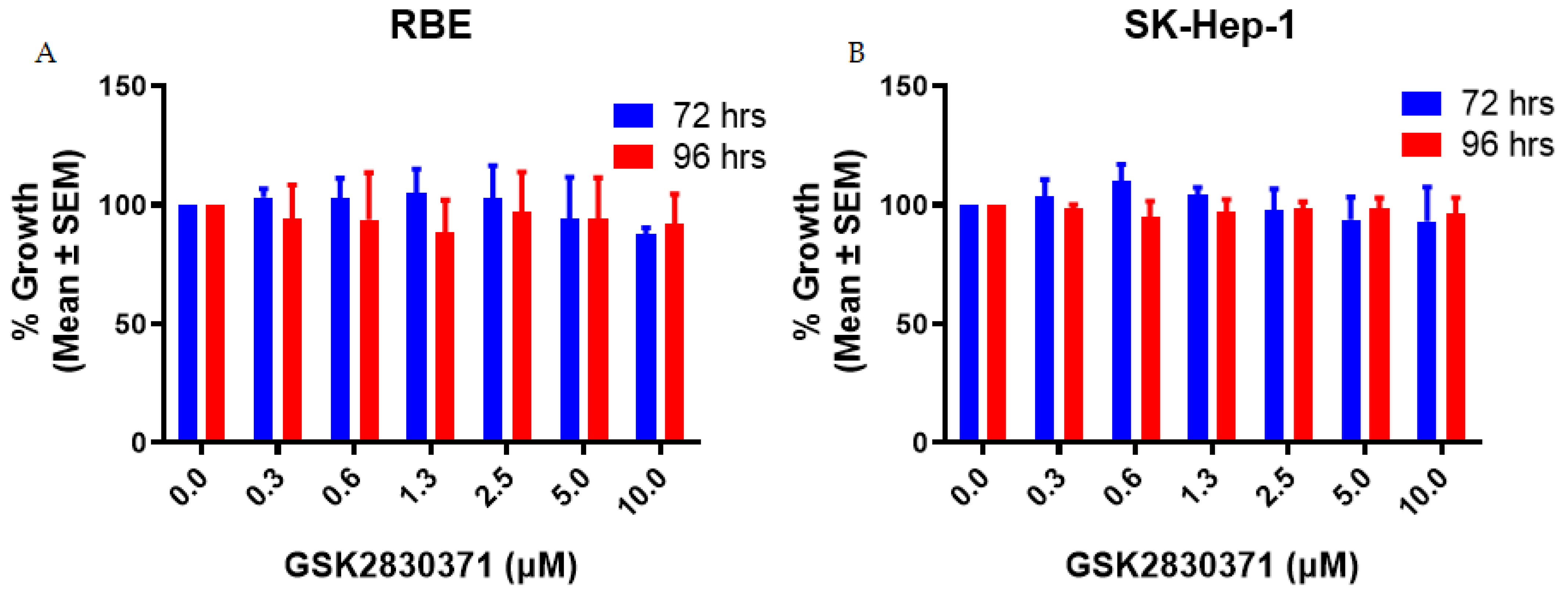
2.1. GSK2830371 Alone Exhibited no Growth Inhibitory Activity
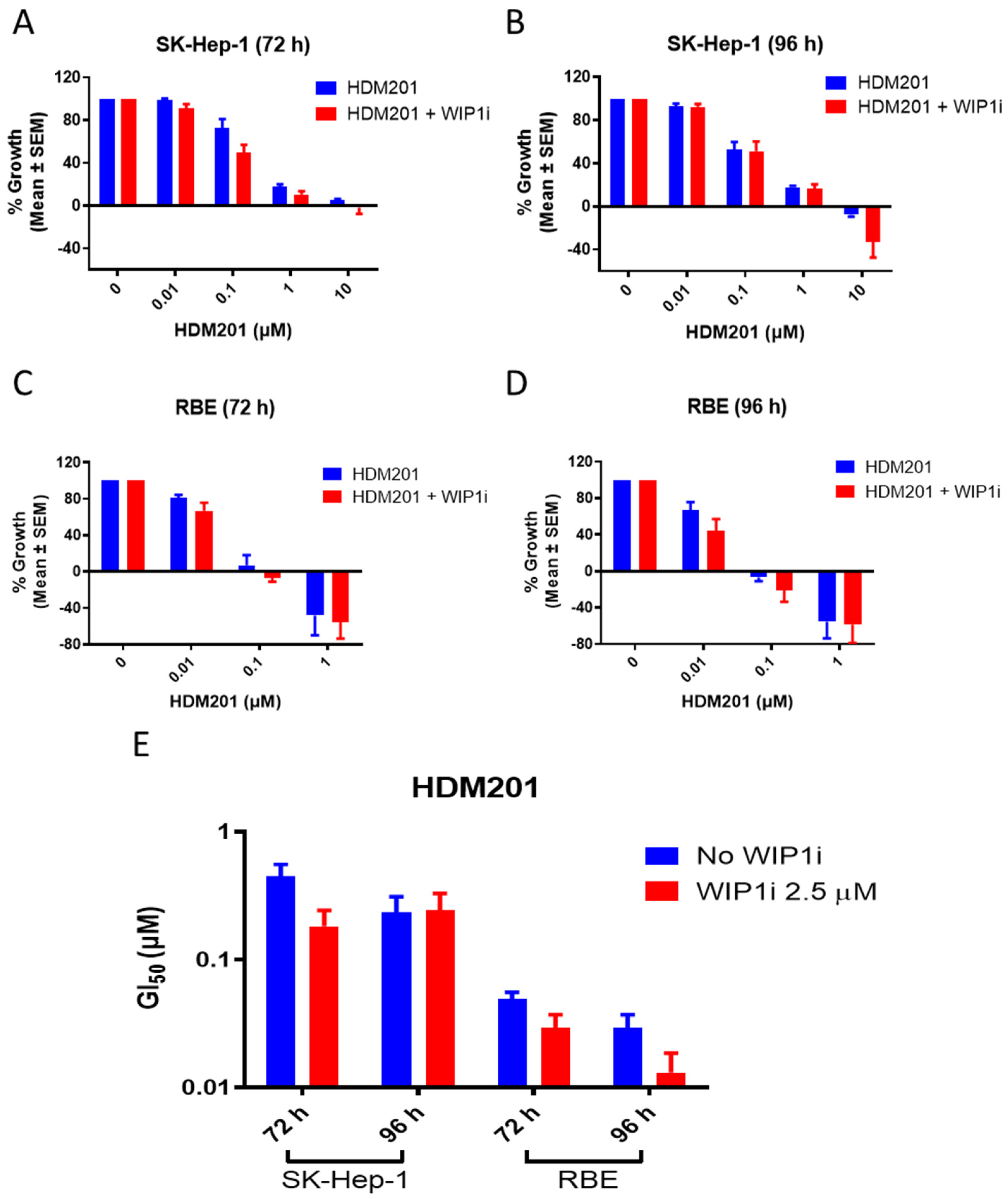
2.2. GSK2830371 Potentiated the Growth Inhibitory Activity of HDM201
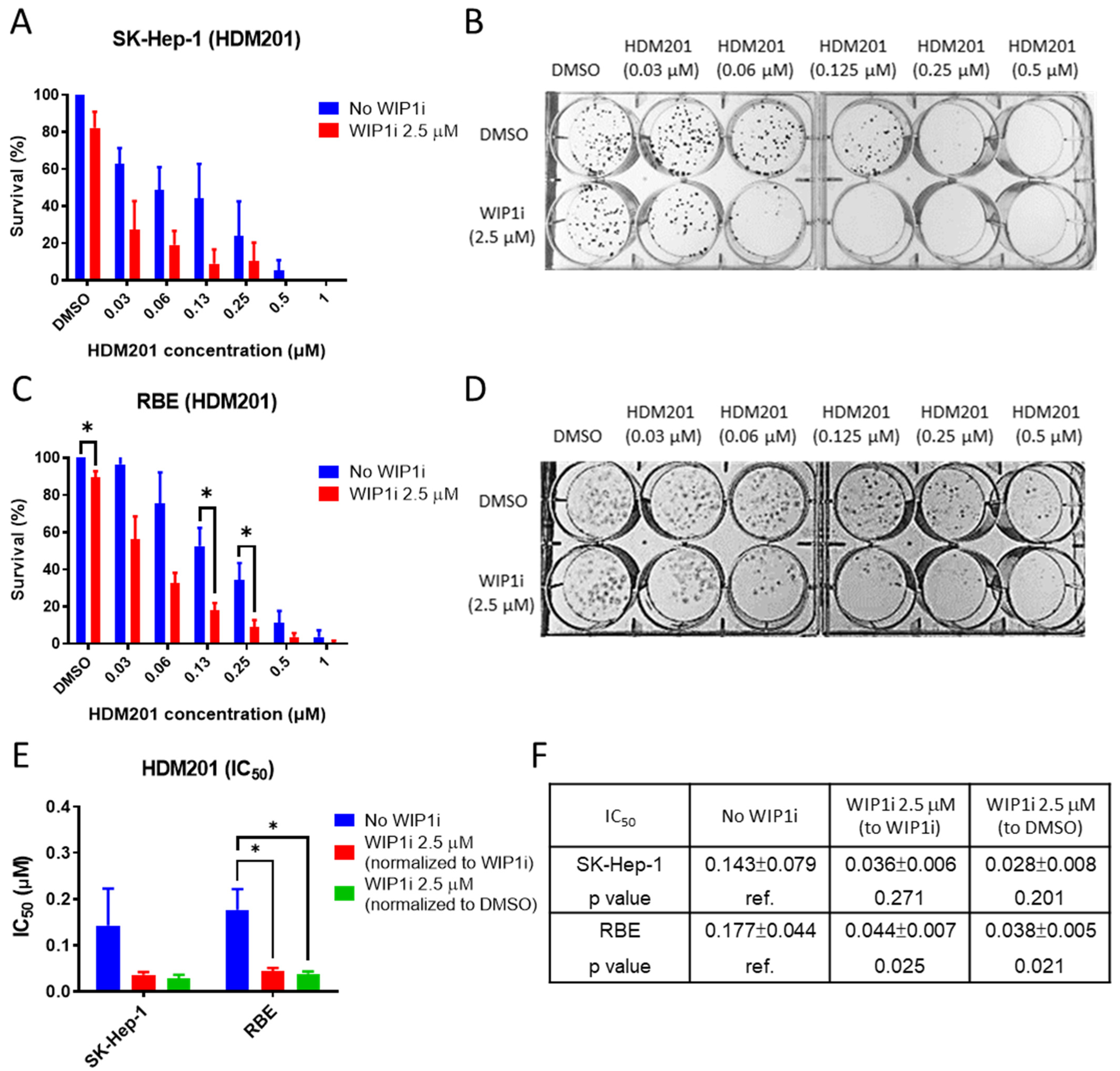
2.3. GSK2830371 Enhanced the Inhibition of Colony Formation by HDM201
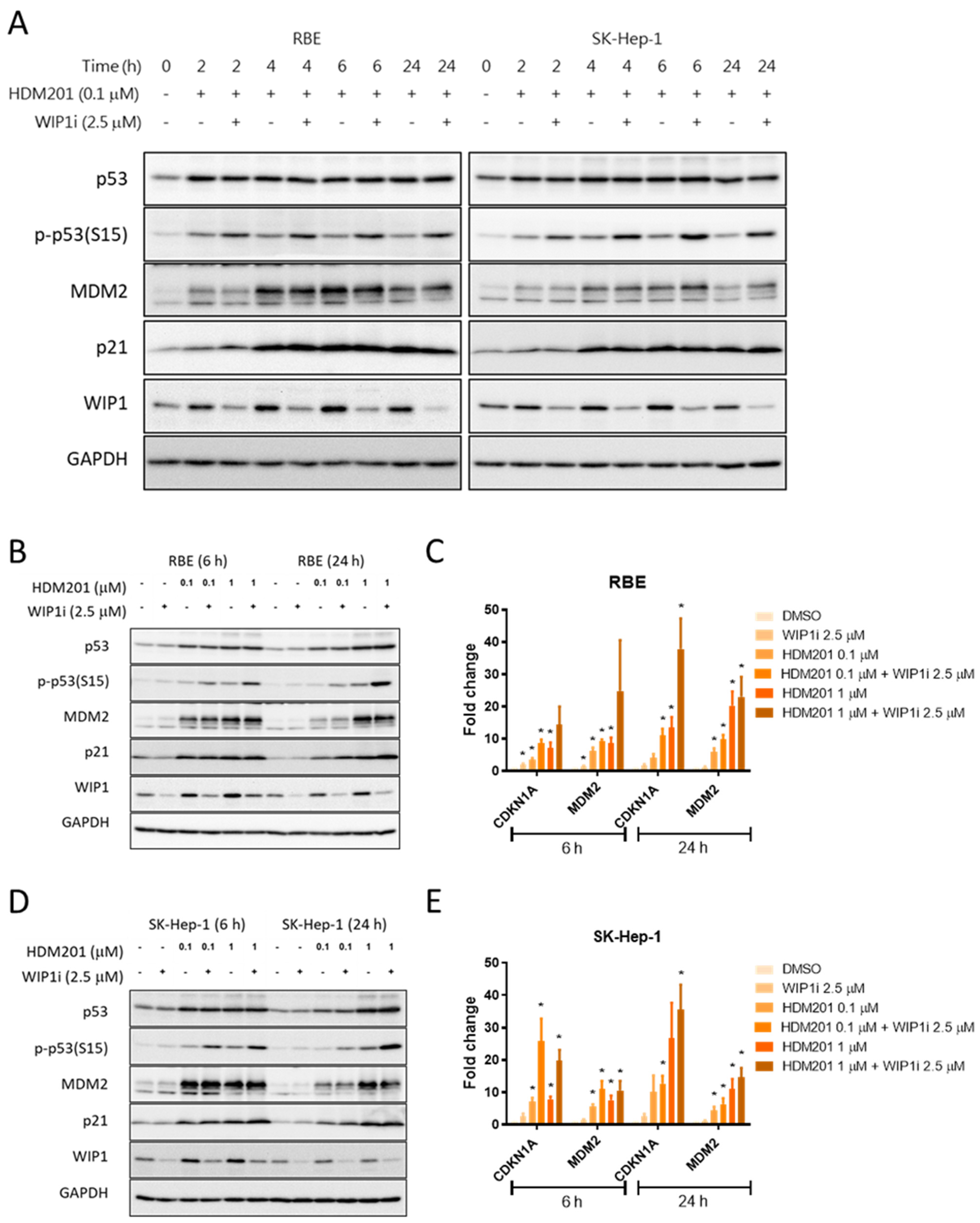
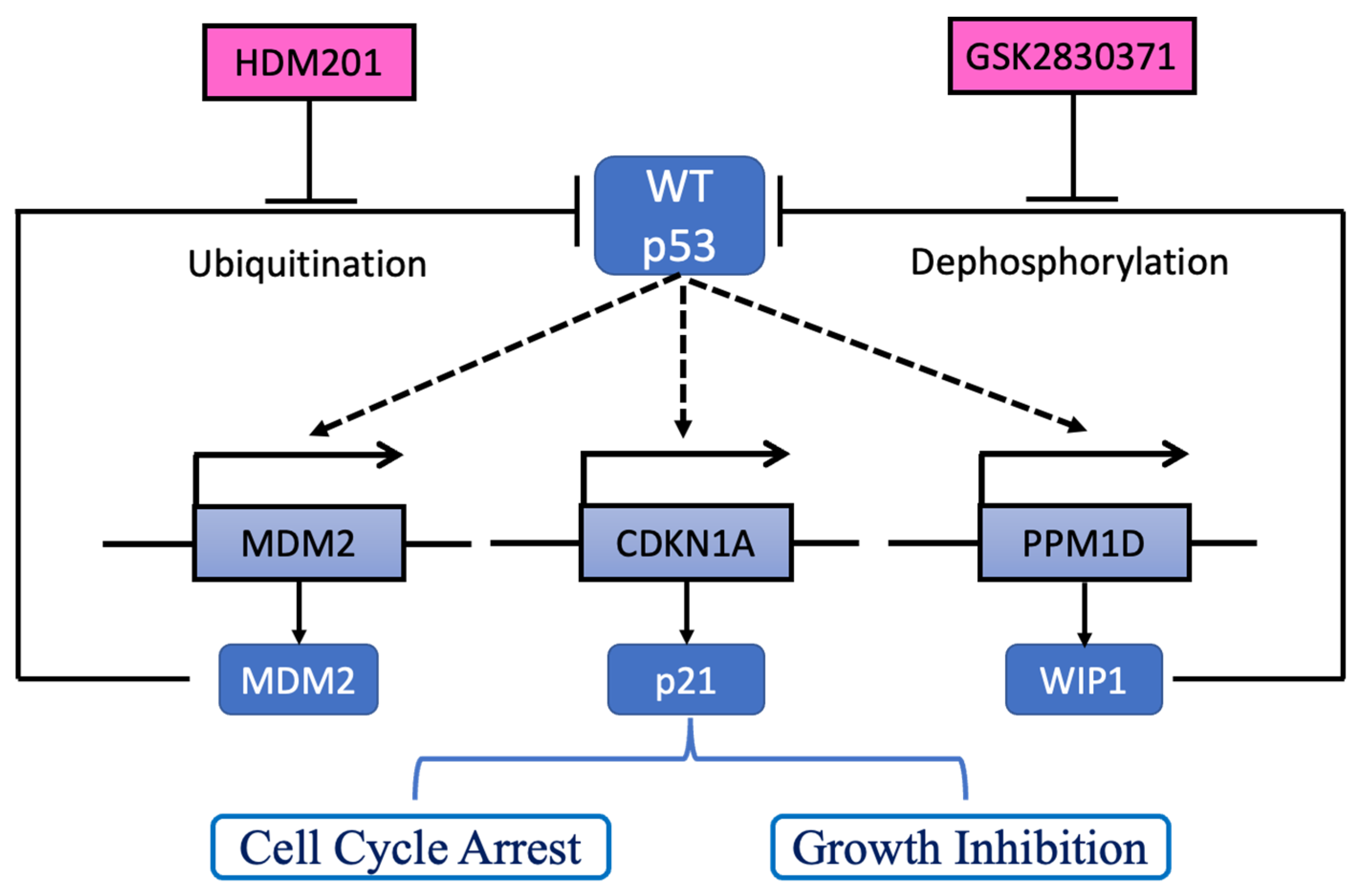
2.4. GSK2830371 Induced p53 Phosphorylation and Stabilization via WIP1 Inhibition
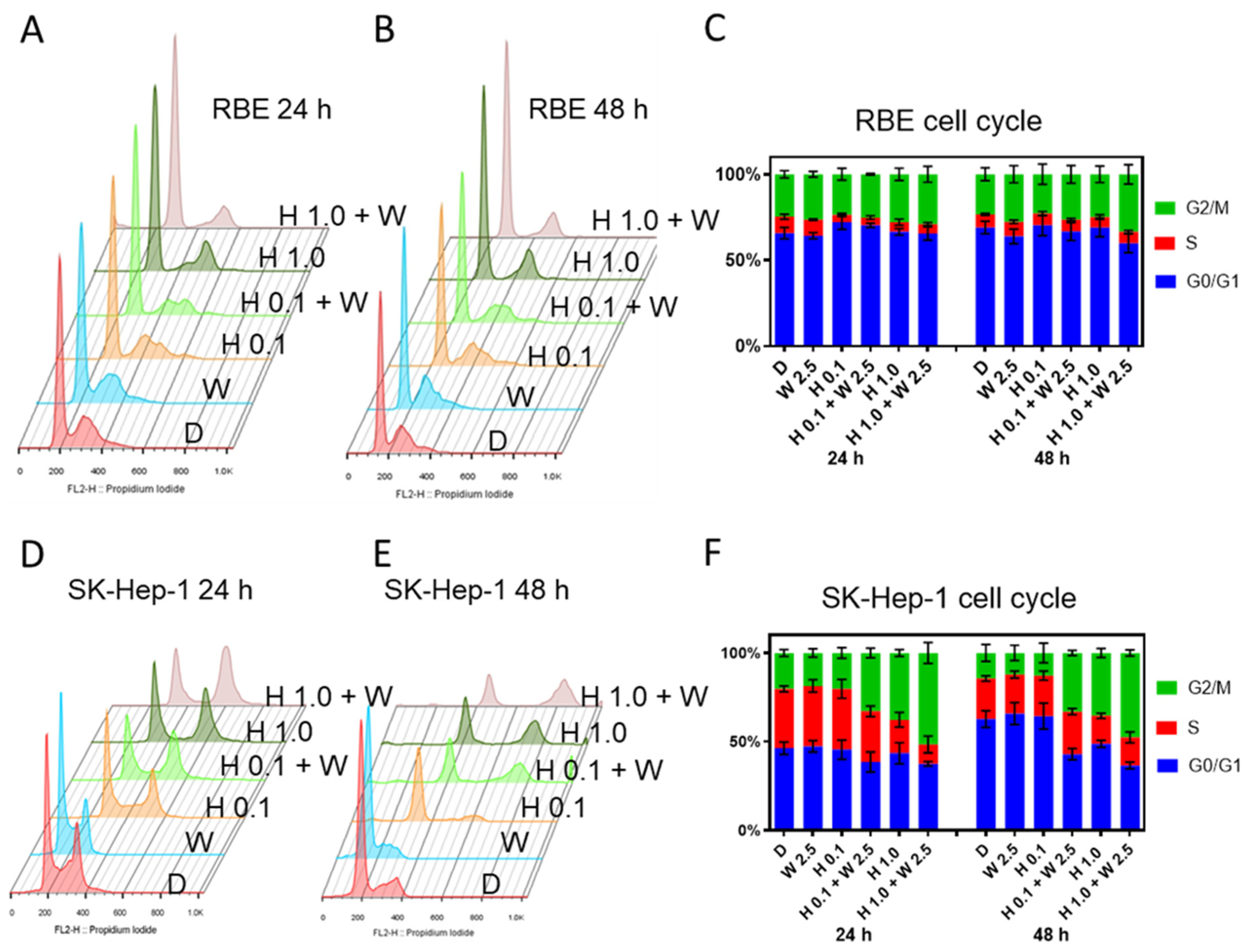
2.5. GSK2830371 Induced Increased G2/M Arrest When Added to HDM201
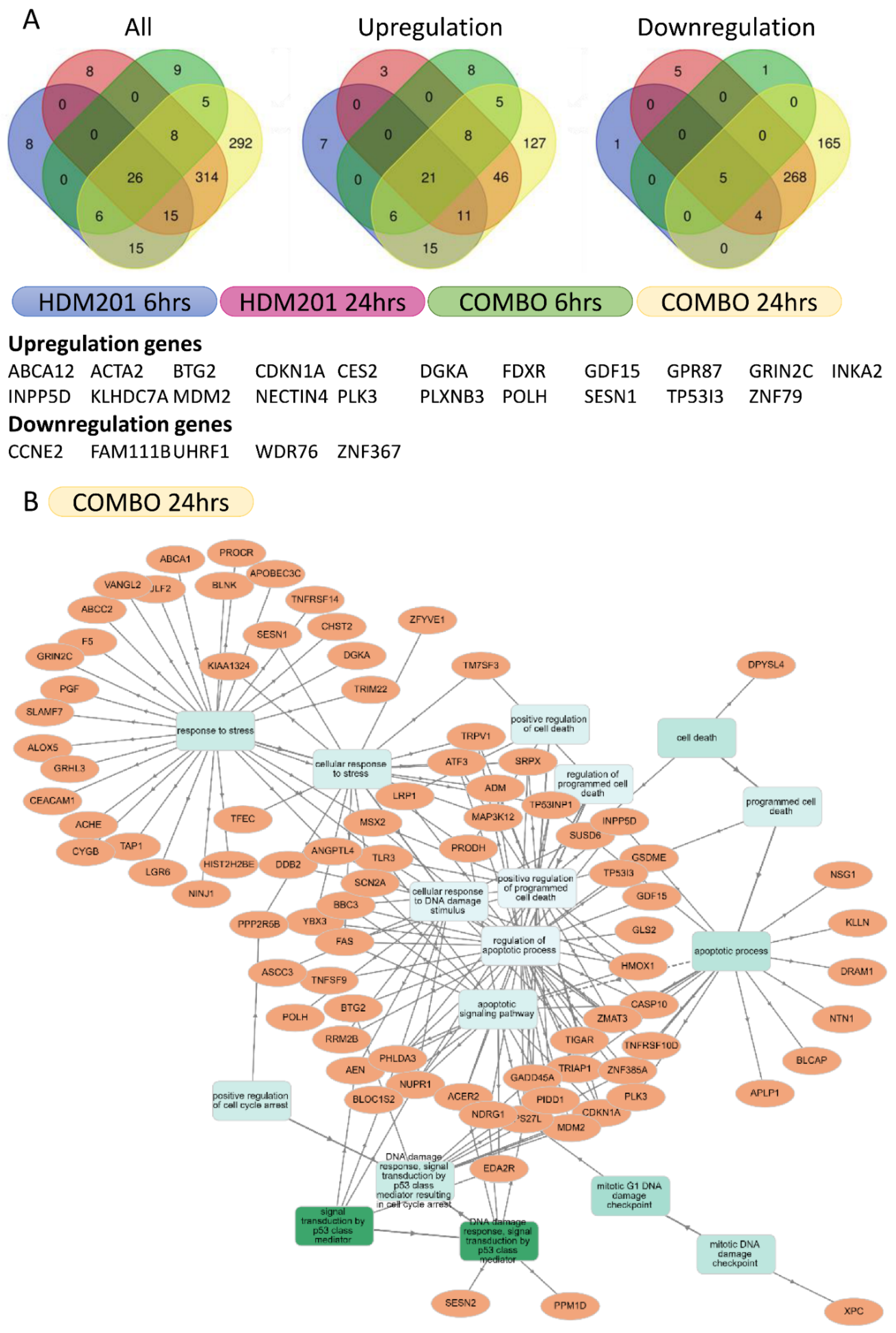
2.6. RNA Sequencing Identified Transcript Changes Resulting from Simultaneous Inhibition of MDM2 and WIP1
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Compounds
4.2. Growth Inhibitory Assay
4.3. Clonogenic Assay
4.4. Immunoblotting
4.5. RNA Extraction and qRT-PCR
4.6. Fluorescence-Activated Cell Sorting (FACS)
4.7. RNA Sequencing
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef]
- Maroni, L.; Pierantonelli, I.; Banales, J.M.; Benedetti, A.; Marzioni, M. The significance of genetics for cholangiocarcinoma development. Ann. Transl. Med. 2013, 1, 28. [Google Scholar]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Seo, J.W.; Kwan, B.S.; Cheon, Y.K.; Lee, T.Y.; Shim, C.S.; Kwon, S.Y.; Choe, W.H.; Yoo, B.C.; Yoon, J.M.; Lee, J.H. Prognostic impact of hepatitis B or C on intrahepatic cholangiocarcinoma. Korean J. Intern. Med. 2020, 35, 566–573. [Google Scholar] [CrossRef]
- Shaib, Y.; El-Serag, H.B. The epidemiology of cholangiocarcinoma. Semin. Liver Dis. 2004, 24, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Deshpande, V.; Zhu, A.X. Genetics of biliary tract cancers and emerging targeted therapies. J. Clin. Oncol. 2010, 28, 3531–3540. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Hezel, A.F. Development of molecularly targeted therapies in biliary tract cancers: Reassessing the challenges and opportunities. Hepatology 2011, 53, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Sun, W. Targeted therapy in biliary tract cancers-current limitations and potentials in the future. J. Gastrointest. Oncol. 2017, 8, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet. Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Mahipal, A.; Tella, S.H.; Kommalapati, A.; Anaya, D.; Kim, R. FGFR2 genomic aberrations: Achilles heel in the management of advanced cholangiocarcinoma. Cancer Treat. Rev. 2019, 78, 1–7. [Google Scholar] [CrossRef]
- Pan, Y.R.; Wu, C.E.; Yeh, C.N. ATM inhibitor suppresses gemcitabine-resistant BTC growth in a polymerase θ deficiency-dependent manner. Biomolecules 2020, 10, 1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-E.; Pan, Y.-R.; Yeh, C.-N.; Lunec, J. Targeting P53 as a future strategy to overcome gemcitabine resistance in biliary tract cancers. Biomolecules 2020, 10, 1474. [Google Scholar] [CrossRef]
- Wu, C.-E.; Yeh, D.-W.; Pan, Y.-R.; Huang, W.-K.; Chen, M.-H.; Chang, J.W.-C.; Chen, J.-S.; Wang, Y.-C.; Yeh, C.-N. Chromosomal instability may not be a predictor for immune checkpoint inhibitors from a comprehensive bioinformatics analysis. Life 2020, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Wang, Y.V.; Wahl, G.M. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010, 20, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.; Chatterjee, M.; Langenberg, M.H.G.; Lin, C.C.; Suárez, C.; Tai, D.; Cassier, P.; Yamamoto, N.; De Weger, V.A.; Jeay, S.; et al. Dose- and regimen-finding phase I study of NVP-HDM201 in patients (pts) with TP53 wild-type (wt) advanced tumors. Eur. J. Cancer 2016, 69, S128–S129. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.E.; Esfandiari, A.; Ho, Y.H.; Wang, N.; Mahdi, A.K.; Aptullahoglu, E.; Lovat, P.; Lunec, J. Targeting negative regulation of p53 by MDM2 and WIP1 as a therapeutic strategy in cutaneous melanoma. Br. J. Cancer 2018, 118, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Esfandiari, A.; Hawthorne, T.A.; Nakjang, S.; Lunec, J. Chemical inhibition of wild-type p53-induced phosphatase 1 (WIP1/PPM1D) by GSK2830371 potentiates the sensitivity to MDM2 inhibitors in a p53-dependent manner. Mol. Cancer Ther. 2016, 15, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedel, B.J.; Singh, D.; Madrigal, A.; Valdovino-Gonzalez, A.G.; White, B.M.; Zapardiel-Gonzalo, J.; Ha, B.; Altay, G.; Greenbaum, J.A.; McVicker, G.; et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 2018, 175, 1701–1715e.16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [Green Version]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016, 7, 14458–14475. [Google Scholar] [CrossRef] [Green Version]
- Sriraman, A.; Radovanovic, M.; Wienken, M.; Najafova, Z.; Li, Y.; Dobbelstein, M. Cooperation of Nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget 2016, 7, 31623–31638. [Google Scholar] [CrossRef] [Green Version]
- Fontana, M.C.; Nanni, J.; Ghelli Luserna di Rorà, A.; Petracci, E.; Padella, A.; Ghetti, M.; Ferrari, A.; Marconi, G.; Soverini, S.; Iacobucci, I.; et al. Pharmacological inhibition of WIP1 sensitizes acute myeloid leukemia cells to the MDM2 inhibitor Nutlin-3a. Biomedicines 2021, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ma, O.; Nguyen, T.-A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L.; et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 2014, 3, e02200. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.; Cogdell, D.; Feng, Y.; Hamilton, S.R.; Zhang, W. Transcriptional activation of the carboxylesterase 2 gene by the p53 pathway. Cancer Biol. Ther. 2006, 5, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Ezra, O.; Rivlin, N.; Molchadsky, A.; Madar, S.; Goldfinger, N.; Rotter, V. p53, a novel regulator of lipid metabolism pathways. J. Hepatol. 2012, 56, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Pourebrahim, R.; Zhang, Y.; Liu, B.; Gao, R.; Xiong, S.; Lin, P.P.; McArthur, M.J.; Ostrowski, M.C.; Lozano, G. Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev. 2017, 31, 1847–1857. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Tanikawa, C.; Ueda, K.; Matsuda, K. INKA2, a novel p53 target that interacts with the serine/threonine kinase PAK4. Int. J. Oncol. 2019, 54, 1907–1920. [Google Scholar] [CrossRef] [Green Version]
- Leszczynska, K.B.; Foskolou, I.P.; Abraham, A.G.; Anbalagan, S.; Tellier, C.; Haider, S.; Span, P.N.; O’Neill, E.E.; Buffa, F.M.; Hammond, E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Investig. 2015, 125, 2385–2398. [Google Scholar] [CrossRef] [Green Version]
- Kerley-Hamilton, J.S.; Pike, A.M.; Li, N.; DiRenzo, J.; Spinella, M.J. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embryonal carcinoma. Oncogene 2005, 24, 6090–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-S.; Schmezer, P.; Popanda, O. Diacylglycerol kinase alpha in radiation-induced fibrosis: Potential as a predictive marker or therapeutic target. Front. Oncol. 2020, 10, 737. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Murata, M.; Shen, C.H.; Furue, M.; Ito, T. NECTIN4: A novel therapeutic target for melanoma. Int. J. Mol. Sci. 2021, 22, 976. [Google Scholar] [CrossRef] [PubMed]
- Fabre-Lafay, S.; Monville, F.; Garrido-Urbani, S.; Berruyer-Pouyet, C.; Ginestier, C.; Reymond, N.; Finetti, P.; Sauvan, R.; Adélaïde, J.; Geneix, J.; et al. Nectin-4 is a new histological and serological tumor associated marker for breast cancer. BMC Cancer 2007, 7, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrba, L.; Junk, D.J.; Novak, P.; Futscher, B.W. p53 induces distinct epigenetic states at its direct target promoters. BMC Genom. 2008, 9, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, J.; Pinto, E.M.; Barnoud, T.; Leung, J.C.; Martynyuk, T.; Kossenkov, A.V.; Philips, A.H.; Desai, H.; Hausler, R.; Kelly, G.; et al. A rare TP53 mutation predominant in Ashkenazi Jews confers risk of multiple cancers. Cancer Res. 2020, 80, 3732–3744. [Google Scholar] [CrossRef]
- Comer, K.A.; Dennis, P.A.; Armstrong, L.; Catino, J.J.; Kastan, M.B.; Kumar, C.C. Human smooth muscle alpha-actin gene is a transcriptional target of the p53 tumor suppressor protein. Oncogene 1998, 16, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Gorjala, P.; Cairncross, J.G.; Gary, R.K. p53-dependent up-regulation of CDKN1A and down-regulation of CCNE2 in response to beryllium. Cell Prolif. 2016, 49, 698–709. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Liu, K.; Huang, J.; Sun, Q.; Shao, C.; Luo, J.; Xu, L.; Shen, Y.; Ren, B. FAM111B, a direct target of p53, promotes the malignant process of lung adenocarcinoma. Onco. Targets Ther. 2019, 12, 2829–2842. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Peng, J.; Mo, R.; Ma, S.; Wang, J.; Zang, L.; Li, W.; Fan, J. Ubiquitin E3 ligase UHRF1 regulates p53 ubiquitination and p53-dependent cell apoptosis in clear cell Renal Cell Carcinoma. Biochem. Biophys. Res. Commun. 2015, 464, 147–153. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Park, J.-C.; Kim, W.-S.; Ro, E.J.; Jeon, S.H.; Lee, S.-K.; Park, Y.N.; Min, D.S.; Choi, K.-Y. WDR76 is a RAS binding protein that functions as a tumor suppressor via RAS degradation. Nat. Commun. 2019, 10, 295. [Google Scholar] [CrossRef] [Green Version]
- Chiang, K.C.; Yeh, T.S.; Wu, R.C.; Pang, J.S.; Cheng, C.T.; Wang, S.Y.; Juang, H.H.; Yeh, C.N. Lipocalin 2 (LCN2) is a promising target for cholangiocarcinoma treatment and bile LCN2 level is a potential cholangiocarcinoma diagnostic marker. Sci. Rep. 2016, 6, 36138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.; Gao, J.-H.; Zhao, C.; Tong, H.; Zheng, S.-P.; Huang, Z.-Y.; Liu, R.; Tang, C.-W.; Li, J. SK-Hep1: Not hepatocellular carcinoma cells but a cell model for liver sinusoidal endothelial cells. Int. J. Clin. Exp. Pathol. 2018, 11, 2931–2938. [Google Scholar] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-E.; Huang, C.-Y.; Chen, C.-P.; Pan, Y.-R.; Chang, J.W.-C.; Chen, J.-S.; Yeh, C.-N.; Lunec, J. WIP1 Inhibition by GSK2830371 Potentiates HDM201 through Enhanced p53 Phosphorylation and Activation in Liver Adenocarcinoma Cells. Cancers 2021, 13, 3876. https://doi.org/10.3390/cancers13153876
Wu C-E, Huang C-Y, Chen C-P, Pan Y-R, Chang JW-C, Chen J-S, Yeh C-N, Lunec J. WIP1 Inhibition by GSK2830371 Potentiates HDM201 through Enhanced p53 Phosphorylation and Activation in Liver Adenocarcinoma Cells. Cancers. 2021; 13(15):3876. https://doi.org/10.3390/cancers13153876
Chicago/Turabian StyleWu, Chiao-En, Chen-Yang Huang, Chiao-Ping Chen, Yi-Ru Pan, John Wen-Cheng Chang, Jen-Shi Chen, Chun-Nan Yeh, and John Lunec. 2021. "WIP1 Inhibition by GSK2830371 Potentiates HDM201 through Enhanced p53 Phosphorylation and Activation in Liver Adenocarcinoma Cells" Cancers 13, no. 15: 3876. https://doi.org/10.3390/cancers13153876