
Ellagic Acid Resensitizes Gemcitabine-Resistant Bladder Cancer Cells by Inhibiting Epithelial-Mesenchymal Transition and Gemcitabine Transporters

 , ,
, , 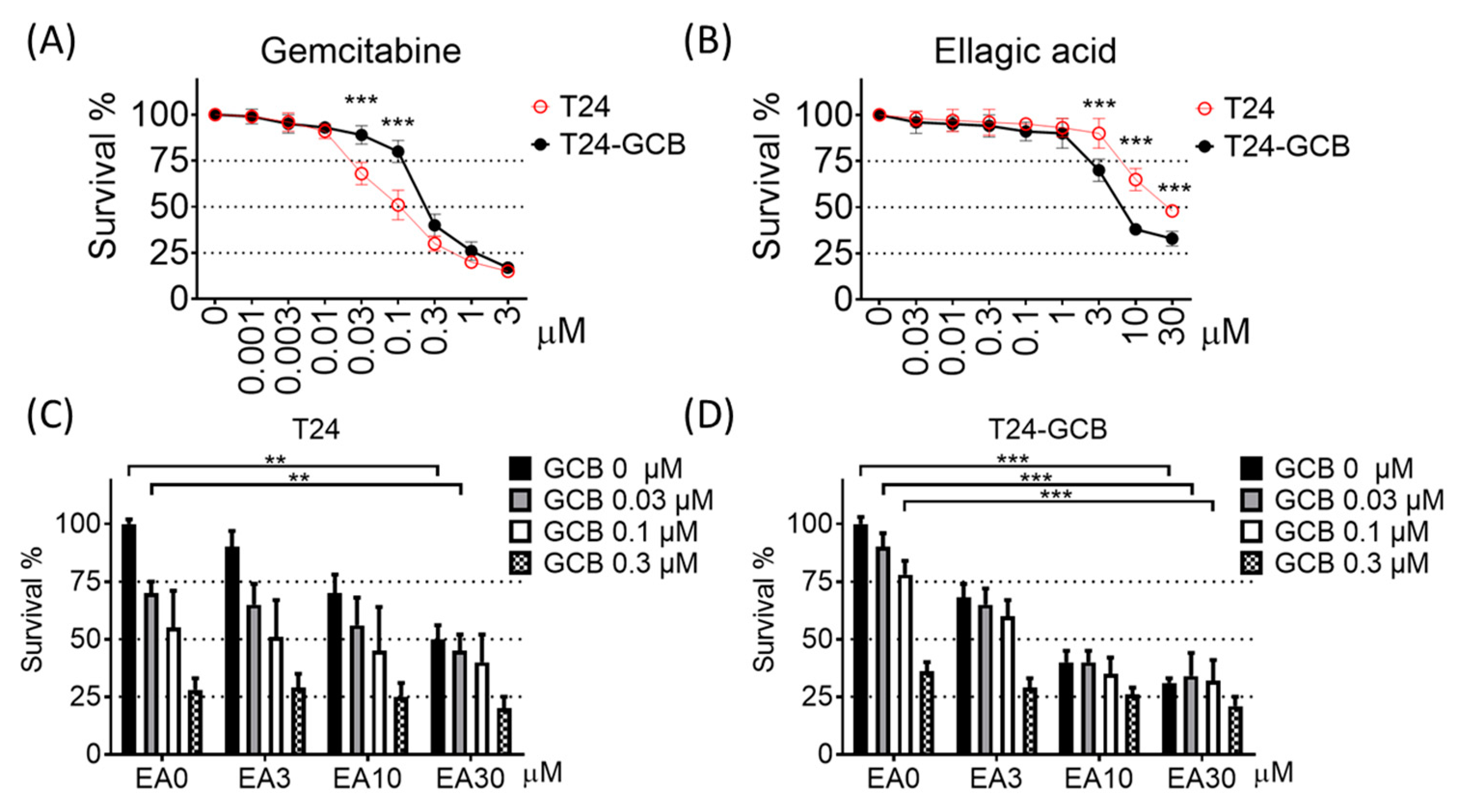{kind=link}
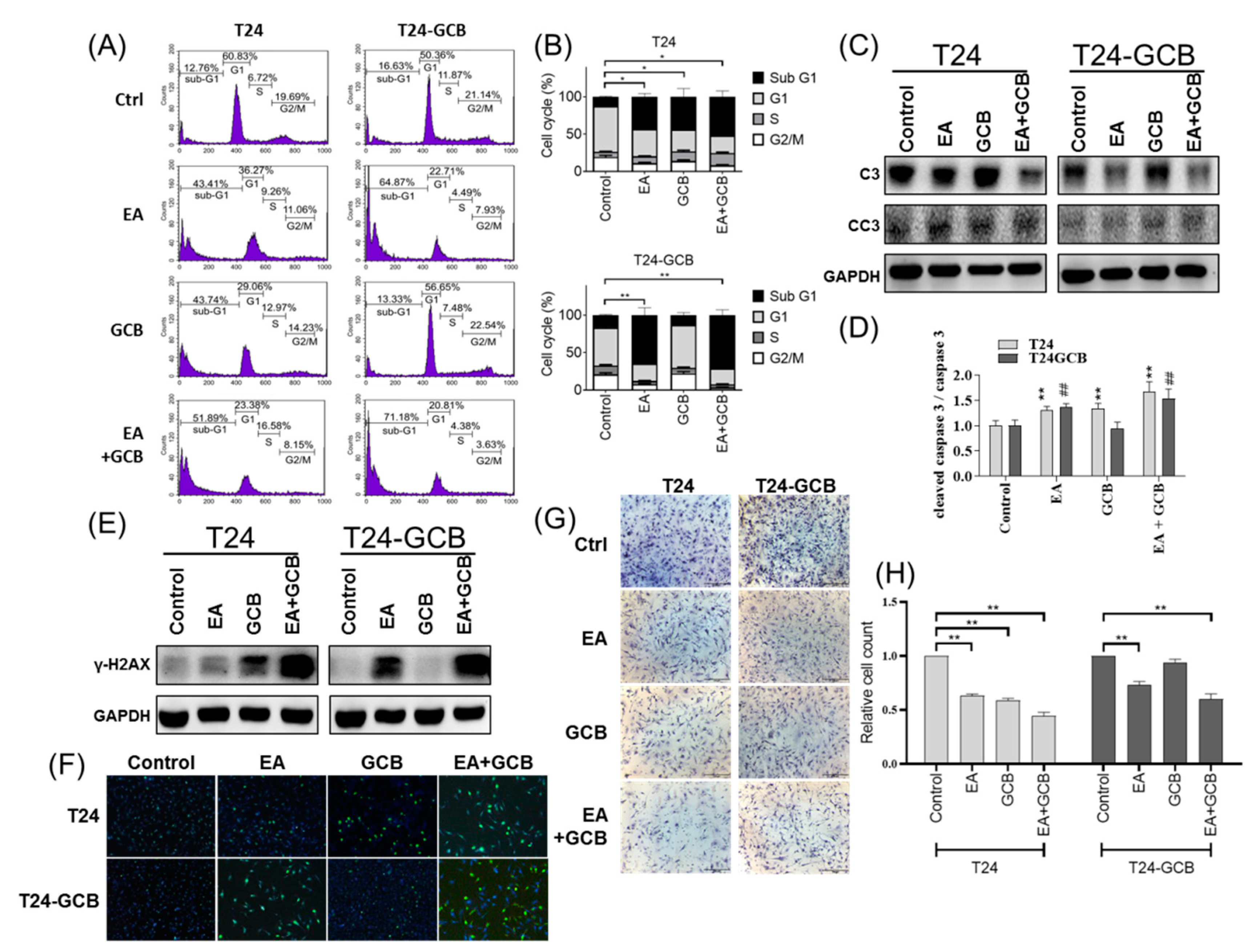{kind=link}
{kind=link}
{kind=link}
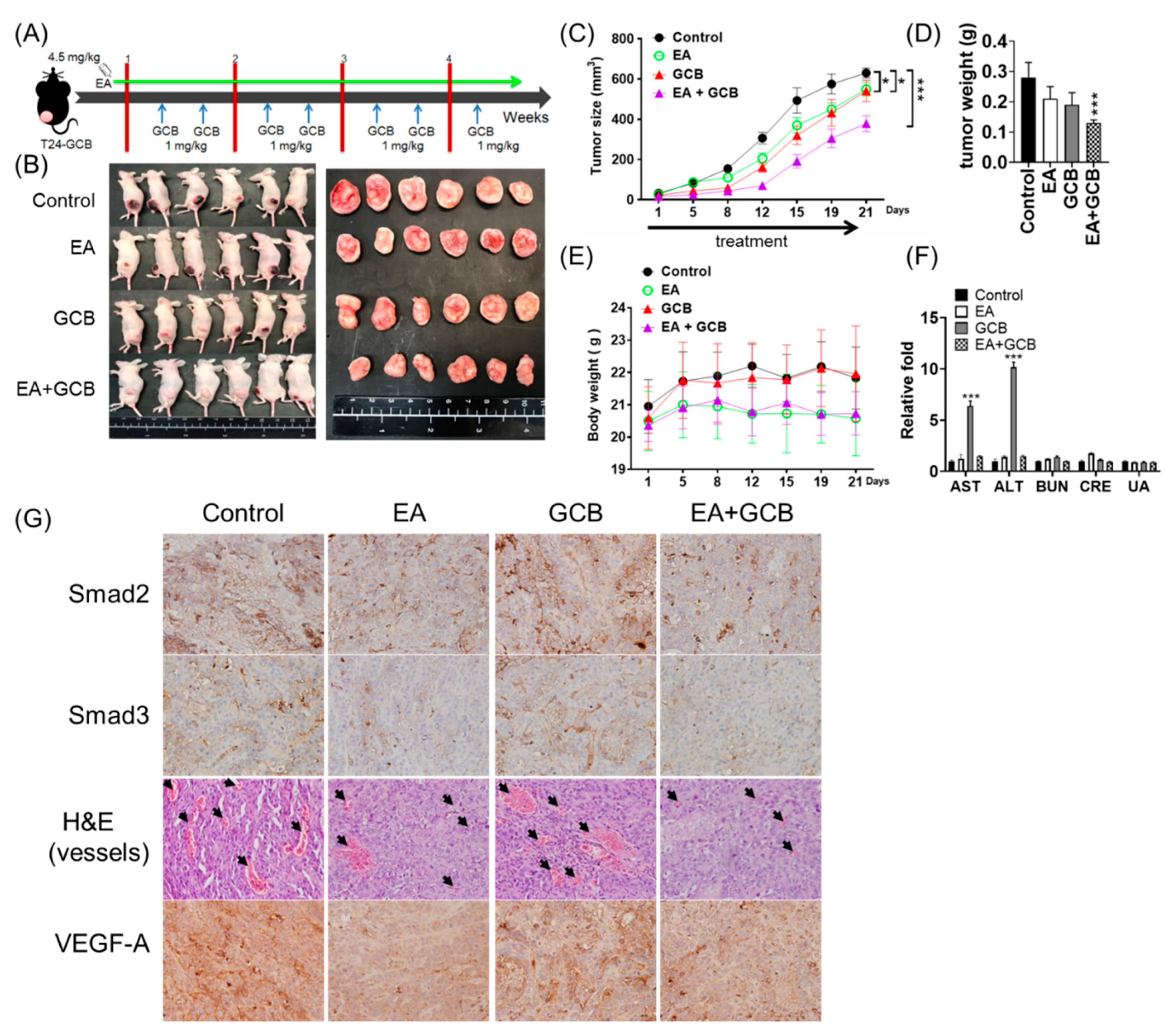{kind=link}
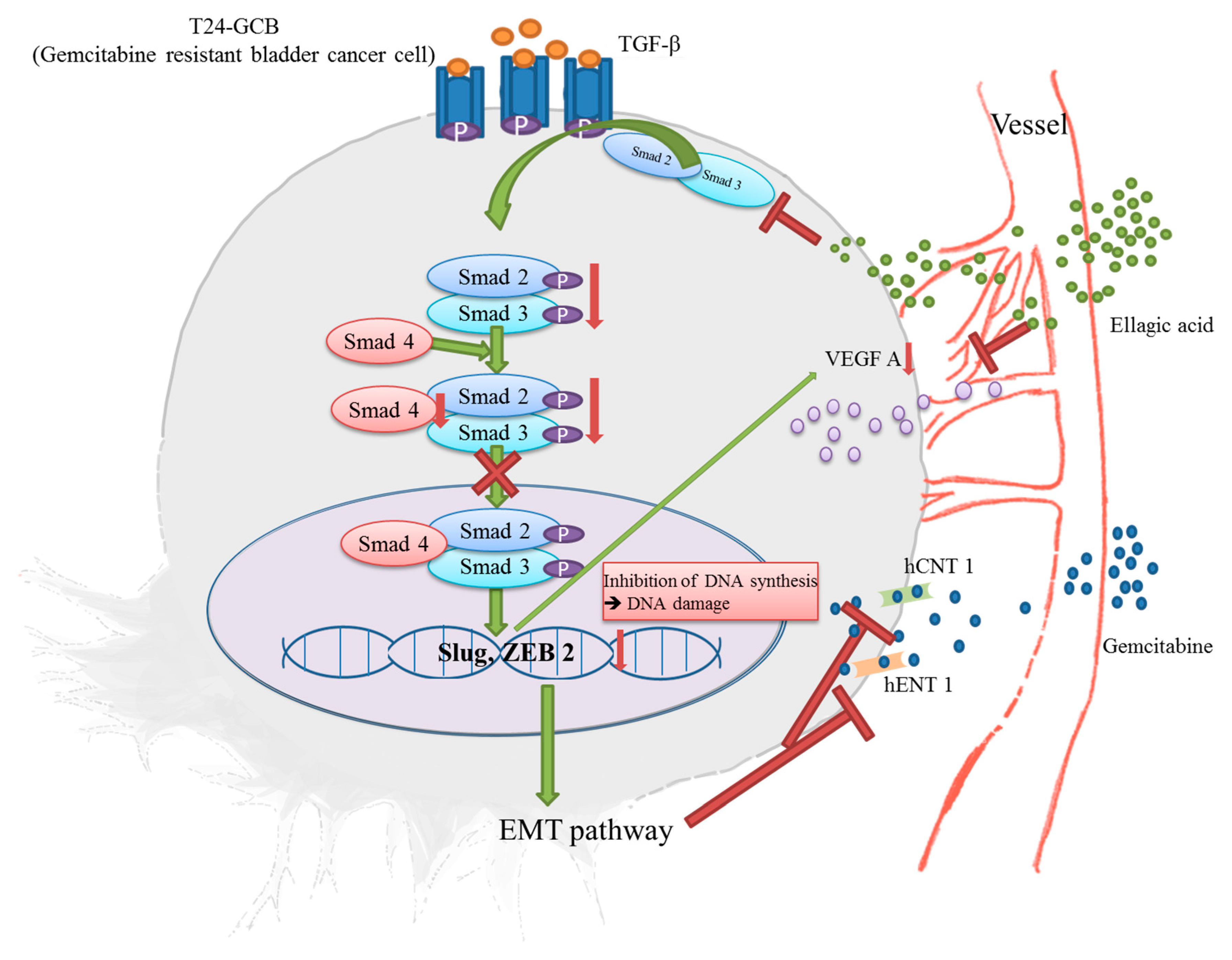{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. EA induces High Cytotoxicity of Gemcitabine in GCB-Resistant Cells
2.2. EA Combined with GCB Treatment Increases Apoptosis and Reduces Cell Motility in GCB Resistant Cells
2.3. EA resensitizes Bladder Cancer Cells to GCB by Reducing Epithelial-Mesenchymal Transition
2.4. EA Reduces EMT by Inhibiting the TGFβ-SMAD2/3 Upward Signaling Pathway
2.5. EA Inhibits the Growth of Bladder Cancer Tumors and Increases the In Vivo Inhibitory Effects of GCB on Tumors
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Viability Assay
4.3. Flow Cytometry
4.4. Cell Migration and Invasion Assays
4.5. Western Blotting
4.6. Quantitative Real-Time PCR
4.7. Xenograft and Treatment of EA and/or GCB
4.8. Immunohistochemistry and γ-H2AX Foci Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Rhijn, B.W.; Burger, M.; Lotan, Y.; Solsona, E.; Stief, C.G.; Sylvester, R.J.; Witjes, J.A.; Zlotta, A.R. Recurrence and progression of disease in non-muscle-invasive bladder cancer: From epidemiology to treatment strategy. Eur. Urol. 2009, 56, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, R.J.; Rodríguez, O.; Hernández, V.; Turturica, D.; Bauerová, L.; Bruins, H.M.; Bründl, J.; van der Kwast, T.H.; Brisuda, A.; Rubio-Briones, J.; et al. European Association of Urology (EAU) Prognostic Factor Risk Groups for Non-muscle-invasive Bladder Cancer (NMIBC) Incorporating the WHO 2004/2016 and WHO 1973 Classification Systems for Grade: An Update from the EAU NMIBC Guidelines Panel. Eur. Urol. 2021, 79, 480–488. [Google Scholar] [CrossRef] [PubMed]
- von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, J.; Kellie, S.J. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr. Hematol. Oncol. 2005, 22, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [Green Version]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of gemcitabine in cancer therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef]
- Zhang, H.M.; Zhao, L.; Li, H.; Xu, H.; Chen, W.W.; Tao, L. Research progress on the anticarcinogenic actions and mechanisms of ellagic acid. Cancer Biol. Med. 2014, 11, 92–100. [Google Scholar]
- Buxton, I.L. Inhibition of Nm23H2 gene product (NDPK-B) by angiostatin, polyphenols and nucleoside analogs. Proc. West Pharmacol. Soc. 2008, 51, 30–34. [Google Scholar] [PubMed]
- Park, S.W.; Kwon, M.J.; Yoo, J.Y.; Choi, H.J.; Ahn, Y.J. Antiviral activity and possible mode of action of ellagic acid identified in Lagerstroemia speciosa leaves toward human rhinoviruses. BMC Complement. Altern. Med. 2014, 14, 171. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, E.L.; Zografov, N.N.; Simeonova, L.S. Comparative study on the antioxidant capacities of synthetic influenza inhibitors and ellagic acid in model systems. Biomed. Pharmacother. 2016, 83, 755–762. [Google Scholar] [CrossRef]
- Pavlova, E.L.; Simeonova, L.S.; Gegova, G.A. Combined efficacy of oseltamivir, isoprinosine and ellagic acid in influenza A(H3N2)-infected mice. Biomed. Pharmacother. 2018, 98, 29–35. [Google Scholar] [CrossRef]
- Saiko, P.; Steinmann, M.T.; Schuster, H.; Graser, G.; Bressler, S.; Giessrigl, B.; Lackner, A.; Grusch, M.; Krupitza, G.; Bago-Horvath, Z.; et al. Epigallocatechin gallate, ellagic acid, and rosmarinic acid perturb dNTP pools and inhibit de novo DNA synthesis and proliferation of human HL-60 promyelocytic leukemia cells: Synergism with arabinofuranosylcytosine. Phytomedicine 2015, 22, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xie, J. Promising molecular mechanisms responsible for gemcitabine resistance in cancer. Genes. Dis. 2015, 2, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, H.J.; Wirth, T.; Beug, H. Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers (Basel) 2010, 2, 2058–2083. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarvepalli, D.; Rashid, M.U.; Rahman, A.U.; Ullah, W.; Hussain, I.; Hasan, B.; Jehanzeb, S.; Khan, A.K.; Jain, A.G.; Khetpal, N.; et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit. Rev. Oncog. 2019, 24, 199–212. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Di Cresce, C.; Figueredo, R.; Rytelewski, M.; Maleki Vareki, S.; Way, C.; Ferguson, P.J.; Vincent, M.D.; Koropatnick, J. siRNA knockdown of mitochondrial thymidine kinase 2 (TK2) sensitizes human tumor cells to gemcitabine. Oncotarget 2015, 6, 22397–22409. [Google Scholar] [CrossRef] [Green Version]
- Weadick, B.; Nayak, D.; Persaud, A.K.; Hung, S.W.; Raj, R.; Campbell, M.J.; Chen, W.; Li, J.; Williams, T.M.; Govindarajan, R. EMT-Induced Gemcitabine Resistance in Pancreatic Cancer Involves the Functional Loss of Equilibrative Nucleoside Transporter 1. Mol. Cancer Ther. 2021, 20, 410–422. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceci, C.; Lacal, P.M.; Tentori, L.; De Martino, M.G.; Miano, R.; Graziani, G. Experimental Evidence of the Antitumor, Antimetastatic and Antiangiogenic Activity of Ellagic Acid. Nutrients 2018, 10, 1756. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Dudas, J.; Ladanyi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algül, H.; Treiber, M.; Lesina, M.; Schmid, R.M. Mechanisms of disease: Chronic inflammation and cancer in the pancreas-A potential role for pancreatic stellate cells? Nat. Clin. Pract. Gastroenterol. Hepatol. 2007, 4, 454. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Yin, H.; Tong, H.; Zhan, K.; Yang, G.; Hossain, M.A.; Li, T.; Gou, X.; He, W. Nucleolar and Spindle Associated Protein 1 (NUSAP1) Promotes Bladder Cancer Progression through the TGF-beta Signaling Pathway. OncoTargets Ther. 2020, 13, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.; Shen, L.; Yang, L.; Huang, X.; Lu, Q.; Cui, Y.; Zheng, X.; Zhao, X.; Zhang, D.; Huang, R.; et al. TGFβ1 Promotes Gemcitabine Resistance through Regulating the LncRNA-LET/NF90/miR-145 Signaling Axis in Bladder Cancer. Theranostics 2017, 7, 3053–3067. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, G.; Yu, D.; Zhu, M. PKCalpha-induced drug resistance in pancreatic cancer cells is associated with transforming growth factor-beta1. J. Exp. Clin. Cancer Res. 2010, 29, 104. [Google Scholar] [CrossRef] [Green Version]
- Xuan, Y.Z.; Jin, C.R.; Yang, K.J. TGF-beta downregulation overcomes gemcitabine resistance in oral squamous cell carcinoma. Cancer Biomark. 2020, 29, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Yamada, D.; Kobayashi, S.; Wada, H.; Kawamoto, K.; Marubashi, S.; Eguchi, H.; Ishii, H.; Nagano, H.; Doki, Y.; Mori, M. Role of crosstalk between interleukin-6 and transforming growth factor-beta 1 in epithelial-mesenchymal transition and chemoresistance in biliary tract cancer. Eur. J. Cancer 2013, 49, 1725–1740. [Google Scholar] [CrossRef] [PubMed]
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. TGF-beta-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016, 37, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Tang, S.N.; Marsh, J.L.; Shankar, S.; Srivastava, R.K. Ellagic acid inhibits human pancreatic cancer growth in Balb c nude mice. Cancer Lett. 2013, 337, 210–217. [Google Scholar] [CrossRef]
- Cheng, H.; Lu, C.; Tang, R.; Pan, Y.; Bao, S.; Qiu, Y.; Xie, M. Ellagic acid inhibits the proliferation of human pancreatic carcinoma PANC-1 cells in vitro and in vivo. Oncotarget 2017, 8, 12301–12310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Chen, Q.; Tan, Y.; Liu, B.; Liu, C. Ellagic acid inhibits human glioblastoma growth in vitro and in vivo. Oncol. Rep. 2017, 37, 1084–1092. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.S.; Bai, M.H.; Zhang, T.; Li, G.D.; Liu, M. Ellagic acid induces cell cycle arrest and apoptosis through TGF-beta/Smad3 signaling pathway in human breast cancer MCF-7 cells. Int. J. Oncol. 2015, 46, 1730–1738. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, H.S.; Wang, L.F.; Bai, M.H.; Wang, Y.C.; Jiang, X.F.; Liu, M. Ellagic acid exerts anti-proliferation effects via modulation of Tgf-beta/Smad3 signaling in MCF-7 breast cancer cells. Asian Pac. J. Cancer Prev. 2014, 15, 273–276. [Google Scholar] [CrossRef]
- Vanella, L.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Cardile, V.; Kim, D.H.; Abraham, N.G.; Sorrenti, V. Apoptotic markers in a prostate cancer cell line: Effect of ellagic acid. Oncol. Rep. 2013, 30, 2804–2810. [Google Scholar] [CrossRef]
- Ricart, A.D. Drug-induced liver injury in Oncology. Ann. Oncol. 2017, 28, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, R.; Sepand, M.R.; Seyednejad, S.A.; Omidi, A.; Akbariani, M.; Gholami, M.; Sabzevari, O. Ellagic acid reduces methotrexate-induced apoptosis and mitochondrial dysfunction via up-regulating Nrf2 expression and inhibiting the IqBalpha/NFqB in rats. DARU J. Pharm. Sci. 2019, 27, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Mehrzadi, S.; Mehrabani, M.; Malayeri, A.R.; Bakhshayesh, M.; Kalantari, H.; Goudarzi, M. Ellagic acid as a potential antioxidant, alleviates methotrexate-induced hepatotoxicity in male rats. Acta Chir. Belg. 2019, 119, 69–77. [Google Scholar] [CrossRef]
- Stojiljković, N.; Ilić, S.; Stojanović, N.; Janković-Veličković, L.; Stojnev, S.; Kocić, G.; Radenković, G.; Arsić, I.; Stojanović, M.; Petković, M. Nanoliposome-encapsulated ellagic acid prevents cyclophosphamide-induced rat liver damage. Mol. Cell Biochem. 2019, 458, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Goyal, Y.; Koul, A.; Ranawat, P. Ellagic acid ameliorates cisplatin induced hepatotoxicity in colon carcinogenesis. Environ. Toxicol. 2019, 34, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Yüce, A.; Ateşşahin, A.; Ceribaşi, A.O.; Aksakal, M. Ellagic acid prevents cisplatin-induced oxidative stress in liver and heart tissue of rats. Basic Clin. Pharmacol. Toxicol. 2007, 101, 345–349. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-S.; Ho, J.-Y.; Yu, C.-P.; Cho, C.-J.; Wu, C.-L.; Huang, C.-S.; Gao, H.-W.; Yu, D.-S. Ellagic Acid Resensitizes Gemcitabine-Resistant Bladder Cancer Cells by Inhibiting Epithelial-Mesenchymal Transition and Gemcitabine Transporters. Cancers 2021, 13, 2032. https://doi.org/10.3390/cancers13092032
Wu Y-S, Ho J-Y, Yu C-P, Cho C-J, Wu C-L, Huang C-S, Gao H-W, Yu D-S. Ellagic Acid Resensitizes Gemcitabine-Resistant Bladder Cancer Cells by Inhibiting Epithelial-Mesenchymal Transition and Gemcitabine Transporters. Cancers. 2021; 13(9):2032. https://doi.org/10.3390/cancers13092032
Chicago/Turabian StyleWu, Ying-Si, Jar-Yi Ho, Cheng-Ping Yu, Chun-Jung Cho, Chia-Lun Wu, Cheng-Shuo Huang, Hong-Wei Gao, and Dah-Shyong Yu. 2021. "Ellagic Acid Resensitizes Gemcitabine-Resistant Bladder Cancer Cells by Inhibiting Epithelial-Mesenchymal Transition and Gemcitabine Transporters" Cancers 13, no. 9: 2032. https://doi.org/10.3390/cancers13092032