
Proteomic Studies of Primary Acute Myeloid Leukemia Cells Derived from Patients Before and during Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid and Valproic Acid

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
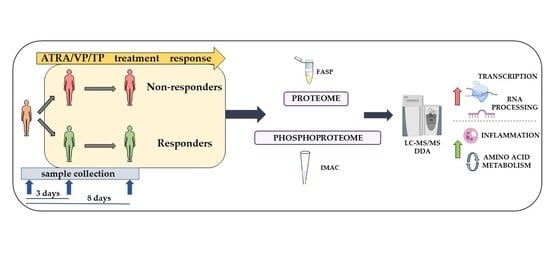
1. Introduction
2. Materials and Methods
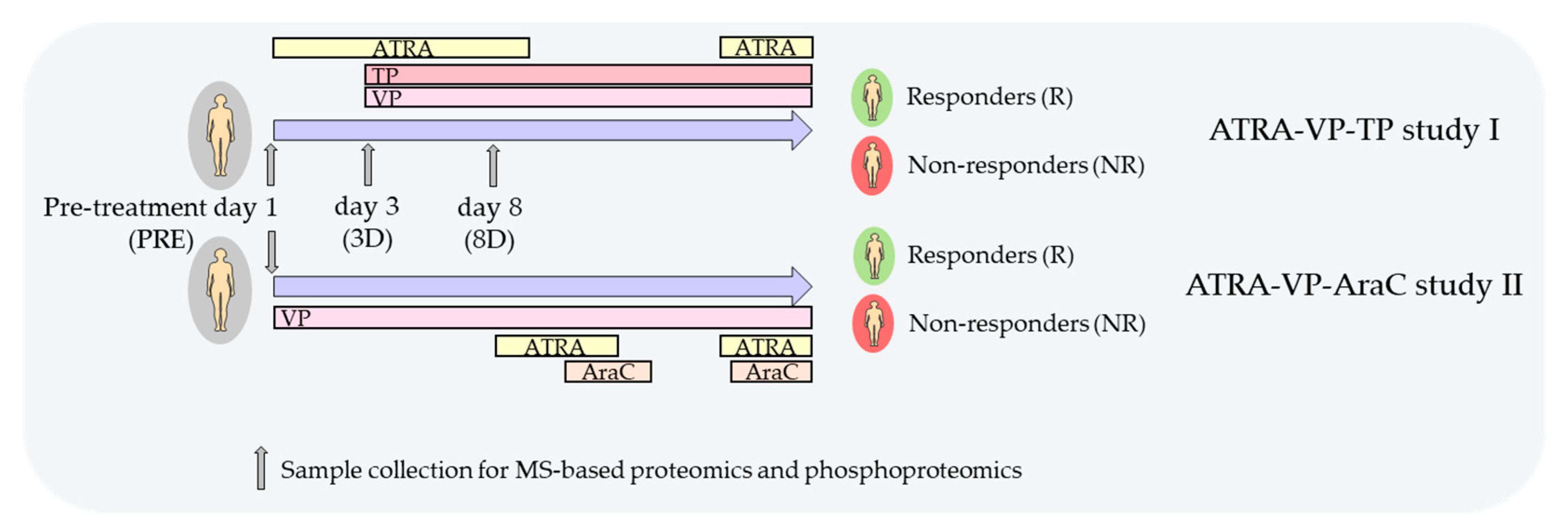
2.1. The Treatment Protocols Based on ATRA, VP and Low-Dose Cytotoxic Therapy
- VP. On day three of the first cycle, the patients received intravenous VP first as a loading dose (5 mg/kg for 60 min) and thereafter as a continuous infusion (28 mg/kg/24 h) until day 8; the patients thereafter received continuous oral treatment with the highest tolerated dose and the target serum level being 300–600 μmol/L.
- TP. The patients received intravenous TP on day 3 with a loading dose (5 mg/kg) and thereafter continuous infusion (0.65 mg/kg/hour) until day 8; they later received continuous oral therapy and the target serum level was 50–100 μM.
- Cytotoxic drugs. Patients with circulating AML blasts > 50 × 109/L at the time of diagnosis or later increasing circulating leukemic blasts received cytotoxic drugs to achieve stable AML blast levels below 50 × 109/L.
2.2. Preparation of Enriched AML Cells
2.3. Patient Sample Preparation for MS-Based Proteomics and Phosphoproteomics Analysis
2.4. LC–MS/MS Measurements
2.5. Data and Bioinformatics Analysis
2.6. DNA Methylation Analysis
3. Results
3.1. AML Patients Included in the Study
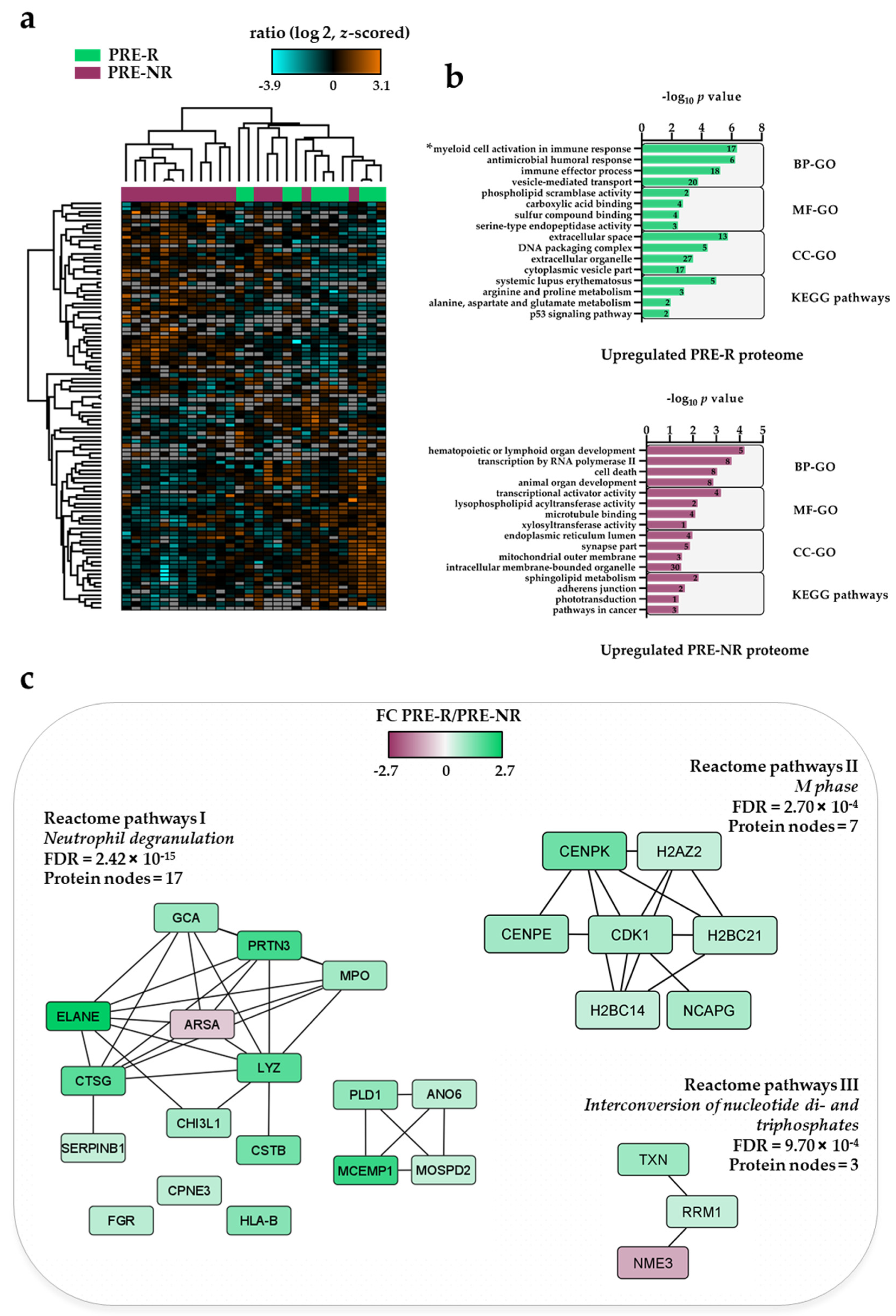
3.2. The Pre-Treatment AML Cell Proteome for Responder (PRE-R) and Non-Responder (PRE-NR) Patients
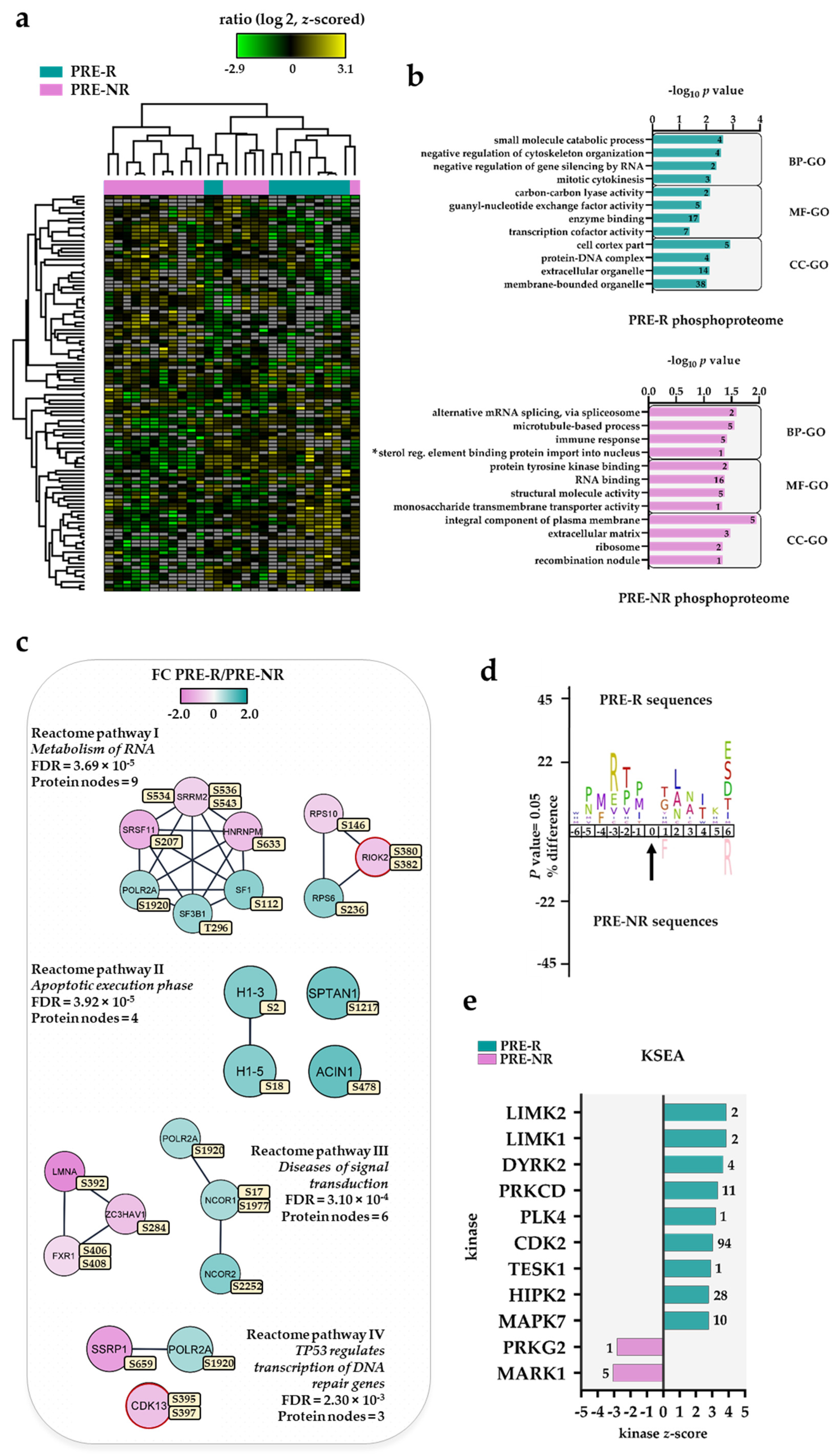
3.3. The Pre-Treatment AML Cell Phosphoproteome for Responder (PRE-R) and Non-Responder (PRE-NR) Patients
3.4. Altered Protein Phosphorylation Levels Are Not Caused by Altered Protein Levels
3.5. Differential Kinase Activity in Pre-Treatment AML Cells Derived from Responders (PRE-R) and Non-Responders (PRE-NR)
3.6. The DNA Methylation of the AML Genome Does Not Differ when Comparing Pre-Treatment AML Cell Samples from Responders (PRE-R) and Non-responders (PRE-NR)
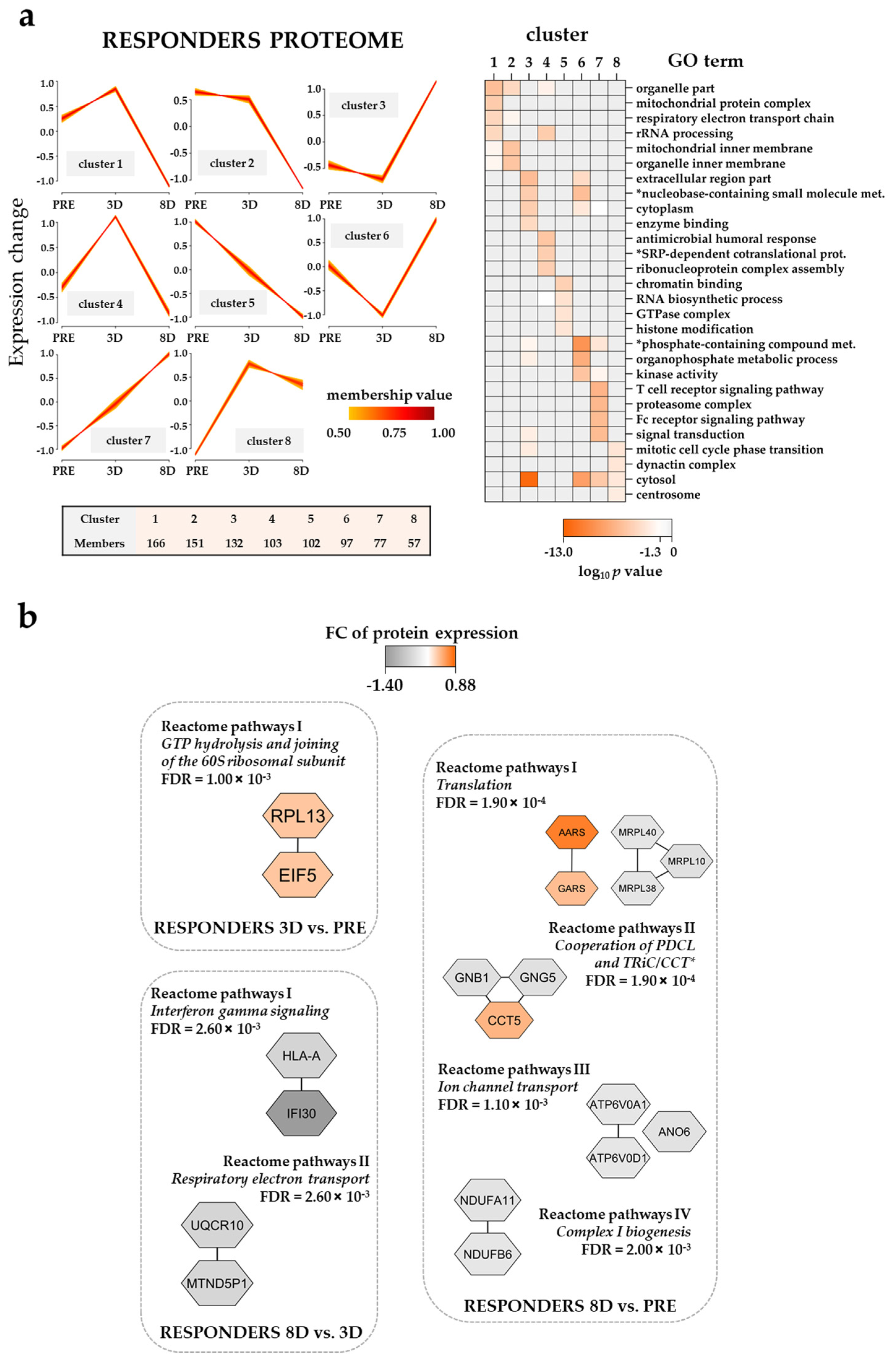
3.7. The Effect of the Triple Combination on AML Proteomic Profiles in Responders; Modulation of Translation, Organophosphate Metabolism, Intracellular Signaling and Mitochondrial Function
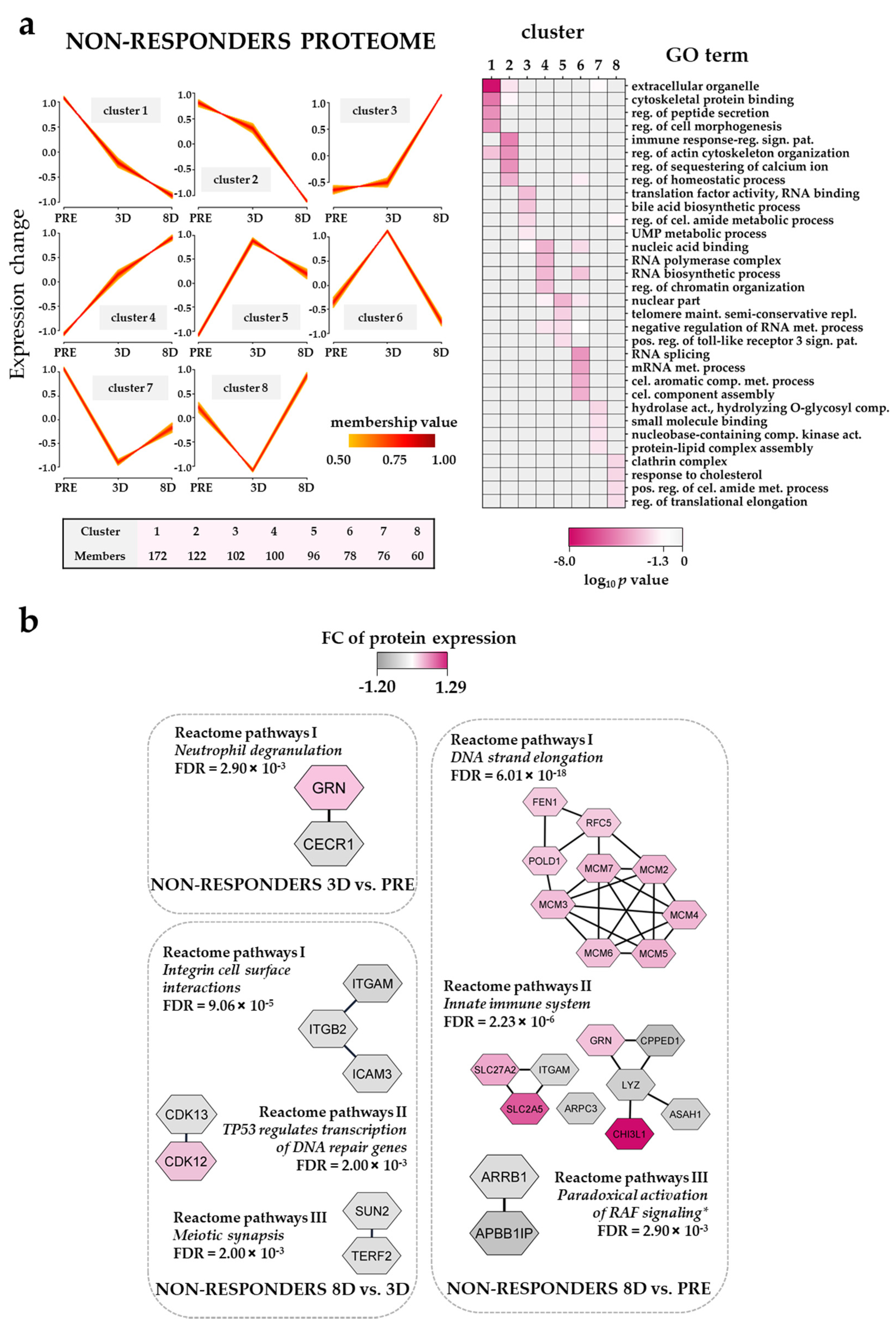
3.8. The Effect of the Triple Combination on AML Cell Proteomic Profiles in Non-Responders; Modulation of DNA Strand Elongation, RNA Processing, Actin/Cytoskeleton and Cholesterol Metabolism
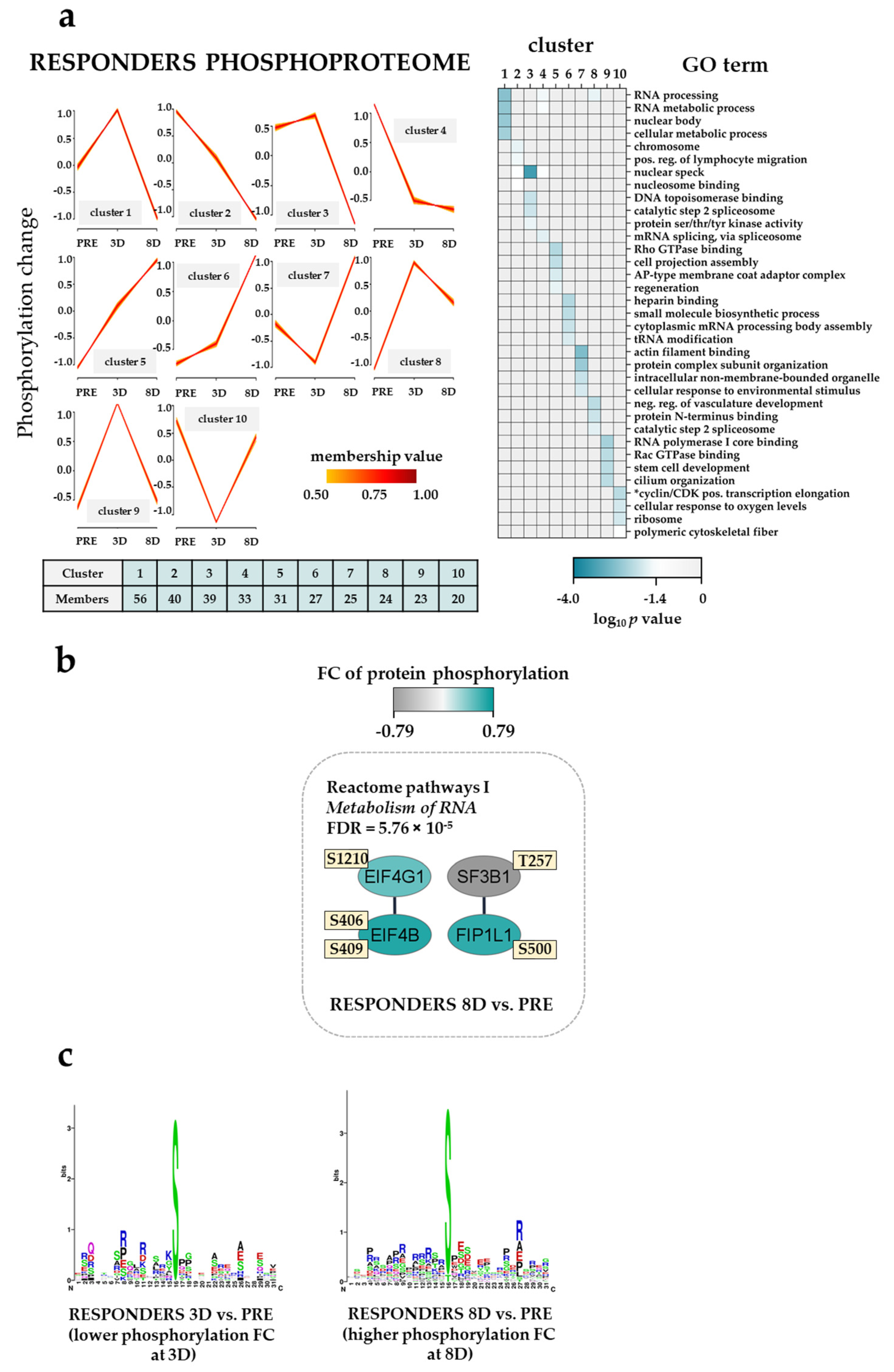
3.9. The Effect of the Triple Combination on AML Cell Phosphoproteomic Profiles in Responders; RNA Processing, Actin/Cytoskeleton and GTPase/Intracellular Signaling
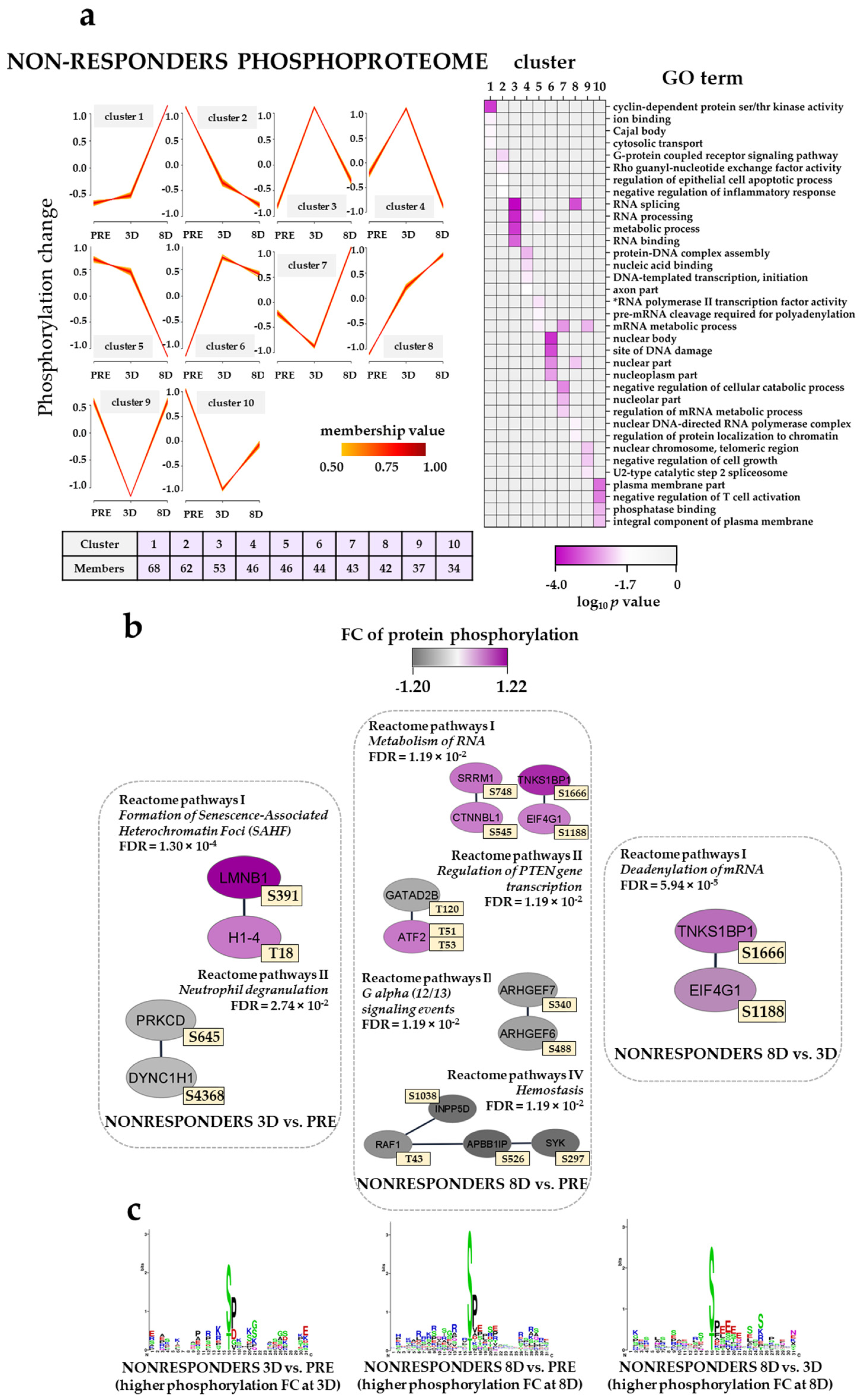
3.10. The Effect of the Triple Combination on AML Cell Phosphoproteomic Profiles in Non-Responders; Modulation of Translation/Transcription/RNA Metabolism and G-Protein Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein Name | Biological Function | FC (P) | FC (T) |
|---|---|---|---|---|
| ELANE | Elastase, neutrophil expressed | Modification the functions of natural killer cells | 2.66 | 4.17 |
| NME3 | NME/NM23 nucleoside diphosphate kinase 3 | Synthesis of nucleoside triphosphates other than ATP | −1.05 | −1.46 |
| HSD17B11 | Hydroxysteroid 17-beta-dehydrogenase 11 | Involvement in androgen metabolism during steroidogenesis | 0.64 | 1.41 |
References
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Lowenberg, B.; Naoe, T.; Lengfelder, E.; Dohner, H.; Burnett, A.K.; Chen, S.J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.K.; Milligan, D.; Prentice, A.G.; Goldstone, A.H.; McMullin, M.F.; Hills, R.K.; Wheatley, K. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 2007, 109, 1114–1124. [Google Scholar] [CrossRef]
- Fredly, H.; Ersvaer, E.; Kittang, A.O.; Tsykunova, G.; Gjertsen, B.T.; Bruserud, O. The combination of valproic acid, all-trans retinoic acid and low-dose cytarabine as disease-stabilizing treatment in acute myeloid leukemia. Clin. Epigenetics 2013, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Fredly, H.; Gjertsen, B.T.; Bruserud, O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: The effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin. Epigenetics 2013, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Ryningen, A.; Stapnes, C.; Lassalle, P.; Corbascio, M.; Gjertsen, B.T.; Bruserud, O. A subset of patients with high-risk acute myelogenous leukemia shows improved peripheral blood cell counts when treated with the combination of valproic acid, theophylline and all-trans retinoic acid. Leuk. Res. 2009, 33, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, J.; van Kann, E.; Rummelt, C.; Koschade, S.; Rollig, C.; Lubbert, M.; Schaich, M.; Parmentier, S.; Sebastian, M.; Chromik, J.; et al. Low-dose melphalan in elderly patients with relapsed or refractory acute myeloid leukemia: A well-tolerated and effective treatment after hypomethylating-agent failure. Leuk. Res. 2019, 85, 106192. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- Lubbert, M.; Grishina, O.; Schmoor, C.; Schlenk, R.F.; Jost, E.; Crysandt, M.; Heuser, M.; Thol, F.; Salih, H.R.; Schittenhelm, M.M.; et al. Valproate and Retinoic Acid in Combination With Decitabine in Elderly Nonfit Patients With Acute Myeloid Leukemia: Results of a Multicenter, Randomized, 2 × 2, Phase II Trial. J. Clin. Oncol. 2020, 38, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Borgmeyer, U.; Heyman, R.A.; Zhou, J.Y.; Ong, E.S.; Oro, A.E.; Kakizuka, A.; Evans, R.M. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992, 6, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangelsdorf, D.J.; Ong, E.S.; Dyck, J.A.; Evans, R.M. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 1990, 345, 224–229. [Google Scholar] [CrossRef]
- Martino, O.D.; Welch, J.S. Retinoic Acid Receptors in Acute Myeloid Leukemia Therapy. Cancers 2019, 11, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Urquiza, A.M.; Liu, S.; Sjoberg, M.; Zetterstrom, R.H.; Griffiths, W.; Sjovall, J.; Perlmann, T. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 2000, 290, 2140–2144. [Google Scholar] [CrossRef] [PubMed]
- Lengqvist, J.; Mata De Urquiza, A.; Bergman, A.C.; Willson, T.M.; Sjovall, J.; Perlmann, T.; Griffiths, W.J. Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid X receptor alpha ligand-binding domain. Mol. Cell. Proteom. 2004, 3, 692–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronningsaeter, I.S.; Fredly, H.K.; Gjertsen, B.T.; Hatfield, K.J.; Bruserud, O. Systemic Metabolomic Profiling of Acute Myeloid Leukemia Patients before and During Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid, Valproic Acid, and Low-Dose Chemotherapy. Cells 2019, 8, 1229. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.H.; Grandits, A.M.; Purton, L.E.; Sill, H.; Wieser, R. All-trans retinoic acid in non-promyelocytic acute myeloid leukemia: Driver lesion dependent effects on leukemic stem cells. Cell Cycle 2020, 19, 2573–2588. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. 2014, 10, 469–478. [Google Scholar] [CrossRef]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [Green Version]
- San Jose-Eneriz, E.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chateauvieux, S.; Morceau, F.; Dicato, M.; Diederich, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010, 479364. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Stapnes, C.; Ryningen, A.; Hatfield, K.; Oyan, A.M.; Eide, G.E.; Corbascio, M.; Kalland, K.H.; Gjertsen, B.T.; Bruserud, O. Functional characteristics and gene expression profiles of primary acute myeloid leukaemia cells identify patient subgroups that differ in susceptibility to histone deacetylase inhibitors. Int. J. Oncol. 2007, 31, 1529–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronningsaeter, I.S.; Reikvam, H.; Aasebo, E.; Bartaula-Brevik, S.; Tvedt, T.H.; Bruserud, O.; Hatfield, K.J. Targeting Cellular Metabolism in Acute Myeloid Leukemia and The Role of Patient Heterogeneity. Cells 2020, 9, 1155. [Google Scholar] [CrossRef] [PubMed]
- Ambinder, A.J.; Norsworthy, K.; Hernandez, D.; Palau, L.; Paun, B.; Duffield, A.; Chandraratna, R.; Sanders, M.; Varadhan, R.; Jones, R.J.; et al. A Phase 1 Study of IRX195183, a RARalpha-Selective CYP26 Resistant Retinoid, in Patients with Relapsed or Refractory AML. Front. Oncol. 2020, 10, 587062. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Berven, F.S.; Bartaula-Brevik, S.; Stokowy, T.; Hovland, R.; Vaudel, M.; Doskeland, S.O.; McCormack, E.; Batth, T.S.; Olsen, J.V.; et al. Proteome and Phosphoproteome Changes Associated with Prognosis in Acute Myeloid Leukemia. Cancers 2020, 12, 709. [Google Scholar] [CrossRef] [Green Version]
- Bruserud, O.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In vitro culture of aml cells--the present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Gjertsen, B.T.; Oyan, A.M.; Marzolf, B.; Hovland, R.; Gausdal, G.; Doskeland, S.O.; Dimitrov, K.; Golden, A.; Kalland, K.H.; Hood, L.; et al. Analysis of acute myelogenous leukemia: Preparation of samples for genomic and proteomic analyses. J. Hematother. Stem Cell Res. 2002, 11, 469–481. [Google Scholar] [CrossRef]
- Hatfield, K.J.; Hovland, R.; Oyan, A.M.; Kalland, K.H.; Ryningen, A.; Gjertsen, B.T.; Bruserud, O. Release of angiopoietin-1 by primary human acute myelogenous leukemia cells is associated with mutations of nucleophosmin, increased by bone marrow stromal cells and possibly antagonized by high systemic angiopoietin-2 levels. Leukemia 2008, 22, 287–293. [Google Scholar] [CrossRef]
- Reikvam, H.; Hovland, R.; Forthun, R.B.; Erdal, S.; Gjertsen, B.T.; Fredly, H.; Bruserud, O. Disease-stabilizing treatment based on all-trans retinoic acid and valproic acid in acute myeloid leukemia—identification of responders by gene expression profiling of pretreatment leukemic cells. Bmc Cancer 2017, 17, 630. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Valladares, M.; Aasebø, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, Ø.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasebo, E.; Vaudel, M.; Mjaavatten, O.; Gausdal, G.; Van der Burgh, A.; Gjertsen, B.T.; Doskeland, S.O.; Bruserud, O.; Berven, F.S.; Selheim, F. Performance of super-SILAC based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics 2014, 14, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Arntzen, M.Ø.; Koehler, C.J.; Barsnes, H.; Berven, F.S.; Treumann, A.; Thiede, B. IsobariQ: Software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. J. Proteome Res. 2011, 10, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Schwämmle, V.; Jensen, O.N. VSClust: Feature-based variance-sensitive clustering of omics data. Bioinformatics 2018, 34, 2965–2972. [Google Scholar] [CrossRef]
- Scholz, C.; Lyon, D.; Refsgaard, J.C.; Jensen, L.J.; Choudhary, C.; Weinert, B.T. Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nat. Methods 2015, 12, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Wiredja, D.D.; Koyuturk, M.; Chance, M.R. The KSEA App: A web-based tool for kinase activity inference from quantitative phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.-M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamsaei, B.; Chojnacki, S.; Pilarczyk, M.; Najafabadi, M.; Niu, W.; Chen, C.; Ross, K.; Matlock, A.; Muhlich, J.; Chutipongtanate, S.; et al. piNET: A versatile web platform for downstream analysis and visualization of proteomics data. Nucleic Acids Res. 2020, 48, W85–W93. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Licata, L.; Lo Surdo, P.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2020, 48, D504–D510. [Google Scholar] [CrossRef]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Bennett, J.M.; Kopecky, K.J.; Buchner, T.; Willman, C.L.; Estey, E.H.; Schiffer, C.A.; Doehner, H.; Tallman, M.S.; Lister, T.A.; et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J. Clin. Oncol. 2003, 21, 4642–4649. [Google Scholar] [CrossRef]
- Cheson, B.D.; Bennett, J.M.; Kantarjian, H.; Pinto, A.; Schiffer, C.A.; Nimer, S.D.; Lowenberg, B.; Beran, M.; de Witte, T.M.; Stone, R.M.; et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood 2000, 96, 3671–3674. [Google Scholar]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Deng, M.; Li, J.; Tong, X.; Wei, Q.; Ye, X. Phosphorylation of right open reading frame 2 (Rio2) protein kinase by polo-like kinase 1 regulates mitotic progression. J. Biol. Chem. 2011, 286, 36352–36360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarg, B.; Helliger, W.; Talasz, H.; Forg, B.; Lindner, H.H. Histone H1 phosphorylation occurs site-specifically during interphase and mitosis: Identification of a novel phosphorylation site on histone H1. J. Biol. Chem. 2006, 281, 6573–6580. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Kanai-Azuma, M.; Kanai, Y.; Moriyama, K.; Yazaki, K.; Hayashi, Y.; Kitamura, N. Cofilin phosphorylation and actin polymerization by NRK/NESK, a member of the germinal center kinase family. Exp. Cell Res. 2003, 287, 219–227. [Google Scholar] [CrossRef]
- Prudent, R.; Demoncheaux, N.; Diemer, H.; Collin-Faure, V.; Kapur, R.; Paublant, F.; Lafanechere, L.; Cianferani, S.; Rabilloud, T. A quantitative proteomic analysis of cofilin phosphorylation in myeloid cells and its modulation using the LIM kinase inhibitor Pyr1. PLoS ONE 2018, 13, e0208979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comino-Mendez, I.; Leandro-Garcia, L.J.; Montoya, G.; Inglada-Perez, L.; de Cubas, A.A.; Curras-Freixes, M.; Tysoe, C.; Izatt, L.; Leton, R.; Gomez-Grana, A.; et al. Functional and in silico assessment of MAX variants of unknown significance. J. Mol. Med. 2015, 93, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.A. Canonical and noncanonical roles of Par-1/MARK kinases in cell migration. Int. Rev. Cell Mol. Biol. 2014, 312, 169–199. [Google Scholar] [CrossRef]
- Rangaswami, H.; Schwappacher, R.; Tran, T.; Chan, G.C.; Zhuang, S.; Boss, G.R.; Pilz, R.B. Protein kinase G and focal adhesion kinase converge on Src/Akt/beta-catenin signaling module in osteoblast mechanotransduction. J. Biol. Chem. 2012, 287, 21509–21519. [Google Scholar] [CrossRef] [Green Version]
- Forthun, R.B.; Hellesoy, M.; Sulen, A.; Kopperud, R.K.; Sjoholt, G.; Bruserud, O.; McCormack, E.; Gjertsen, B.T. Modulation of phospho-proteins by interferon-alpha and valproic acid in acute myeloid leukemia. J. Cancer Res. Clin. Oncol. 2019, 145, 1729–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrikov, M.; Dobrikova, E.; Shveygert, M.; Gromeier, M. Phosphorylation of eukaryotic translation initiation factor 4G1 (eIF4G1) by protein kinase C{alpha} regulates eIF4G1 binding to Mnk1. Mol. Cell Biol. 2011, 31, 2947–2959. [Google Scholar] [CrossRef] [Green Version]
- Kreitz, J.; Schonfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnutgen, F.; Kurrle, N.; et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [Green Version]
- Latagliata, R.; Bongarzoni, V.; Carmosino, I.; Mengarelli, A.; Breccia, M.; Borza, P.A.; D’Andrea, M.; D’Elia, G.M.; Mecarocci, S.; Morano, S.G.; et al. Acute myelogenous leukemia in elderly patients not eligible for intensive chemotherapy: The dark side of the moon. Ann. Oncol. 2006, 17, 281–285. [Google Scholar] [CrossRef]
- Rucker, F.G.; Lang, K.M.; Futterer, M.; Komarica, V.; Schmid, M.; Dohner, H.; Schlenk, R.F.; Dohner, K.; Knudsen, S.; Bullinger, L. Molecular dissection of valproic acid effects in acute myeloid leukemia identifies predictive networks. Epigenetics 2016, 11, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Aasebo, E.; Berven, F.S.; Hovland, R.; Doskeland, S.O.; Bruserud, O.; Selheim, F.; Hernandez-Valladares, M. The Progression of Acute Myeloid Leukemia from First Diagnosis to Chemoresistant Relapse: A Comparison of Proteomic and Phosphoproteomic Profiles. Cancers 2020, 12, 1466. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Bruserud, O.; Selheim, F. The Implementation of Mass Spectrometry-Based Proteomics Workflows in Clinical Routines of Acute Myeloid Leukemia: Applicability and Perspectives. Int. J. Mol. Sci. 2020, 21, 6830. [Google Scholar] [CrossRef]
- Mer, A.S.; Lindberg, J.; Nilsson, C.; Klevebring, D.; Wang, M.; Gronberg, H.; Lehmann, S.; Rantalainen, M. Expression levels of long non-coding RNAs are prognostic for AML outcome. J. Hematol. Oncol. 2018, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Staubert, C.; Bhuiyan, H.; Lindahl, A.; Broom, O.J.; Zhu, Y.; Islam, S.; Linnarsson, S.; Lehtio, J.; Nordstrom, A. Rewired metabolism in drug-resistant leukemia cells: A metabolic switch hallmarked by reduced dependence on exogenous glutamine. J. Biol. Chem. 2015, 290, 8348–8359. [Google Scholar] [CrossRef] [Green Version]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Gronbaek, K.; Muller-Tidow, C.; Perini, G.; Lehmann, S.; Bach Treppendahl, M.; Mills, K.; Plass, C.; Schlegelberger, B.; European, G.; Epigenomics Study on, M.D.S.; et al. A critical appraisal of tools available for monitoring epigenetic changes in clinical samples from patients with myeloid malignancies. Haematologica 2012, 97, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, V.; Horstedt, A.S.; Johansson, B.; Antunovic, P.; Billstrom, R.; Derolf, A.; Lehmann, S.; Mollgard, L.; Peterson, S.; Stockelberg, D.; et al. Failure matters: Unsuccessful cytogenetics and unperformed cytogenetics are associated with a poor prognosis in a population-based series of acute myeloid leukaemia. Eur. J. Haematol. 2015, 94, 419–423. [Google Scholar] [CrossRef]
- Cimino, G.; Lo-Coco, F.; Fenu, S.; Travaglini, L.; Finolezzi, E.; Mancini, M.; Nanni, M.; Careddu, A.; Fazi, F.; Padula, F.; et al. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Res. 2006, 66, 8903–8911. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.K.; Noh, E.K.; Yoon, D.J.; Jo, J.C.; Park, J.H.; Kim, H. Dasatinib accelerates valproic acid-induced acute myeloid leukemia cell death by regulation of differentiation capacity. PLoS ONE 2014, 9, e98859. [Google Scholar] [CrossRef] [Green Version]
- Deluche, E.; Bessette, B.; Durand, S.; Caire, F.; Rigau, V.; Robert, S.; Chaunavel, A.; Forestier, L.; Labrousse, F.; Jauberteau, M.O.; et al. CHI3L1, NTRK2, 1p/19q and IDH Status Predicts Prognosis in Glioma. Cancers 2019, 11, 544. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.Y.; Lin, L.L.; Zhang, C.; Yang, M.; Zhong, H.C.; Wei, R.X. The Potential of Five Immune-Related Prognostic Genes to Predict Survival and Response to Immune Checkpoint Inhibitors for Soft Tissue Sarcomas Based on Multi-Omic Study. Front. Oncol. 2020, 10, 1317. [Google Scholar] [CrossRef]
- Hao, Y.; Hu, P.; Zhang, J. Genomic analysis of the prognostic effect of tumor-associated neutrophil-related genes across 15 solid cancer types: An immune perspective. Ann. Transl. Med. 2020, 8, 1507. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, Q. Weighted gene co-expression network analysis of gene modules for the prognosis of esophageal cancer. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 319–325. [Google Scholar] [CrossRef]
- Yagi, Y.; Fushida, S.; Harada, S.; Kinoshita, J.; Makino, I.; Oyama, K.; Tajima, H.; Fujita, H.; Takamura, H.; Ninomiya, I.; et al. Effects of valproic acid on the cell cycle and apoptosis through acetylation of histone and tubulin in a scirrhous gastric cancer cell line. J. Exp. Clin. Cancer Res. 2010, 29, 149. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Satyamoorthy, K.; Herlyn, M.; Rosdahl, I. All-trans retinoic acid (atRA) differentially induces apoptosis in matched primary and metastatic melanoma cells—A speculation on damage effect of atRA via mitochondrial dysfunction and cell cycle redistribution. Carcinogenesis 2003, 24, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, M.; Argiropoulos, B.; Kuchenbauer, F.; Yung, E.; Piper, J.; Fung, S.; Schlenk, R.F.; Dohner, K.; Hinrichsen, T.; Rudolph, C.; et al. MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in patients with AML. Blood 2007, 110, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, M.H.; Dezaki, Z.R. Prognostic Value of EVI1 Expression in Pediatric Acute Myeloid Leukemia: A Systematic Review. Iran. J. Pathol. 2018, 13, 294–300. [Google Scholar] [PubMed]
- Wu, X.; Wang, H.; Deng, J.; Zheng, X.; Ling, Y.; Gong, Y. Prognostic significance of the EVI1 gene expression in patients with acute myeloid leukemia: A meta-analysis. Ann. Hematol. 2019, 98, 2485–2496. [Google Scholar] [CrossRef]
- Kahl, M.; Brioli, A.; Bens, M.; Perner, F.; Kresinsky, A.; Schnetzke, U.; Hinze, A.; Sbirkov, Y.; Stengel, S.; Simonetti, G.; et al. The acetyltransferase GCN5 maintains ATRA-resistance in non-APL AML. Leukemia 2019, 33, 2628–2639. [Google Scholar] [CrossRef]
- Hernandez-Valladares, M.; Aasebo, E.; Berven, F.; Selheim, F.; Bruserud, O. Biological characteristics of aging in human acute myeloid leukemia cells: The possible importance of aldehyde dehydrogenase, the cytoskeleton and altered transcriptional regulation. Aging 2020, 12, 24734–24777. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Dohner, K.; Kneba, M.; Gotze, K.; Hartmann, F.; Del Valle, F.; Kirchen, H.; Koller, E.; Fischer, J.T.; Bullinger, L.; et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica 2009, 94, 54–60. [Google Scholar] [CrossRef]
- Massaro, F.; Corrillon, F.; Stamatopoulos, B.; Meuleman, N.; Lagneaux, L.; Bron, D. Aging of Bone Marrow Mesenchymal Stromal Cells: Hematopoiesis Disturbances and Potential Role in the Development of Hematologic Cancers. Cancers 2020, 13, 68. [Google Scholar] [CrossRef]
- Zjablovskaja, P.; Florian, M.C. Acute Myeloid Leukemia: Aging and Epigenetics. Cancers 2019, 12, 103. [Google Scholar] [CrossRef] [Green Version]
- Aasebo, E.; Birkeland, E.; Selheim, F.; Berven, F.; Brenner, A.K.; Bruserud, O. The Extracellular Bone Marrow Microenvironment-A Proteomic Comparison of Constitutive Protein Release by In Vitro Cultured Osteoblasts and Mesenchymal Stem Cells. Cancers 2020, 13, 62. [Google Scholar] [CrossRef]
- Hernandez, D.; Palau, L.; Norsworthy, K.; Anders, N.M.; Alonso, S.; Su, M.; Petkovich, M.; Chandraratna, R.; Rudek, M.A.; Smith, B.D.; et al. Overcoming microenvironment-mediated protection from ATRA using CYP26-resistant retinoids. Leukemia 2020, 34, 3077–3081. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.T.; Oh, S.; Ro, D.H.; Yoo, H.; Kwon, Y.W. The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis. J. Lipid Atheroscler. 2020, 9, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.G.; Otterson, G.A. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem. Anticancer Agents 2003, 3, 187–199. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shallis, R.; Stahl, M.; Zeidan, A.M. Epigenetic therapy combinations in acute myeloid leukemia: What are the options? Adv. Hematol. 2019, 10, 2040620718816698. [Google Scholar] [CrossRef] [Green Version]
- Dhall, A.; Zee, B.M.; Yan, F.; Blanco, M.A. Intersection of Epigenetic and Metabolic Regulation of Histone Modifications in Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.; Carlet, M.; Schlenk, R.F.; Weber, A.; Kress, J.; Brunner, I.; Slabicki, M.; Grill, G.; Weisemann, S.; Cheng, Y.Y.; et al. Requirement for LIM kinases in acute myeloid leukemia. Leukemia 2020, 34, 3173–3185. [Google Scholar] [CrossRef]
- Ishaq, M.; Lin, B.R.; Bosche, M.; Zheng, X.; Yang, J.; Huang, D.; Lempicki, R.A.; Aguilera-Gutierrez, A.; Natarajan, V. LIM kinase 1—dependent cofilin 1 pathway and actin dynamics mediate nuclear retinoid receptor function in T lymphocytes. BMC Mol. Biol. 2011, 12, 41. [Google Scholar] [CrossRef] [Green Version]
- Brattas, M.K.; Reikvam, H.; Tvedt, T.H.A.; Bruserud, O. Dasatinib as an investigational drug for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia in adults. Expert Opin. Investig. Drugs 2019, 28, 411–420. [Google Scholar] [CrossRef]
- Hoelzer, D.; Ludwig, W.D.; Thiel, E.; Gassmann, W.; Loffler, H.; Fonatsch, C.; Rieder, H.; Heil, G.; Heinze, B.; Arnold, R.; et al. Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 1996, 87, 495–508. [Google Scholar] [CrossRef] [Green Version]
- Alanazi, B.; Munje, C.R.; Rastogi, N.; Williamson, A.J.K.; Taylor, S.; Hole, P.S.; Hodges, M.; Doyle, M.; Baker, S.; Gilkes, A.F.; et al. Integrated nuclear proteomics and transcriptomics identifies S100A4 as a therapeutic target in acute myeloid leukemia. Leukemia 2020, 34, 427–440. [Google Scholar] [CrossRef]
- Lee, J.S.; Cheong, H.S.; Koh, Y.; Ahn, K.S.; Shin, H.D.; Yoon, S.S. MCM7 polymorphisms associated with the AML relapse and overall survival. Ann. Hematol. 2017, 96, 93–98. [Google Scholar] [CrossRef]
- Simonetti, G.; Padella, A.; do Valle, I.F.; Fontana, M.C.; Fonzi, E.; Bruno, S.; Baldazzi, C.; Guadagnuolo, V.; Manfrini, M.; Ferrari, A.; et al. Aneuploid acute myeloid leukemia exhibits a signature of genomic alterations in the cell cycle and protein degradation machinery. Cancer 2019, 125, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [Green Version]
- Akanuma, H.; Qin, X.Y.; Nagano, R.; Win-Shwe, T.T.; Imanishi, S.; Zaha, H.; Yoshinaga, J.; Fukuda, T.; Ohsako, S.; Sone, H. Identification of Stage-Specific Gene Expression Signatures in Response to Retinoic Acid during the Neural Differentiation of Mouse Embryonic Stem Cells. Front. Genet. 2012, 3, 141. [Google Scholar] [CrossRef] [Green Version]
- Falker-Gieske, C.; Mott, A.; Franzenburg, S.; Tetens, J. Multi-species transcriptome meta-analysis of the response to retinoic acid in vertebrates and comparative analysis of the effects of retinol and retinoic acid on gene expression in LMH cells. BMC Genom. 2021, 22, 146. [Google Scholar] [CrossRef] [PubMed]
- Rochette-Egly, C. Retinoic Acid-Regulated Target Genes During Development: Integrative Genomics Analysis. Subcell. Biochem. 2020, 95, 57–85. [Google Scholar] [CrossRef] [PubMed]
- Menzin, J.; Lang, K.; Earle, C.C.; Kerney, D.; Mallick, R. The outcomes and costs of acute myeloid leukemia among the elderly. Arch. Intern. Med. 2002, 162, 1597–1603. [Google Scholar] [CrossRef] [Green Version]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andra, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grotzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Biol. 2019, 11, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Kmit, A.; van Kruchten, R.; Ousingsawat, J.; Mattheij, N.J.; Senden-Gijsbers, B.; Heemskerk, J.W.; Schreiber, R.; Bevers, E.M.; Kunzelmann, K. Calcium-activated and apoptotic phospholipid scrambling induced by Ano6 can occur independently of Ano6 ion currents. Cell Death Dis. 2013, 4, e611. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, K.; Nilius, B.; Owsianik, G.; Schreiber, R.; Ousingsawat, J.; Sirianant, L.; Wanitchakool, P.; Bevers, E.M.; Heemskerk, J.W. Molecular functions of anoctamin 6 (TMEM16F): A chloride channel, cation channel, or phospholipid scramblase? Pflug. Arch 2014, 466, 407–414. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Ousingsawat, J.; Benedetto, R.; Cabrita, I.; Schreiber, R. Contribution of Anoctamins to Cell Survival and Cell Death. Cancers 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Roh, J.; Woo, J.H.; Kim, S.J.; Nam, J.H. TMEM16F/ANO6, a Ca(2+)-activated anion channel, is negatively regulated by the actin cytoskeleton and intracellular MgATP. Biochem. Biophys. Res. Commun. 2018, 503, 2348–2354. [Google Scholar] [CrossRef]
- Veit, M.; Koyro, K.I.; Ahrens, B.; Bleibaum, F.; Munz, M.; Rovekamp, H.; Andra, J.; Schreiber, R.; Kunzelmann, K.; Sommer, A.; et al. Anoctamin-6 regulates ADAM sheddase function. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1598–1610. [Google Scholar] [CrossRef]
- Kovacs, Z.; Jung, I.; Gurzu, S. Arylsulfatases A and B: From normal tissues to malignant tumors. Pathol. Res. Pr. 2019, 215, 152516. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Yin, S.; Liu, T.; Riabov, V.; Mitrofanova, I. Role of chitinase-like proteins in cancer. Biol. Chem. 2016, 397, 231–247. [Google Scholar] [CrossRef]
- Fu, L.; Fu, H.; Qiao, J.; Pang, Y.; Xu, K.; Zhou, L.; Wu, Q.; Li, Z.; Ke, X.; Xu, K.; et al. High expression of CPNE3 predicts adverse prognosis in acute myeloid leukemia. Cancer Sci. 2017, 108, 1850–1857. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Wang, H.X.; Wang, J.; Zhang, J.Y.; Liu, B.Y.; Li, A.L.; Lv, M.; Hu, M.R.; Yu, M.; Feng, J.N.; et al. Proteomic analysis of interleukin 6-induced differentiation in mouse myeloid leukemia cells. Int. J. Biochem. Cell Biol. 2005, 37, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Haznedaroglu, I.C.; Malkan, U.Y. Local bone marrow renin-angiotensin system in the genesis of leukemia and other malignancies. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4089–4111. [Google Scholar]
- Makaryan, V.; Zeidler, C.; Bolyard, A.A.; Skokowa, J.; Rodger, E.; Kelley, M.L.; Boxer, L.A.; Bonilla, M.A.; Newburger, P.E.; Shimamura, A.; et al. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr. Opin. Hematol. 2015, 22, 3–11. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of myeloid Src-family kinases is associated with poor prognosis in AML and influences Flt3-ITD kinase inhibitor acquired resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Moroco, J.A.; Patel, R.K.; Shi, H.; Engen, J.R.; Dorman, H.R.; Smithgall, T.E. The Src family kinase Fgr is a transforming oncoprotein that functions independently of SH3-SH2 domain regulation. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Weir, M.C.; Shu, S.T.; Patel, R.K.; Hellwig, S.; Chen, L.; Tan, L.; Gray, N.S.; Smithgall, T.E. Selective Inhibition of the Myeloid Src-Family Kinase Fgr Potently Suppresses AML Cell Growth in Vitro and in Vivo. ACS Chem. Biol. 2018, 13, 1551–1559. [Google Scholar] [CrossRef]
- Han, S.H.; Korm, S.; Han, Y.G.; Choi, S.Y.; Kim, S.H.; Chung, H.J.; Park, K.; Kim, J.Y.; Myung, K.; Lee, J.Y.; et al. GCA links TRAF6-ULK1-dependent autophagy activation in resistant chronic myeloid leukemia. Autophagy 2019, 15, 2076–2090. [Google Scholar] [CrossRef]
- Kim, T.W.; Hong, S.; Talukder, A.H.; Pascual, V.; Liu, Y.J. Grancalcin (GCA) modulates Toll-like receptor 9 (TLR9) mediated signaling through its direct interaction with TLR9. Eur. J. Immunol. 2016, 46, 712–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liu, N.; Wang, S.; Wang, L.; Zhao, J.; Su, L.; Zhang, Y.; Zhang, S.; Xu, Z.; Zhao, B.; et al. Identification of a small molecule targeting annexin A7. Biochim. Biophys. Acta 2013, 1833, 2092–2099. [Google Scholar] [CrossRef] [Green Version]
- Marra, J.; Greene, J.; Hwang, J.; Du, J.; Damon, L.; Martin, T.; Venstrom, J.M. KIR and HLA genotypes predictive of low-affinity interactions are associated with lower relapse in autologous hematopoietic cell transplantation for acute myeloid leukemia. J. Immunol. 2015, 194, 4222–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ucar, F.; Sonmez, M.; Erkut, N.; Balci, M.; Yucel, B.; Yilmaz, M.; Erduran, E.; Ovali, E. Relation of HLA-A, -B, -DRB1 alleles and haplotypes in patients with acute leukemia: A case control study. Arch. Med. Res. 2011, 42, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Strobl, H.; Knapp, W. Myeloid cell-associated lysosomal proteins as flow cytometry markers for leukocyte lineage classification. J. Biol. Regul. Homeost. Agents 2004, 18, 335–339. [Google Scholar]
- Kumar, M.; Birdi, A.; Gupta, Y.N.; Gupta, S. Serum lactic dehydrogenase isoenzymes alteration in carcinoma cervix uteri. Int. J. Gynaecol. Obs. 1988, 27, 91–95. [Google Scholar] [CrossRef]
- Di Mattia, T.; Martinet, A.; Ikhlef, S.; McEwen, A.G.; Nomine, Y.; Wendling, C.; Poussin-Courmontagne, P.; Voilquin, L.; Eberling, P.; Ruffenach, F.; et al. FFAT motif phosphorylation controls formation and lipid transfer function of inter-organelle contacts. Embo J. 2020, 39, e104369. [Google Scholar] [CrossRef]
- Di Mattia, T.; Wilhelm, L.P.; Ikhlef, S.; Wendling, C.; Spehner, D.; Nomine, Y.; Giordano, F.; Mathelin, C.; Drin, G.; Tomasetto, C.; et al. Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. Embo Rep. 2018, 19. [Google Scholar] [CrossRef]
- Li, C.H.; Chen, J.; Nie, L.; Chen, J. MOSPD2 is a receptor mediating the LEAP-2 effect on monocytes/macrophages in a teleost, Boleophthalmus pectinirostris. Zool. Res. 2020, 41, 644–655. [Google Scholar] [CrossRef]
- Kamijo, R.; Itonaga, H.; Kihara, R.; Nagata, Y.; Hata, T.; Asou, N.; Ohtake, S.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; et al. Distinct gene alterations with a high percentage of myeloperoxidase-positive leukemic blasts in de novo acute myeloid leukemia. Leuk. Res. 2018, 65, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Roberson, J.R.; Onciu, M.; Pounds, S.; Rubnitz, J.E.; Pui, C.H.; Razzouk, B.I. Prognostic significance of myeloperoxidase expression in childhood acute myeloid leukemia. Pediatr. Blood Cancer 2008, 50, 542–548. [Google Scholar] [CrossRef]
- Brown, F.D.; Thompson, N.; Saqib, K.M.; Clark, J.M.; Powner, D.; Thompson, N.T.; Solari, R.; Wakelam, M.J. Phospholipase D1 localises to secretory granules and lysosomes and is plasma-membrane translocated on cellular stimulation. Curr. Biol. 1998, 8, 835–838. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhou, J.; Pei, R.; Li, F.; Jin, J.; Jiang, L. Expression and clinical significance of phospholipase D1 in de novo acute myeloid leukemia. Hematology 2020, 25, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Ichikawa, T.; Matsukura, M.; Sueda, M.; Takashima, M.; Ikeda, R.; Nishikawa, A.; Shinoda, T. Genetic diversity and biochemical characteristics of Trichosporon asahii isolated from clinical specimens, houses of patients with summer-type-hypersensitivity pneumonitis, and environmental materials. J. Clin. Microbiol. 2001, 39, 2405–2411. [Google Scholar] [CrossRef] [Green Version]
- Greiner, J.; Schmitt, M.; Li, L.; Giannopoulos, K.; Bosch, K.; Schmitt, A.; Dohner, K.; Schlenk, R.F.; Pollack, J.R.; Dohner, H.; et al. Expression of tumor-associated antigens in acute myeloid leukemia: Implications for specific immunotherapeutic approaches. Blood 2006, 108, 4109–4117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Ansari, D.; Zhou, Q.; Sasor, A.; Said Hilmersson, K.; Andersson, R. Low P4HA2 and high PRTN3 expression predicts poor survival in patients with pancreatic cancer. Scand. J. Gastroenterol. 2019, 54, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.E. Primary neoplasms in vesical diverticula. Reports of two cases. Scand. J. Urol. Nephrol. 1988, 22, 347–348. [Google Scholar] [CrossRef]
- Karatepe, K.; Zhu, H.; Zhang, X.; Guo, R.; Kambara, H.; Loison, F.; Liu, P.; Yu, H.; Ren, Q.; Luo, X.; et al. Proteinase 3 Limits the Number of Hematopoietic Stem and Progenitor Cells in Murine Bone Marrow. Stem Cell Rep. 2018, 11, 1092–1105. [Google Scholar] [CrossRef] [Green Version]
- Yau, C.; Esserman, L.; Moore, D.H.; Waldman, F.; Sninsky, J.; Benz, C.C. A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res. 2010, 12, R85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, I.; Ma, X.; Seger, C.; Maolake, A.; Garcia-Hernandez, M.L.; Rangel-Moreno, J.; Ackerman, J.; Nastiuk, K.L.; Susiarjo, M.; Hammes, S.R. Epigenetic Suppression of SERPINB1 Promotes Inflammation-Mediated Prostate Cancer Progression. Mol. Cancer Res. 2019, 17, 845–859. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Rios, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]







| ID | Sex | Age | Previous Disease, Present Status | FAB | Membrane Molecule Expression 2 | Karyotype | FLT3 | NPM1 | Additional Mutations | WBC Counts | Survival (Days) 3 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANPEP | CD14 | FUT4 | CD33 | CD34 | |||||||||||
| Responders | |||||||||||||||
| R1 | M | 74 | de novo | M0 | + | - | - | + | + | multiple | wt | wt | TP53 | 18.7 | 151 |
| R2 | M | 73 | de novo | M1 | nt | wt | INS | IDH2, SRSF1 | 12.1 | 383 | |||||
| R3 * | F | 72 | MDS | M2 | + | - | - | + | - | t (1;5), t (2;3) | ITD | wt | SETD2, RUNX1 | 42.6 | 132 |
| R4 | M | 81 | polycytemia vera | M2 | - | - | - | - | + | del (7) | wt | wt | ASXL1, SRSF2, RAD21 | 22.3 | 610 |
| R5 | F | 77 | MDS | M2 | + | - | - | + | + | normal | wt | wt | NRAS, TET2, ASXL1, RUNX1, SRSF1, STAG2, BCOR | 142.0 | 132 |
| R6 * | M | 80 | de novo | M1 | + | - | - | + | + | multiple | wt | wt | nt | 8.0 | 58 |
| R7 * | M | 78 | MDS | M1 | + | - | - | + | + | nt | nt | nt | nt | 142.0 | 69 |
| R8 * | F | 68 | 1st relapse | M1 | + | - | + | + | + | normal | wt | wt | TET2, ASXL1, BCOR | 15.6 | 105 |
| R9 * | M | 86 | de novo | M4 | + | + | + | + | - | nt | nt | nt | nt | 18.7 | 59 |
| R10 * | F | 61 | 1st relapse | M1 | + | - | + | + | + | multiple | wt | wt | NRAS, SF3B1 | 55.8 | 644 |
| R11 * | M | 62 | 2nd relapse | M2 | + | - | + | + | + | del (7) | wt | wt | nt | 4.9 | 350 |
| Non-Responders | |||||||||||||||
| NR1 | F | 82 | de novo | M5 | + | - | + | + | + | normal | ITD, TKD | wt | WT1, DNMT3A | 142.0 | 37 |
| NR2 | F | 60 | relapse | M4 | + | - | + | + | - | normal | ITD | INS | DNMT3A, TET2 | 16.7 | 12 |
| NR3 | F | 77 | de novo | M1 | - | - | - | - | - | normal | ITD | INS | DNMT3A | 68.5 | 32 |
| NR4 | F | 78 | de novo | M0 | + | - | - | - | + | nt | wt | wt | PTPN11, ASXL1, RUNX1, SRSF2 | 21.0 | 18 |
| NR5 | F | 82 | polycytemia vera | M4 | + | - | - | + | - | der (18); trisomy 8 | wt | wt | JAK2, GATA2 | 32.5 | 19 |
| NR6 | M | 71 | chemotherapy | M4 | + | - | - | + | + | normal | wt | INS | KRAS, DNMT3A, TET2 | 104.0 | 2 |
| NR7 | M | 48 | relapse | M4 | + | nt | - | + | + | normal | ITD, TKD | INS | DNMT3A, IDH1 | 30.4 | 8 |
| NR8 | F | 86 | de novo | M0 | - | - | - | + | + | del (5q) | wt | wt | GATA2 | 249.0 | 17 |
| NR9 | M | 68 | MDS | M0 | - | - | - | - | + | normal | wt | wt | TET2, ASXL1, CEBPA, SRSF2, STAG2 | 1.5 | 24 |
| NR10 | F | 77 | de novo | M2 | + | - | - | + | - | normal | ITD | INS | DNMT3A | 77.8 | 17 |
| NR11 | F | 70 | MDS | M2 | nt | nt | + | + | + | del (12) | wt | wt | NRAS, KRAS, PTPN11, ASXL1, STAG2 | 81.0 | 21 |
| NR12 * | F | 70 | chemotherapy | M4 | + | - | + | + | - | normal | wt | INS | NRAS, DNMT3A, IDH1 | 73.7 | 7 |
| NR13 * | M | 60 | 2nd relapse | M4 | + | - | + | + | + | normal | ITD | wt | WT1 | 66.0 | 6 |
| NR14 * | M | 67 | 1st relapse | M1 | + | - | - | - | + | normal | TKD | wt | none | 15.6 | 73 |
| NR15 * | M | 68 | myelofibrosis | M1 | + | - | - | + | + | normal | wt | wt | KRAS | 34.3 | 56 |
| NR16 * | F | 53 | Li Fraumeni | M0 | + | - | - | + | - | multiple | wt | wt | TP53 | 16.2 | 28 |
| NR17 * | M | 74 | de novo | M0 | + | - | - | - | + | multiple | wt | wt | IDH2 | 13.3 | 112 |
| Gene | Protein Name | BP-GO 1 |
|---|---|---|
| ERG | ETS transcription factor ERG | Transcription by RNA polymerase II |
| NCBP2 | Nuclear cap binding protein subunit 2 | |
| NFKB2 | Nuclear factor kappa B subunit 2 | |
| SMAD4 | SMAD family member 4 | |
| CBFB | Core-binding factor subunit beta | |
| INTS12 | Integrator complex subunit 12 | |
| POLB | DNA polymerase beta | Cell death |
| NME3 | NME/NM23 nucleoside diphosphate kinase 3 | |
| CASP8 | Caspase 8 | |
| BRAT1 | BRCA1 associated ATM activator 1 | |
| RMDN3 | Regulator of microtubule dynamics 3 | |
| VAPA | VAMP associated protein A | |
| ARRB1 | Arrestin beta 1 | Both terms |
| MEF2D | Myocyte enhancer factor 2D |
| Gene | Protein Name | BP/MF-GO 1 |
|---|---|---|
| ALDOA | Aldolase, fructose-bisphosphate A | Small molecule catabolic process |
| BCKDHA | Branched chain keto acid dehydrogenase E1 subunit alpha | |
| PGM2L1 | Phosphoglucomutase 2 like 1 | |
| TRERF1 | Transcriptional regulating factor 1 | |
| SPTBN1 | Spectrin beta, non-erythrocytic 1 | Guanyl-nucleotide exchange factor activity |
| AKAP13 | A-kinase anchoring protein 13 | |
| SPTAN1 | Spectrin alpha, non-erythrocytic 1 | |
| RASGRP2 | RAS guanyl releasing protein 2 | |
| FLCN | Folliculin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Valladares, M.; Wangen, R.; Aasebø, E.; Reikvam, H.; Berven, F.S.; Selheim, F.; Bruserud, Ø. Proteomic Studies of Primary Acute Myeloid Leukemia Cells Derived from Patients Before and during Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid and Valproic Acid. Cancers 2021, 13, 2143. https://doi.org/10.3390/cancers13092143
Hernandez-Valladares M, Wangen R, Aasebø E, Reikvam H, Berven FS, Selheim F, Bruserud Ø. Proteomic Studies of Primary Acute Myeloid Leukemia Cells Derived from Patients Before and during Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid and Valproic Acid. Cancers. 2021; 13(9):2143. https://doi.org/10.3390/cancers13092143
Chicago/Turabian StyleHernandez-Valladares, Maria, Rebecca Wangen, Elise Aasebø, Håkon Reikvam, Frode S. Berven, Frode Selheim, and Øystein Bruserud. 2021. "Proteomic Studies of Primary Acute Myeloid Leukemia Cells Derived from Patients Before and during Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid and Valproic Acid" Cancers 13, no. 9: 2143. https://doi.org/10.3390/cancers13092143