
Classification of Non-Small Cell Lung Cancer’s Tumor Immune Micro-Environment and Strategies to Augment Its Response to Immune Checkpoint Blockade

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. TIME Classification Applicable to NSCLC and Its Correlation with Response to ICB
3. TIME Subtype Classification Based on Analysis of Immunogenomic Data from the Cancer Genome Atlas (TCGA)
4. Potential Biomarkers and Therapeutic Targets in ICI Responsive vs. Nonresponsive TIME with a Focus on NSCLC
5. Therapeutic Strategies to Augment Response to ICI in NSCLC with “Unresponsive” Types of TIMEs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Comprehensive Cancer Network. Non-Small Cell Lung Cancer (Version 4. 2021). Available online: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 10 April 2021).
- Herbst, R.S.; Baas, P.; Kim, D.; Felip, E.; Pérez-Gracia, J.L.; Han, J.; Molina, J.; Kim, J.; Arvis, C.D.; Ahn, M.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomized controlled trial. Lancet 2015, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.K.; Wu, Y.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomized, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for first-line treatment of PD-L1-selected patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: Two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef]
- Herbst, R.S.; Garon, E.B.; Kim, D.; Cho, B.C.; Perez-Gracia, J.L.; Han, J.; Arvis, C.D.; Majem, M.; Forster, M.D.; Monnet, I.; et al. Long-term outcomes and retreatment among patients with previously treated, programmed death-ligand 1-positive, advanced non-small-cell lung cancer in the Keynote-010 study. J. Clin. Oncol. 2020, 38, 1580–1591. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 immunohistochemistry assays for lung cancer: Results from phase 1 of the Blueprint PD-L1 IHC assay comparison project. J. Thor. Oncol. 2016, 12, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenhacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generatoin sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilumumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomized controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Soria, J.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussiotis, V.A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell. Mol. Immunol. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kleinovink, J.W.; Marijt, K.A.; Schoonderwoerd, M.J.A.; van Hall, T.; Ossendorp, F.; Fransen, M.F. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology 2017, 6, e1294299. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for NSCLC patients with EGFR mutations. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [Green Version]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Rosell, R. Systemic treatment in EGFR-ALK NSCLC patients: Second line therapy and beyond. Cancer Biol. Med. 2014, 11, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediates STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [Green Version]
- Song, T.L.; Nairismägi, M.; Laurensia, Y.; Lim, J.; Tan, J.; Li, Z.; Pang, W.; Kizhakeyil, A.; Wijaya, G.; Huang, D.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toki, M.I.; Mani, N.; Smithy, J.W.; Liu, Y.; Phil, M.; Altan, M.; Wasserman, B.; Tuktamyshov, R.; Schalper, K.; Syrigos, K.N.; et al. Immune marker profiling and programmed death ligand 1 expression across NSCLC mutations. J. Thorac. Oncol. 2018, 13, 1884–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: A retrospective analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [Green Version]
- Heigener, D.F.; Reck, M. PD-1 axis inhibition in EGFR positives: A blunt sword? J. Thorac. Oncol. 2017, 12, 171–172. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Disc. 2016, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Borczuk, A.; Janakiram, M.; Ren, X.; Lin, J.; Assal, A.; Halmos, B.; Perez-Soler, R.; Zang, X. Wide expression and significance of alternative immune checkpoint molecules, B7x and HHLA2, in PD-L1-negative human lung cancers. Clin. Cancer Res. 2018, 24, 1954–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The immune landscape of cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arina, A.; Karrison, T.; Galka, E.; Schreiber, K.; Weichselbaum, R.R.; Schreiber, H. Transfer of allogeneic CD4+ T cells rescues CD8+ T cells in anti-PD-L1-resistant tumors leading to tumor eradication. Cancer Immunol. Res. 2017, 5, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Marraco, S.A.F.; Calderon-Copete, S.; Ferreira, D.P.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+ PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019, 50, 195–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, D.; Mazzotta, M.; Scalera, S.; Terrenato, I.; Sperati, F.; D’Ambrosio, L.; Pallocca, M.; Corleone, G.; Krasniqi, E.; Pizzuti, L.; et al. KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann. Oncol. 2020, 31, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin. Cancer. Res. 2017, 23, 5024–5033. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Niemeijer, A.N.; Sahba, S.; Smit, E.F.; Lissenberg-Witte, B.I.; de Langen, A.J.; Thunnissen, E. Association of tumour and stroma PD-1, PD-L1, CD3, CD4, CD8 expression with DCB and OS to nivolumab treatment in NSCLC patients pre-treated with chemotherapy. Br. J. Cancer 2020, 123, 392–402. [Google Scholar] [CrossRef]
- Liu, Y.; Zugazagoitia, J.; Ahmed, F.S.; Henick, B.S.; Gettinger, S.N.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Immune cell PD-L1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin. Cancer Res. 2020, 26, 970–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Gonzalez-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Gimenez-Capitan, A.; Teixido, C.; Molina-Vila, M.A.; Viteri, S.; De Los Llanos Gil, M.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hurkmans, D.P.; Kuipers, M.E.; Smit, J.; van Marion, R.; Mathijssen, R.H.J.; Postmus, P.E.; Hiemstra, P.S.; Aerts, J.G.J.V.; von der Thüsen, J.H.; van der Burg, S.H.; et al. Tumor mutational load, CD8+ T cells, expression of PD-L1 and HLA class I to guide immunotherapy decisions in NSCLC patients. Cancer Immunol. Immunother. 2020, 69, 771–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapara, M.Y.; Sykes, M. Tolerance and cancer: Mechanisms of tumor evasion and strategies for breaking tolerance. J. Clin. Oncol. 2004, 22, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.; Liljeström, P.; Uyttenhove, C.; van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Comm. 2017, 8, 1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol. Res. 2017, 6, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T regulatory cells and priming the suppressive tumor microenvironment. Front. Immunol. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.A.; Kim, E.Y.; Marangoni, F.; Carrizosa, E.; Claudio, N.M.; Mempel, T.R. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Investig. 2014, 124, 2425–2440. [Google Scholar] [CrossRef]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual faces of IFNγ in cancer progression: A role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theelen, W.S.M.E.; Kuilman, T.; Schulze, K.; Zou, W.; Krijgsman, O.; Peters, D.D.G.C.; Cornelissen, S.; Monkhorst, K.; Sarma, P.; Sumiyoshi, T.; et al. Absence of PD-L1 expression on tumor cells in the context of an activated immune infiltrate may indicate impaired IFNγ signaling in non-small cell lung cancer. PLoS ONE 2019, 14, e0216864. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Aspeslagh, S.; Postel-Vinay, S.; Soria, J. JAK mutations as escape mechanisms to anti-PD-1 therapy. Cancer Discov. 2017, 7, 128–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, C.M.; Sen, T.; Gay, C.M.; Ramkumar, K.; Diao, L.; Cardnell, R.J.; Rodriguez, B.L.; Stewart, C.A.; Papadimitrakopoulou, V.A.; Gibson, L.; et al. STING pathway expression identifies NSCLC with an immune-responsive phenotype. J. Thorac. Oncol. 2020, 15, 777–791. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Abosy, R.A.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1−CD8+ tumor-infiltrating T cells. Immunity 2019, 50, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Tian, Q.; Amar, N.; Yu, A.; Rivard, C.J.; Caldwell, C.; Ng, T.L.; Tu, M.; Liu, Y.; Gao, D.; et al. The immune checkpoint HVEM may contribute to immune escape in non-small cell lung cancer lacking PD-L1 expression. Lung Cancer 2018, 125, 115–120. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol. Res. 2015, 3, 1344–1355. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Comm. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yu, H.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Suda, K.; Ren, S.; Wu, C.; Hou, L.; et al. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J. Thorac. Oncol. 2017, 12, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Datar, I.J.; Sanmamed, M.F.; Wang, J.; Henick, B.S.; Choi, J.; Badri, T.; Dong, W.; Mani, N.; Toki, M.; Mejías, L.D.; et al. Expression analysis and significance of PD-1, LAG-3 and TIM-3 in human non-small cell lung cancer using spatially-resolved and multiparametric single-cell analysis. Clin. Cancer Res. 2019, 25, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Carvajal-Hausdorf, D.; McLaughlin, J.; Altan, M.; Velcheti, V.; Gaule, P.; Sanmamed, M.F.; Chen, L.; Herbst, R.S.; Rimm, D.L. Differential expression and significance of PD-L1, IDO-1, and B7-H4 in human lung cancer. Clin. Cancer Res. 2016, 23, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Chang, M.Y.; Parker, K.H.; Beury, D.W.; DuHadaway, J.B.; Flick, H.E.; Boulden, J.; Sutanto-Ward, E.; Soler, A.P.; Laury-Kleintop, L.D.; et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Disc. 2012, 2, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Guo, Z.S.; Gregory, A.D.; Shapiro, S.D.; Xiao, G.; Qu, Z. Dual but not single PD-1 or TIM-3 blockade enhances oncolytic virotherapy in refractory lung cancer. J. Immunother. Cancer 2020, 8, e000294. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Zhu, X.; Zhang, H.; Yang, G.H.Y.; Friesen, T.J.; Shipley, M.; Maeda, D.Y.; Zebala, J.A.; McKay-Fleisch, J.; Meredith, G.; et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. J. Cancer Investig. Insight 2019, 4, e130850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, J.L.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signaling prevents anti-tumor immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Fu, C.; Liang, X.; Cui, W.; Ober-Blöbaum, J.L.; Vazzana, J.; Shrikant, P.A.; Lee, K.P.; Clausen, B.E.; Mellman, I.; Jiang, A. β-Catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc. Natl. Acad. Sci. USA 2015, 112, 2823–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, S.; Shui, X.; Craig, K.P.; Park, J.; Wang, W.; Brown, B.D.; Abrams, M.T. RNAi-mediated β-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol. Ther. 2018, 26, 2567–2579. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold tumors: A therapeutic challenge for immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on tumor neoantigens and their utility: Why it is good to be different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, A.M.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Theresa, Z.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Reuben, A.; Zhang, J.; Chiou, S.; Gittelman, R.M.; Li, J.; Lee, W.; Fujimoto, J.; Behrens, C.; Liu, X.; Wang, F.; et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Comm. 2020, 11, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Muto, S.; Ozaki, Y.; Yamaguchi, H.; Mine, H.; Takagi, H.; Watanabe, M.; Inoue, T.; Yamaura, T.; Fukuhara, M.; Okabe, N.; et al. Tumor β-catenin expression is associated with immune evasion in non-small cell lung cancer with high tumor mutation burden. Oncol. Lett. 2021, 21, 203. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Gimenez-Xavier, P.; Pros, E.; Pajares, M.J.; Moro, M.; Gomez, A.; Navarro, A.; Condom, E.; Moran, S.; Gomez-Lopez, G.; et al. Genomic profiling of patient-derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin. Cancer Res. 2016, 23, 3203–3213. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, F.; Bernal, M.; Sanchez-Palencia, A.; Carretero, J.; Torres, C.; Bayarri, C.; Gomez-Morales, M.; Garrido, F.; Ruiz-Cabello, F. The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. Int. J. Cancer 2017, 140, 888–899. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019, 36, 385–401. [Google Scholar] [CrossRef] [Green Version]
- Lai, Q.; Wang, H.; Li, A.; Xu, Y.; Tang, L.; Chen, Q.; Zhang, C.; Gao, Y.; Song, J.; Du, Z. Decitibine improve the efficiency of anti-PD-1 therapy via activating the response to IFN/PD-L1 signal of lung cancer cells. Oncogene 2018, 37, 2302–2312. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Tessema, M.; Klinge, D.M.; Yingling, C.M.; Do, K.; van Neste, L.; Belinsky, S.A. Re-expression of CXCL14, a common target for epigenetic silencing in lung cancer, induces tumor necrosis. Oncogene 2010, 29, 5159–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of Th1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Chung, L.; Tang, S.; Sun, G.; Chou, T.; Yeh, T.; Yu, S.; Sun, K. Galectin-1 promotes lung cancer progression and chemoresistance by upregulating p38 MAPK, ERK, and cyclooxygenase-2. Clin. Cancer Res. 2012, 18, 4037–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; von Eyben, R.; et al. Galectin-1-driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Investig. 2019, 129, 5553–5567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowanetz, M.; Zou, W.; Gettinger, S.N.; Koeppen, H.; Kockx, M.; Schmid, P.; Kadel, E.E., III; Wistuba, I.; Chaft, J.; Rizvi, N.A.; et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc. Natl. Acad. Sci. USA 2018, 115, E10119–E10126. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2015, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; König, K.; Michels, S.; Fischer, R.; et al. Clinical and pathological characteristics of KEAP1- and NFE2L2-mutated non-small cell lung carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Nadal, E.; Palmero, R.; Muñoz-Pinedo, C. Mutations in the antioxidant KEAP1/NRF2 pathway define an aggressive subset of NSCLC resistant to conventional treatments. J. Thorac. Oncol. 2019, 14, 1881–1883. [Google Scholar] [CrossRef] [PubMed]
- Truica, M.I.; Burns, M.C.; Han, H.; Abdulkadir, S.A. Turning up the heat on Myc: Progress in small-molecule inhibitors. Cancer Res. 2021, 81. in press. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.F.; De Marchi, F.; Lokhandwala, P.M.; Belchis, D.; Xian, R.; Gocke, C.D.; Eshleman, J.R.; Illei, P.; Li, M. IDH1 and IDH2 mutations in lung adenocarcinomas: Evidences of subclonal evolution. Cancer Med. 2020, 9, 4386–4394. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Ricca, J.; van Oudenhove, E.; Olvera, N.; Merghoub, T.; Levine, D.A.; Zamarin, D. Immune-active microenvironment in small cell carcinoma of the ovary, hypercalcemic type: Rationale for immune checkpoint blockade. J. Natl. Cancer Inst. 2018, 110, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Bandlamudi, C.; Lavery, J.A.; Montecalvo, J.; Namakydoust, A.; Rizvi, H.; Egger, J.; Concepcion, C.P.; Paul, S.; Arcila, M.E.; et al. The genomic landscape of SMARCA4 alterations and associations with outcomes in patients with lung cancer. Clin. Cancer. Res. 2020, 26, 5701–5708. [Google Scholar] [CrossRef]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Jeanson, A.; Tomasini, P.; Souquet-Bressand, M.; Brandone, N.; Boucekine, M.; Grangeon, M.; Chaleat, S.; Khobta, N.; Milia, J.; Mhanna, L.; et al. Efficacy of immune checkpoint inhibitors in KRAS-mutant non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 2019, 14, 1095–1101. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Suda, K.; Wiens, J.; Bunn, P.A., Jr. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 2016, 388, 1012–1024. [Google Scholar] [CrossRef]
- Swanton, C.; Govindan, R. Clinical implications of genomic discoveries in lung cancer. N. Engl. J. Med. 2016, 374, 1864–1873. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Guan, X.; Jiang, P. Cytokine and chemokine signals of T-cell exclusion in tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Frezzetti, D.; Gallo, M.; Maiello, M.R.; D’Alessio, A.; Esposito, C.; Chicchinelli, N.; Normanno, N.; De Luca, A. VEGF as a potential target in lung cancer. Expert Opin. Ther. Targets 2017, 21, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Toonkel, R.L.; Borczuk, A.C.; Powell, C.A. TGF-β signaling pathway in lung adenocarcinoma invasion. J. Thorac. Oncol. 2010, 5, 153–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NK-κB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Lawrence, D.; McDermott, D.; Zhou, J.; Rodig, S.; Hodi, F.S. VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol. Res. 2016, 4, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Transforming growth factor beta (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstructed colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF-β attenuates tumor response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Seed, R.I.; Cormier, A.; Bondesson, A.J.; Lou, J.; Elattma, A.; Ito, S.; Yanagisawa, H.; Hashimoto, M.; Ma, R.; et al. Integrin αvβ8-expressing tumor cells evade host immunity by regulating TGF-β activation in immune cells. JCI Insight 2018, 3, e122591. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef]
- Demaria, S.; Coleman, C.N.; Formenti, S.C. Radiotherapy: Changing the game in immunotherapy. Trends Cancer 2016, 2, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Durante, M.; Brenner, D.J.; Formenti, S.C. Does heavy ion therapy work through the immune system? Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 934–936. [Google Scholar] [CrossRef]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ruiz, M.E.; Vanpouille-Box, C.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol. 2018, 39, 644–655. [Google Scholar] [CrossRef]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019, 29, 1236–1248. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; Tariveranmoshabad, M.; Hakimi, K.; Kremer, S.; Campbell, K.M.; Funes, J.M.; Vega-Crespo, A.; Parisi, G.; Champekar, A.; Nguyen, C.; et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci. Transl. Med. 2020, 12, eabb0152. [Google Scholar] [CrossRef]
- Yamamoto, T.N.; Lee, P.; Vodnala, S.K.; Gurusamy, D.; Kishton, R.J.; Yu, Z.; Eidizadeh, A.; Eil, R.; Fioravanti, J.; Gattinoni, L.; et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Investig. 2019, 129, 1551–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burova, E.; Hermann, A.; Dai, J.; Ullman, E.; Halasz, G.; Potocky, T.; Hong, S.; Liu, M.; Allbritton, O.; Woodruff, A.; et al. Preclinical development of the anti-LAG-3 antibody REGN3767: Characterization and activity in combination with the anti-PD-1 antibody cemiplimab in human PD-1xLAG-3-knockin mice. Mol. Cancer Ther. 2019, 18, 2051–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preillon, J.; Cuende, J.; Rabolli, V.; Garnero, L.; Mercier, M.; Wald, N.; Pappalardo, A.; Denies, S.; Jamart, D.; Michaux, A.; et al. Restoration of T-cell effector function, depletion of Tregs, and direct killing of tumor cells: The multiple mechanisms of action of a-TIGIT antagonist antibodies. Mol. Cancer Ther. 2021, 20, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, H.; Chien, C.; Lai, Y.; Sun, W.; Chen, C.; Cheng, W. BTLA blockade enhances cancer therapy by inhibiting IL-6/IL-10-induced CD19high B lymphocytes. J. Immunother. Cancer 2019, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Pfannenstiel, L.W.; Diaz-Montero, C.M.; Tian, Y.F.; Scharpf, J.; Ko, J.S.; Gastman, B.R. Immune-checkpoint blockade opposes CD8+ T-cell suppression in human and murine cancer. Cancer Immunol. Res. 2019, 7, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.; Kim, S.; St. Jean, N.; O’Brien, S.; Lian, L.; Sun, J.; Verona, R.I.; Moon, E. Addition of anti-TIM3 or anti-TIGIT antibodies to anti-PD1 blockade augments human T cell adoptive cell transfer. Oncoimmunology 2021, 10, 1873607. [Google Scholar] [CrossRef]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 9, 160. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, D.; Elez, E.; Tabernero, J.; Seoane, J. Clinical development of therapies targeting TGFβ: Current knowledge and future perspectives. Ann. Oncol. 2020, 31, 1336–1349. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A novel bifunctional anti-PD-L1/TGF-β trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. OncoImmunology 2017, 6, e1349589. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [Green Version]
- Onda, M.; Kobayashi, K.; Pastan, I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 4575–4582. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Clinton, J.; Wang, Q.; Chang, S.H. Targeting ST2 expressing activated regulatory T cells in Kras-mutant lung cancer. Oncoimmunology 2020, 9, e1682380. [Google Scholar] [CrossRef] [Green Version]
- Ji, D.; Song, C.; Li, Y.; Xia, J.; Wu, Y.; Jia, J.; Cui, X.; Yu, S.; Gu, J. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J. Immunother. Cancer. 2020, 8, e000826. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Fukumoto, T.; Zhang, R.; Gabrilovich, D. Selective targeting of different populations of myeloid-derived suppressor cells by histone deacetylase inhibitors. Cancer Immunol. Immunother. 2020, 69, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Fultang, L.; Panetti, S.; Ng, M.; Collins, P.; Graef, S.; Rizkalla, N.; Booth, S.; Lenton, R.; Noyvert, B.; Shannon-Lowe, C.; et al. MDSC targeting with Gemtuzumab ozogammicin restores T cell immunity and immunotherapy against cancer. EBioMedicine 2019, 47, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, A.; Cammarata, I.; Martire, C.; Focaccetti, C.; Piconese, S.; Buccilli, M.; Mancone, C.; Buzzacchino, F.; Berrios, J.R.G.; D’Alessandris, N.; et al. Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun. Biol. 2020, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional upregulation of NLRC5 by radiation drives STING- and interferon-independent MHC-I expression on cancer cells and T cell cytotoxicity. Sci. Rep. 2020, 10, 7376. [Google Scholar] [CrossRef]
- Herter-Sprie, G.S.; Koyama, S.; Korideck, H.; Hai, J.; Deng, J.; Li, Y.Y.; Buczkowski, K.A.; Grant, A.K.; Ullas, S.; Rhee, K.; et al. Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 2016, 1, e87415. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.L.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated radiation therapy stimulates anti-tumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD1 blockade. Clin. Cancer Res. 2017, 23, 5514–5526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formenti, S.C.; Rudqvist, N.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; de Andrade, L.F.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/ TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 2020, 8, e000433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wei, J.; Dong, J.; Lin, Q.; Tang, H.; Jia, Y.; Tan, W.; Chen, Q.; Zeng, T.; Xing, S.; et al. Laminin γ2-mediating T cell exclusion attenuates response to anti-PD-1 therapy. Sci. Adv. 2021, 7, eabc8346. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, X.; Negrao, M.V.; Reuben, A.; Federico, L.; Diao, L.; McGrail, D.; Nilsson, M.; Robichaux, J.; Munoz, I.G.; Patel, S.; et al. Characterization of the immune landscape of EGFR-mutant NSCLC identifies CD73/adenosine pathway as a potential therapeutic target. J. Thorac. Oncol. 2021, 16, 583–600. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Offin, M.; Guo, R.; Wu, S.L.; Sabari, J.; Land, J.D.; Ni, A.; Montecalvo, J.; Halpenny, D.F.; Buie, L.W.; Pak, T.; et al. Immunophenotype and response to immunotherapy of RET-rearranged lung cancers. JCO Precis. Oncol. 2019, 3, PO.18.00386. [Google Scholar] [CrossRef] [PubMed]
- Guisier, F.; Dubos-Arvis, C.; Viñas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J. Thorac. Oncol. 2020, 15, 628–636. [Google Scholar] [CrossRef]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J.M.; Amann, J.M.; Park, K.; Arasada, R.R.; Li, H.; Shyr, Y.; Carbone, D.P. LKB1 loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. J. Thorac. Oncol. 2014, 9, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Olagnier, D.; Brandtoft, A.M.; Gunderstofte, C.; Villadsen, N.L.; Krapp, C.; Thielke, A.L.; Laustsen, A.; Peri, S.; Hansen, A.L.; Bonefeld, L.; et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Comm. 2018, 9, 3506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goeman, F.; De Nicola, F.; Scalera, S.; Sperati, F.; Gallo, E.; Ciuffreda, L.; Pallocca, M.; Pizzuti, L.; Krasniqi, E.; Barchiesi, G.; et al. Mutations in the KEAP1-NFE2L2 pathway define a molecular subset of rapidly progressing lung adenocarcinoma. J. Thorac. Oncol. 2019, 14, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Binkley, M.S.; Jeon, Y.; Nesselbush, M.; Moding, E.J.; Nabet, B.Y.; Almanza, D.; Kunder, C.; Stehr, H.; Yoo, C.H.; Rhee, S.; et al. KEAP1/NFE2L2 mutations predict lung cancer radiation resistance that can be targeted by glutaminase inhibition. Cancer Discov. 2020, 10, 1826–1841. [Google Scholar] [CrossRef] [PubMed]
- Sitthideatphaiboon, P.; Galan-Cobo, A.; Negrao, M.V.; Qu, X.; Poteete, A.; Zhang, F.; Liu, D.D.; Lewis, W.E.; Kemp, H.N.; Lewis, J.; et al. STK11/LKB1 mutations in NSCLC are associated with KEAP1/NRF2-dependent radiotherapy resistance targetable by glutaminase inhibition. Clin. Cancer Res. 2021, 27, 1720–1733. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; June, B.J.; Lee, S.H.; Yoo, H.S.; Shin, E.A.; Ko, H.J.; Chang, S.; Kim, S.Y.; Jeon, S.M. A clinical drug library screen identifies clobetasol propionate as an NRF2 inhibitor with potential therapeutic efficacy in KEAP1 mutant lung cancer. Oncogene 2017, 36, 5285–5295. [Google Scholar] [CrossRef]
- Stakheev, D.; Taborska, P.; Strizova, Z.; Podrazil, M.; Bartunkova, J.; Smrz, D. The Wnt/β-catenin signaling inhibitor XAV939 enhances the elimination of LNCaP and PC-3 prostate cancer cells by prostate cancer patient lymphocytes in vitro. Sci. Rep. 2019, 9, 4761. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef]
- Chaft, J.E.; Arcila, M.E.; Paik, P.K.; Lau, C.; Riely, G.J.; Pietanza, M.C.; Zakowski, M.F.; Rusch, V.; Sima, C.S.; Ladanyi, M.; et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol. Cancer Ther. 2011, 11, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.Y.; Zhang, J.T.; Liu, S.Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.Y.; Xu, C.R.; Yan, L.X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology 2017, 6, e1356145. [Google Scholar] [CrossRef] [Green Version]
- Lisberg, A.; Cummings, A.; Goldman, J.W.; Bornazyan, K.; Reese, N.; Wang, T.; Coluzzi, P.; Ledezma, B.; Mendenhall, M.; Hunt, J.; et al. A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, tyrosine kinase inhibitor naïve patients with advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Hastings, K.; Yu, H.A.; Wei, W.; Sanchez-Vega, F.; DeVeaux, M.; Choi, J.; Rizvi, H.; Lisberg, A.; Truini, A.; Lydon, C.A.; et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Pyo, K.; Lim, S.M.; Park, C.; Jo, H.; Kim, J.H.; Yun, M.; Kim, D.; Xin, C.; Lee, W.; Gheorghiu, B.; et al. Comprehensive analyses of immunodynamics and immunoreactivity in response to treatment in ALK-positive non-small-cell lung cancer. J. Immunother. Cancer 2020, 8, e000970. [Google Scholar] [CrossRef]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 2017, 28, 1532–1539. [Google Scholar] [CrossRef]
- Gettinger, S.; Hellmann, M.D.; Chow, L.Q.M.; Borghaei, H.; Antonia, S.; Brahmer, J.R.; Goldman, J.W.; Gerber, D.E.; Juergens, R.A.; Shepherd, F.A.; et al. Nivolumab plus erlotinib in patients with EGFR-mutant advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, A.J.; Arbour, K.C.; Rizvi, H.; Iqbal, A.N.; Gadgeel, S.M.; Girshman, J.; Kris, M.G.; Riely, G.J.; Yu, H.A.; Hellmann, M.D.; et al. Severe immune-related adverse events are common with sequential PD-(L)1 blockade and Osimertinib. Ann. Oncol. 2019, 30, 839–844. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.; Yu, H.; Kim, S.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Spigel, D.R.; Reynolds, C.; Waterhouse, D.; Garon, E.B.; Chandler, J.; Babu, S.; Thurmes, P.; Spira, A.; Jotte, R.; Zhu, J.; et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation-positive advanced non-small cell lung cancer (CheckMate 370). J. Thorac. Oncol. 2018, 13, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (Impower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomized, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]


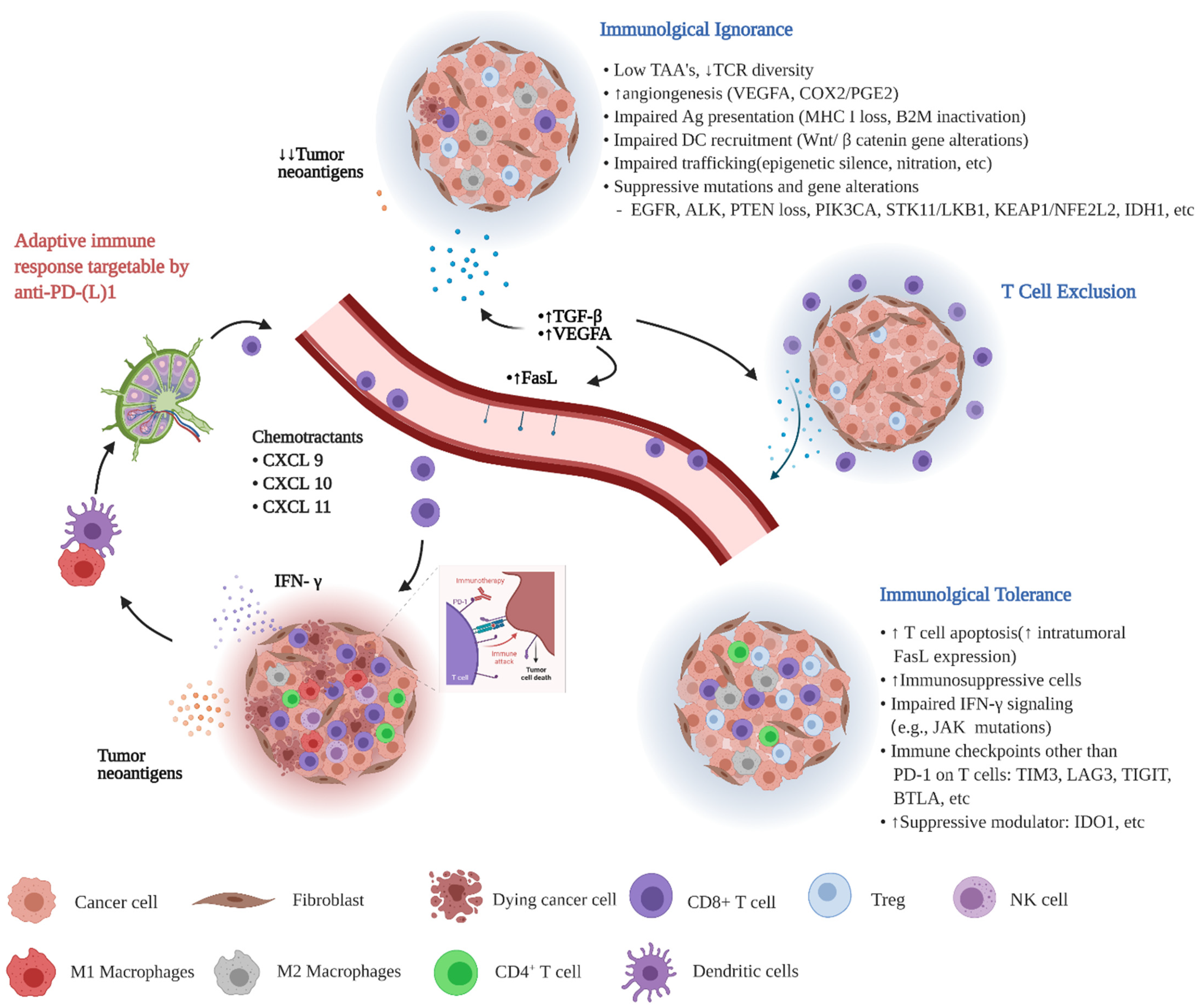{kind=link}
| References | Method | Criteria | TIME Classification | Major Features | Additional Features |
|---|---|---|---|---|---|
| Herbst et al. [19] | IHC | PD-L1 expression | Responsive | Before Rx | Before Rx |
| (TC and IC) | Increased PD-L1 expression | Increased expression of another checkpoint (NSCLC): | |||
| CD8+ T cell infiltration | (TC, IC) | B7-H3, CTLA-4, TIM3, LAG3, IDO1, PD-L2 | |||
| Decreased CX3CL1; increased CTLA-4 | |||||
| Increased IFN-γ and IFN-γ-inducible genes (e.g., IDO1 and CXCL9) | |||||
| After Rx | After Rx | ||||
| Increased PD-L1 expression | Increased tumor IFN-γ expression | ||||
| (TC, IC) | Gene expression pattern of immune activation: | ||||
| CD8 and Th1 T cell activation | granzyme-A, B; Perforin, EOMES, IFN-γ, TNF | ||||
| CXCL10, CD8A, CTLA 4 | |||||
| Non-Responsive | Pre-Rx and After Rx | After Rx | |||
| Immunological ignorance | Little or no TILs | No overexpression of genes associated with immune activation | |||
| Non-functional immune response | TIL without PD-L1 expression | No overexpression of genes associated with immune activation | |||
| (with pre-treatment CD 8 T cell infiltrate) | |||||
| Excluded infiltrate | Immune infiltrate at tumor margin | Same as the two types above, except with increased CTLA-4 expression | |||
| Proliferation and PD-L1 expression in immune cells at tumor margin | |||||
| Teng et al. [24] | IHC | PD-L1 expression (TC) | Type I (adaptive immune resistance) | PD-L1 (+), TIL (+) | Immunogenic mutations associated with |
| TILs | increased TILs of higher PD-1, CTLA-4 expression | ||||
| Type II (immune ignorance) | PD-L1 (-), TIL (-) | No pre-existing T cell infiltration | |||
| Type III (intrinsic induction) | PD-L1 (+), TIL (-) | More common in oncogenic mutation-driven NSCLC | |||
| LUAD: PD-L1 expression-associated EGFR mutations | |||||
| Type IV (tolerance) | PD-L1 (-), TIL (+) | Increased myeloid cells | |||
| Activation of other immune checkpoints and suppressive pathways |
| TIME Subtypes | Wound Healing ǂ | IFN-γ Dominant | Inflammatory | Lymphocyte Depleted | Immunologically Quiet | TGF-β Dominant |
|---|---|---|---|---|---|---|
| Leukocyte fraction * | Intermed. | High | Intermed. | Low | Low | Highest |
| Lymphocyte fraction (25–55%) | High | Highest | High | Intermed. low | Lowest | Intermed. |
| TIL (H and E) | High | Highest | Intermed. low | Low | Lowest | Intermed. |
| Immune cell composition | ||||||
| T cells | ||||||
| CD8 T cells (<15%) | Intermed. high | Highest | High | Intermed. low | Lowest | Intermed. |
| CD4 T cells (<35%) | ||||||
| Th1 | Lowest | Elevated | Elevated | Elevated | ||
| Th2 | Highest | Highest | Lowest | Intermed. | Low | Intermed. high |
| Tfh (<10%) | High | Highest | Intermed. | Low | Lowest | Intermed. low |
| Tregs (<5%) | High | Highest | Intermed. high | Low | Lowest | High |
| Macrophages (38–60%) | Elevated | Most elevated | Elevated | |||
| M0 (<15%) | Highest | High | Intermed. low | Intermed. | Lowest | High |
| M1 (<10%) | Intermed. | Highest | Intermed. | Intermed. low | Lowest | Intermed. |
| M2 (>20%) | Intermed. low | Lowest | Intermed. | High | Highest | High |
| Tumor proliferation rate | Highest | Highest | Low | High | Lowest | High |
| Survival | ||||||
| OS | Intermediate | Intermediate | Best | Worst | Worse | Worst |
| PFI | Intermediate | Intermediate | Best | Worst | Worse | Worst |
| NSCLC subtype | Predom. in LUSC; third common in LUAD ** | Second most common in LUAD and LUSC | Predom. In LUAD *** | LUSC ** | ||
| Factors of immunogenecity | ||||||
| DNA damage | ||||||
| Tumor neoantigen load | ||||||
| SNVs | Highest | Second highest | Lowest | |||
| Indels | Highest | Second highest | Lowest | |||
| ITH | Elevated | Elevated | Lowest | |||
| Enriched oncogenic driver mutations | APC, JAK1, PIK3CA, FGFR3 | PIK3CA, FGFR3 | CDH1, PIK3CA, FGFR3 | EGFR | ||
| TCR diversity | Intermediate | Highest | Intermediate | Low | Lowest | Highest |
| Immunomodulators | ||||||
| Expression | ||||||
| CXCL10 | Highest | Lowest | Second Highest | |||
| EDNRB | Low | Lowest | Highest | |||
| BTLA | High | High | ||||
| Networks modulating the immune response | ||||||
| Predominant immune cells | CD8 T cells | CD8 T cells, CD4 T cells | CD4 T cells | CD4 T cells | ||
| Intracellular regulatory networks | ||||||
| TGF-β (somatic mut+) | ↓Leuk Fract. | ↑Leuk Fract. | ↓Leuk Fract. | |||
| ↑r DC, M0, M1, M2, r NK, plasma cells | ↑E, a Mast, M0/2, a DC, r NK, TγΔ | ↑M1, M2, N, CD4, Treg | ↑M0,M1, a DC | ↑M0, Treg, mr CD4 | ↑r DC | |
| ↓a NK, Treg, Tfh, CD8 | ↓CD8, Treg, Tfh, a NK | ↓DC, M0, Tfh, m B cells, plamsa cells | ↓monocytes | ↓n CD4, CD8 | ||
| Extracellular comm. networks | ||||||
| IFN-γ (+) | IFN-γ (+) | |||||
| TGF-β (+) | TGF-β, TGF-βR(+) | TGF-β, TGF-βR(+) | ||||
| T cell and macrophage-related signaling | CD80-CTLA4 | LAG-3, CD27/28 | CD27, PD-1 | TLR4, VEGFB | TLR4 | TLR4 |
| CD70-CD27 | TIGIT, ICOS, CTLA, PD-1 | CCR4, 5; CXCR3 DARC | EDN3-EDNRB, CX3CL1-CX3CR1 | ITGB2 | ||
| IL1A/1B-IL1R2 | CXCR3, CCR1,4,5 | CD276 | ||||
| CXCL9-CXCR3 | BTLA |
| TIME Subtype | Neoantigen-Related Driver Mutations | Enrichment |
|---|---|---|
| Wound healing | KRAS, KRAS G12, PIKC3A, TP53 | APC (OM), JAK1 (OM), TP53 *, FAT1, PPP2R1A, BRCA1, RB1, PIK3CA (OM), PTPRD, SPTA1, CTNNB1 *, FGFR3 * (OM), SMARCA4, KRAS G12, DACH1, PTEN *, SMARCA1, JAK1, KRAS *, MSH3 |
| IFN-γ-dominant | PIKC3A, TP53 | CASP8, HLA-A, HLA-B, ZNF750, TP53 *, MLH1, NF1 *, FAT1, PPP2R1A, BRCA1, RB1 *, PIK3CA(OM), PTPRD, SPTA1, DACH1 |
| Inflammatory | BRAF | BRAF, CDH1 (OM), PBRM1 * |
| Lymphocyte-depleted | IDH1 | EGFR (OM), CTNNB1 * |
| Immunologically quiet | TP53, IDH1 | IDH1 R132H, ATRX, CIC *, TP53 * |
| TGF-β-dominant | KRAS G12 | KRAS G12 |
| TIME Classification | Characteristics/Alterations | Proposed Treatment |
|---|---|---|
| Tolerance | JAK mutations | ICI + RT, chemotherapy, STING agonists, dsDNA sensing nano-therapy, and/or adoptive T cell transfer |
| ↑Tregs | ICI + Treg suppressors (e.g., anti-CD25, anti-ST2) ± RT | |
| ↑MDSCs | ICI + MDSC suppressors (targeted therapies, HDAC inhibitors, CXCR1/2 inhibitors, etc.) | |
| ↑FasL (MDSC) | ICI + adoptive T cell therapy or antibodies to FasL | |
| ↑VEGF | ICI + VEGF inhibitors | |
| ↑TGF-β | ICI + TGF-β inhibitors, fused anti-PD-L1/TGF-β trap | |
| ↑IDO1 | ICI + IDO1 inhibitors | |
| Terminal T cell exhaustion through other immune checkpoints | ICI of multiple/or alternative immune checkpoints, such as LAG-3, TIM-3, TIGIT, and BTLA ± RT ± chemotherapy | |
| Immunological Ignorance/Exclusion | ||
| Lack of TAAs/immuno-editing | ICI + RT and/or chemotherapy | |
| STK11/LKB1, KEAP1 mutations | ICI + glutaminase inhibitors and/or NRF2 inhibitors ± RT, chemotherapy, or other STING activators | |
| Wnt/β-catenin mutations | Wnt/β-catenin inhibitors | |
| PTEN loss/PIK3CA mutations | PIK3CA inhibitors | |
| EGFR mutation (exon 21) | EGFR TKI with ICI at progression; ICI/chemo/inhibitors of other targets/RT combinations | |
| EGFR mutation (other) | ICI/chemo/inhibitors of other targets/RT combinations | |
| ALK or RET re-arranged | ICI/chemo/inhibitors of other targets/RT combinations | |
| ↑Angiogenesis | ICI + VEGF inhibitors, COX-2 inhibitors, or FasL antibodies ± chemotherapy | |
| ↑TGF-β | As above | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, A.; He, X.; Hou, L.; Nguyen, N.P.; Zhu, G.; Cameron, R.B.; Lee, J.M. Classification of Non-Small Cell Lung Cancer’s Tumor Immune Micro-Environment and Strategies to Augment Its Response to Immune Checkpoint Blockade. Cancers 2021, 13, 2924. https://doi.org/10.3390/cancers13122924
Chi A, He X, Hou L, Nguyen NP, Zhu G, Cameron RB, Lee JM. Classification of Non-Small Cell Lung Cancer’s Tumor Immune Micro-Environment and Strategies to Augment Its Response to Immune Checkpoint Blockade. Cancers. 2021; 13(12):2924. https://doi.org/10.3390/cancers13122924
Chicago/Turabian StyleChi, Alexander, Xia He, Lin Hou, Nam P. Nguyen, Guangying Zhu, Robert B. Cameron, and Jay M. Lee. 2021. "Classification of Non-Small Cell Lung Cancer’s Tumor Immune Micro-Environment and Strategies to Augment Its Response to Immune Checkpoint Blockade" Cancers 13, no. 12: 2924. https://doi.org/10.3390/cancers13122924