
Anti-PD-1/PD-L1 Based Combination Immunotherapy to Boost Antigen-Specific CD8+ T Cell Response in Hepatocellular Carcinoma

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
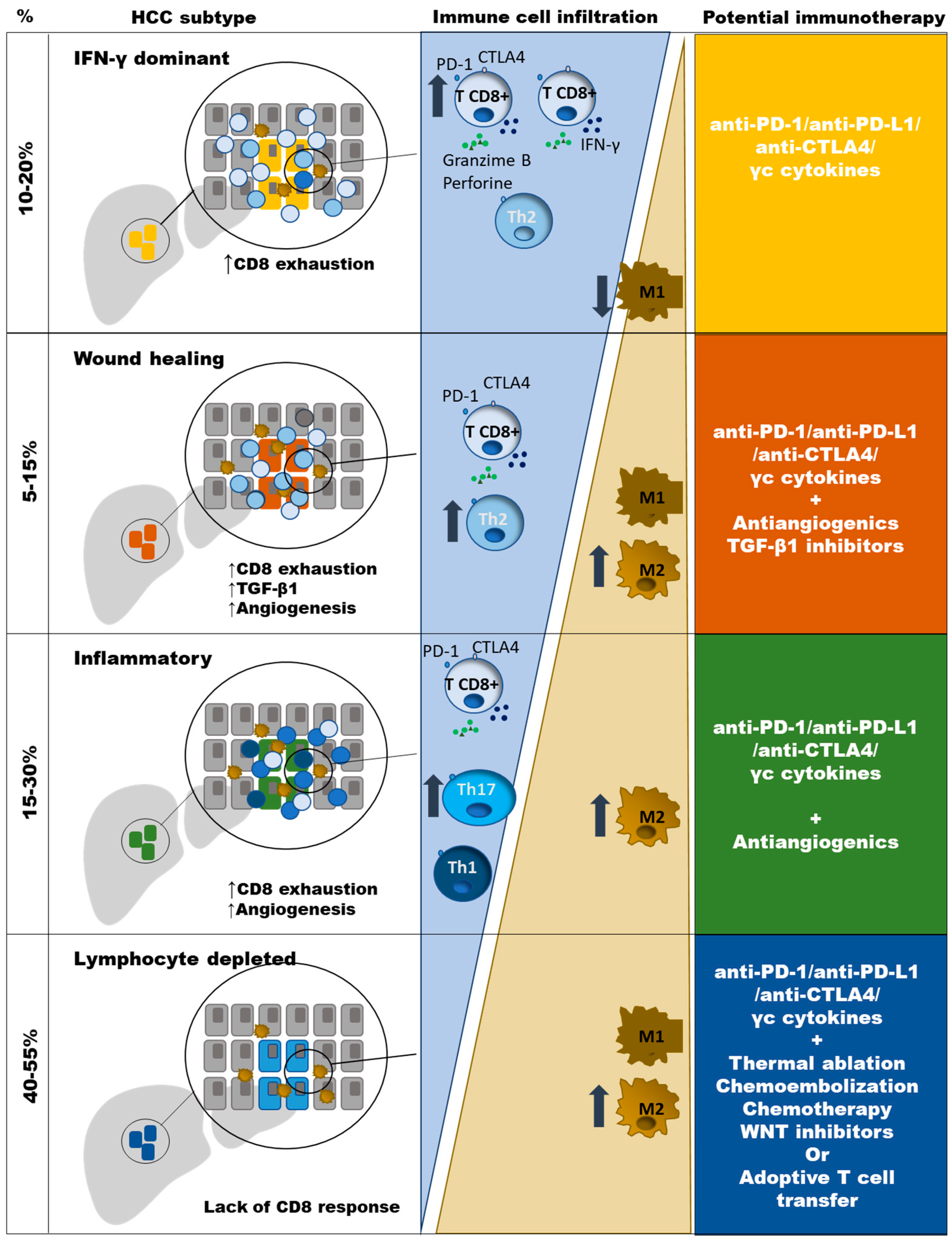
2. Types of HCC According to Potential Sensitivity to PD-1 Based Immunotherapy
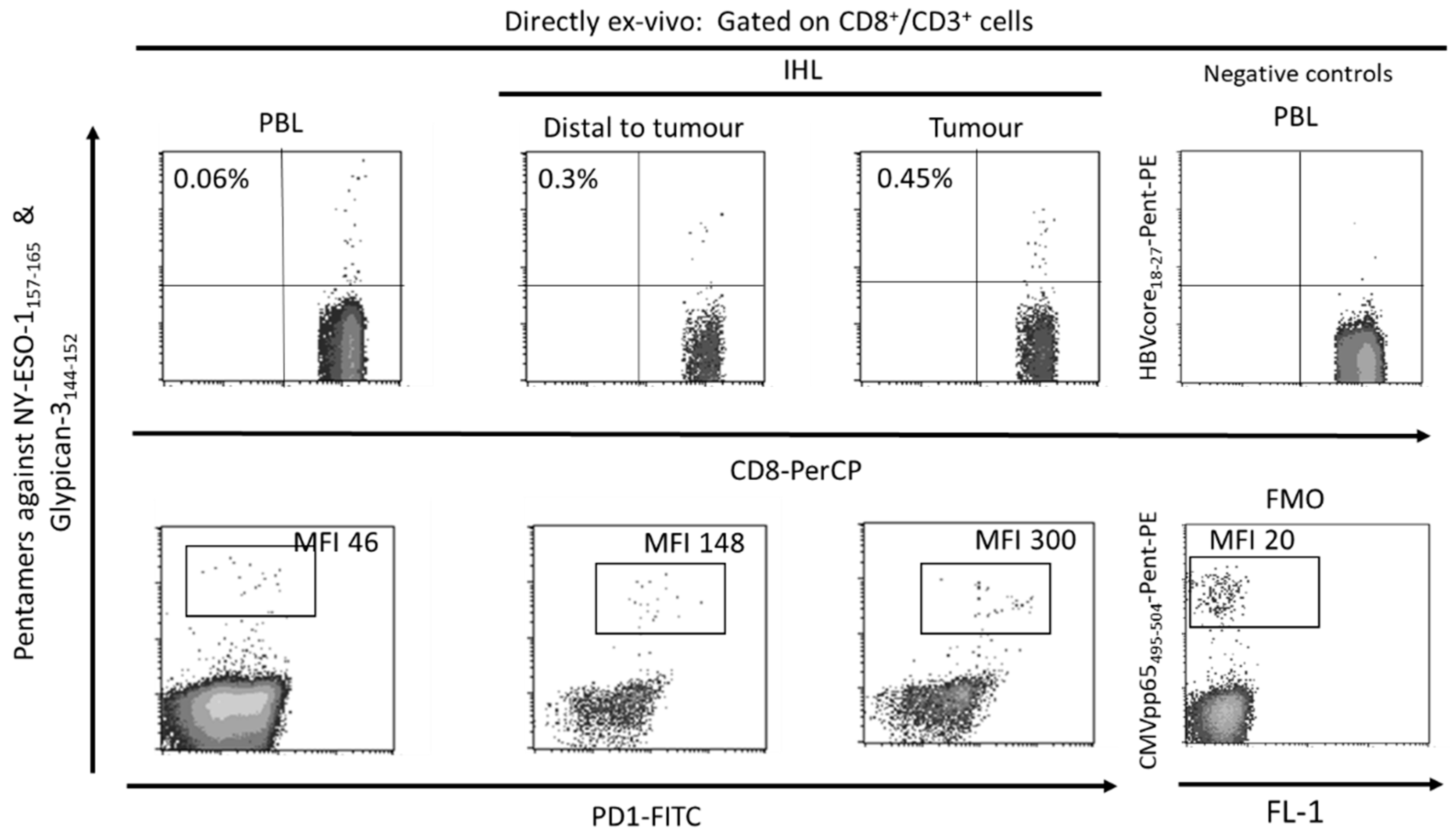
3. Role of PD-1-Expressing HCC-Specific CD8 T Cell Response in HCC Control
4. Targeting the Progenitor HCC-Specific CD8 T Cell Pool for Immunomodulation
5. PD-1 Mechanisms Involved in CD8 T Cell Exhaustion
6. Results of PD-1 Blocking Based Combination Therapy on CD8+ Cells in HCC
6.1. PD-1/PD-L1 Blockade Monotherapy
6.2. PD-1/PD-L1 Blockade Plus Antiangiogenics
6.3. PD-1/PD-L1 Blockade Plus Anti-TGF-β1
6.4. PD-1/PD-L1 Blockade Plus Other Immune Checkpoint Modulators
6.5. PD-1/PD-L1 Blockade Plus Strategies to Increase HCC Antigen Presentation
6.6. PD-1/PD-L1 Blockade Plus γc Cytokines
6.7. PD-1/PD-L1 Blockade Plus Other Immunotherapies
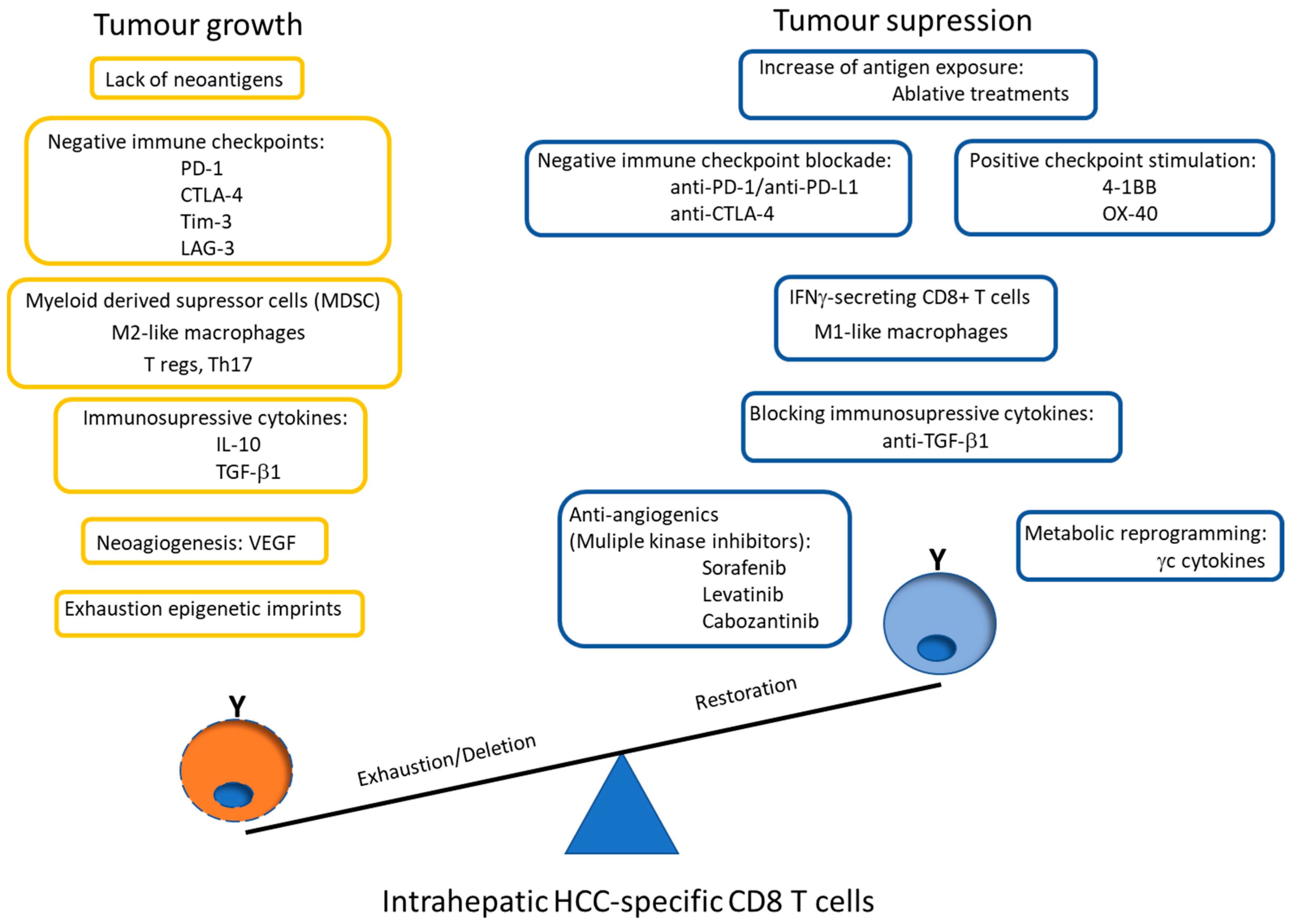
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Hartke, J.; Johnson, M.; Ghabril, M. The diagnosis and treatment of hepatocellular carcinoma. Semin. Diagn. Pathol. 2017, 34, 153–159. [Google Scholar] [CrossRef]
- Li, S.; Yang, F.; Ren, X. Immunotherapy for hepatocellular carcinoma. Drug Discov. Ther. 2015, 9, 363–371. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- El Dika, I.; Khalil, D.N.; Abou-Alfa, G.K. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer 2019, 125, 3312–3319. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Wherry, E.J.; Ahmed, R.; Sharpe, A.H. Reinvigorating exhausted HIV-specific T cells via PD-1–PD-1 ligand blockade. J. Exp. Med. 2006, 203, 2223–2227. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8 + T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.-L.; Hsu, C.; Chan, S.L.; Choo, S.-P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inada, Y.; Mizukoshi, E.; Seike, T.; Tamai, T.; Iida, N.; Kitahara, M.; Yamashita, T.; Arai, K.; Terashima, T.; Fushimi, K.; et al. Characteristics of Immune Response to Tumor-Associated Antigens and Immune Cell Profile in Patients with Hepatocellular Carcinoma. Hepatology 2019, 69, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.; Wolf, D.; Bortone, D.S.; Ouyang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilmi, M.; Neuzillet, C.; Calderaro, J.; Lafdil, F.; Pawlotsky, J.-M.; Rousseau, B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: Current knowledge and future research directions. J. Immunother. Cancer 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Roth, G.S.; Decaens, T. Liver immunotolerance and hepatocellular carcinoma: Patho-physiological mechanisms and therapeutic perspectives. Eur. J. Cancer 2017, 87, 101–112. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-D.; Song, G.-W.; Park, S.; Jung, M.K.; Kim, M.H.; Kang, H.J.; Yoo, C.; Yi, K.; Kim, K.H.; Eo, S.; et al. Association between expression level of PD1 by tumor-infiltrating CD8+ T cells and features of hepatocellular carcinoma. Gastroenterology 2018, 155, 1936.e17–1950.e17. [Google Scholar] [CrossRef]
- Mühlbauer, M.; Fleck, M.; Schütz, C.; Weiss, T.; Froh, M.; Blank, C.; Schölmerich, J.; Hellerbrand, C. PD-L1 is induced in hepatocytes by viral infection and by interferon-α and -γ and mediates T cell apoptosis. J. Hepatol. 2006, 45, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Wang, X.-Y.; Qiu, S.-J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.-Z.; Shi, Y.-H.; Xiao, Y.-S.; et al. Overexpression of PD-L1 Significantly Associates with Tumor Aggressiveness and Postoperative Recurrence in Human Hepatocellular Carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Gabrielson, A.; Wu, Y.; Wang, H.; Jiang, J.; Kallakury, B.; Gatalica, Z.; Reddy, S.; Kleiner, D.; Fishbein, T.; Johnson, L.; et al. Intratumoral CD3 and CD8 T-cell Densities Associated with Relapse-Free Survival in HCC. Cancer Immunol. Res. 2016, 4, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.-S.; Gu, X.; Xiong, W.; Guo, W.; Han, L.; Bai, Y.; Peng, C.; Cui, M.; Xie, M. Increased programmed death ligand-1 expression predicts poor prognosis in hepatocellular carcinoma patients. OncoTargets Ther. 2016, 9, 4805–4813. [Google Scholar] [CrossRef] [Green Version]
- Gandini, S.; Massi, D.; Mandalà, M. PD-L1 expression in cancer patients receiving anti PD-1/PD-L1 antibodies: A systematic review and meta-analysis. Crit. Rev. Oncol. 2016, 100, 88–98. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef]
- Yarchoan, M.; Xing, D.; Luan, L.; Xu, H.; Sharma, R.B.; Popovic, A.; Pawlik, T.M.; Kim, A.K.; Zhu, Q.; Jaffee, E.M.; et al. Characterization of the Immune Microenvironment in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 7333–7339. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauber, C.; Schultheiss, M.; De Luca, R.; Buettner, N.; Llewellyn-Lacey, S.; Emmerich, F.; Zehe, S.; Price, D.A.; Neumann-Haefelin, C.; Schmitt-Graeff, A.; et al. Inefficient induction of circulating TAA-specific CD8+ T-cell responses in hepatocellular carcinoma. Oncotarget 2019, 10, 5194–5206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.J.; Lee, Y.H.; Pan, L.; Lai, L.; Chua, C.; Wasser, M.; Lim, T.K.H.; Yeong, J.; Toh, H.C.; Lee, S.Y.; et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019, 68, 916–927. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Eynde, B.J.V.D.; van der Bruggen, P. T cell defined tumor antigens. Curr. Opin. Immunol. 1997, 9, 684–693. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, Y.; Nakashima, O.; Kutami, R.; Yamamoto, O.; Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 1998, 27, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Ueda, T.; Arihara, F.; Kagaya, T.; Yamashita, T.; Fushimi, K.; Kaneko, S. Enhancement of tumor-associated antigen-specific T cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology 2013, 57, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Chang, B.; Shen, L.; Wang, K.; Jin, J.; Huang, T.; Chen, Q.; Li, W.; Wu, P. High number of PD-1 positive intratumoural lymphocytes predicts survival benefit of cytokine-induced killer cells for hepatocellular carcinoma patients. Liver Int. 2018, 38, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.; Flecken, T.; Thimme, R. Tumor-associated antigen specific CD8+T cells in hepatocellular carcinoma-a promising target for immunotherapy. OncoImmunology 2014, 3, e954919. [Google Scholar] [CrossRef] [Green Version]
- Gehring, A.J.; Ho, Z.Z.; Tan, A.T.; Aung, M.O.; Lee, K.H.; Tan, K.C.; Lim, S.G.; Bertoletti, A. Profile of Tumor Antigen-Specific CD8 T Cells in Patients with Hepatitis B Virus-Related Hepatocellular Carcinoma. Gastroenterology 2009, 137, 682–690. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, R.R.; Im, S.J.; Hu, B.; Hashimoto, M.; Li, P.; Lin, J.-X.; Leonard, W.J.; Greenleaf, W.J.; Ahmed, R.; Goronzy, J.J. Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 14113–14118. [Google Scholar] [CrossRef] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; Van Der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Sprengers, D.; Boor, P.P.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e10. [Google Scholar] [CrossRef]
- Yu, M.-C.; Chen, C.-H.; Liang, X.; Wang, L.; Gandhi, C.R.; Fung, J.J.; Lu, L.; Qian, S. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology 2004, 40, 1312–1321. [Google Scholar] [CrossRef]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2007, 47, 296–305. [Google Scholar] [CrossRef]
- Kassel, R.; Cruise, M.W.; Iezzoni, J.C.; Taylor, N.A.; Pruett, T.L.; Hahn, Y.S. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology 2009, 50, 1625–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer Cell Suppression of CD8+ T Cells in Human Hepatocellular Carcinoma Is Mediated by B7-H1/Programmed Death-1 Interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [Green Version]
- Mocan, T.; Sparchez, Z.; Craciun, R.; Bora, C.N.; Leucuta, D.C. Programmed cell death protein-1 (PD-1)/programmed death-ligand-1 (PD-L1) axis in hepatocellular carcinoma: Prognostic and therapeutic perspectives. Clin. Transl. Oncol. 2019, 21, 702–712. [Google Scholar] [CrossRef]
- Shi, F.; Shi, M.; Zeng, Z.; Qi, R.-Z.; Liu, Z.-W.; Zhang, J.-Y.; Yang, Y.-P.; Tien, P.; Wang, F.-S. PD-1 and PD-L1 upregulation promotes CD8+ T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2010, 128, 887–896. [Google Scholar] [CrossRef]
- Otano, I.; Escors, D.; Schurich, A.; Singh, H.; Robertson, F.; Davidson, B.R.; Fusai, G.; Vargas, F.A.; Tan, Z.M.; Aw, J.Y.; et al. Molecular Recalibration of PD-1+ Antigen-Specific T Cells from Blood and Liver. Mol. Ther. 2018, 26, 2553–2566. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; He, Q.; Shen, H.; Xia, A.; Tian, W.; Yu, W.; Sun, B. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 2019, 71, 731–741. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.-M.; Zhang, Z.; Hsu, J.L.; Li, C.-W.; Lim, S.-O.; Sheng, Y.-Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Freitas-Lopes, M.A.; Mafra, K.; David, B.A.; Carvalho-Gontijo, R.; Menezes, G.B. Differential Location and Distribution of Hepatic Immune Cells. Cells 2017, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 Macrophages Induce PD-L1 Expression in Hepatocellular Carcinoma Cells Through IL-1β Signaling. Front. Immunol. 2019, 10, 1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, J.; Yu, G.; Hu, P.; Chao, Y.; Li, X.; Chen, X.; Wei, Q.; Wang, J. PD-1 expression is elevated in monocytes from hepatocellular carcinoma patients and contributes to CD8 T cell suppression. Immunol. Res. 2020, 68, 436–444. [Google Scholar] [CrossRef]
- Yasuoka, H.; Asai, A.; Ohama, H.; Tsuchimoto, Y.; Fukunishi, S.; Higuchi, K. Increased both PD–L1 and PD–L2 expressions on monocytes of patients with hepatocellular carcinoma was associated with a poor prognosis. Sci. Rep. 2020, 10, 10377. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Q.; Xu, J.; Zhou, Z.-G.; Jin, L.-L.; Yu, X.-J.; Xiao, G.; Lin, J.; Zhuang, S.-M.; Zhang, Y.-J.; Zheng, L. Expression patterns of programmed death ligand 1 correlate with different microenvironments and patient prognosis in hepatocellular carcinoma. Br. J. Cancer 2018, 119, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Wang, Y.; Luo, G.-Y.; Han, F.; Li, Y.-Q.; Zhou, Z.-G.; Xu, G.-L. Relationship Between PD-L1 Expression and CD8+ T-cell Immune Responses in Hepatocellular Carcinoma. J. Immunother. 2017, 40, 323–333. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nat. Cell Biol. 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Kim, H.; Park, S.; Jeong, S.; Lee, Y.J.; Lee, H.; Kim, C.G.; Kim, K.H.; Hong, S.; Lee, J.; Kim, S.; et al. 4-1BB Delineates Distinct Activation Status of Exhausted Tumor-Infiltrating CD8 + T Cells in Hepatocellular Carcinoma. Hepatology 2020, 71, 955–971. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, H.; Wei, M.; Mou, T.; Shi, T.; Ma, Y.; Cai, X.; Li, Y.; Dong, J.; Wei, J. Recombinant Adenovirus Expressing a Soluble Fusion Protein PD-1/CD137L Subverts the Suppression of CD8+ T Cells in HCC. Mol. Ther. 2019, 27, 1906–1918. [Google Scholar] [CrossRef]
- Chew, V.; Lai, L.; Pan, L.; Lim, C.J.; Li, J.; Chong, T.H.; Chua, C.; Leong, J.Y.; Lim, K.H.; Toh, H.C.; et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc. Natl. Acad. Sci. USA 2017, 114, E5900–E5909. [Google Scholar] [CrossRef] [Green Version]
- Escobar, G.; Mangani, D.; Anderson, A.C. T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 2020, 5, eabb9726. [Google Scholar] [CrossRef]
- Abdelsamed, H.A.; Zebley, C.C.; Youngblood, B. Epigenetic Maintenance of Acquired Gene Expression Programs during Memory CD8 T Cell Homeostasis. Front. Immunol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Reading, J.L.; Gálvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8+T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Borsa, M.; Barnstorf, I.; Baumann, N.S.; Pallmer, K.; Yermanos, A.; Gräbnitz, F.; Barandun, N.; Hausmann, A.; Sandu, I.; Barral, Y.; et al. Modulation of asymmetric cell division as a mechanism to boost CD8+ T cell memory. Sci. Immunol. 2019, 4, eaav1730. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Kratchmarov, R.; Lin, W.-H.W.; Rothman, N.J.; Yen, B.; Adams, W.C.; Nish, S.A.; Rathmell, J.C.; Reiner, S.L. Asymmetric PI3K Activity in Lymphocytes Organized by a PI3K-Mediated Polarity Pathway. Cell Rep. 2018, 22, 860–868. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Li, Y.; Tang, H.; Liu, W.; Chen, Y.; Dai, T.; Liang, R.; Shi, M.; Yi, S.; Chen, G.; et al. CD8+CXCR5+T cells infiltrating hepatocellular carcinomas are activated and predictive of a better prognosis. Aging 2019, 11, 8879–8891. [Google Scholar] [CrossRef]
- Jin, Y.; Lang, C.; Tang, J.; Geng, J.; Song, H.K.; Sun, Z.; Wang, J. CXCR5 + CD8 + T cells could induce the death of tumor cells in HBV-related hepatocellular carcinoma. Int. Immunopharmacol. 2017, 53, 42–48. [Google Scholar] [CrossRef]
- Panjideh, H.; Müller, G.; Koch, M.; Wilde, F.; Scheu, S.; Moldenhauer, G.; Lipp, M. Immunotherapy of B-cell non-Hodgkin lymphoma by targeting the chemokine receptor CXCR5 in a preclinical mouse model. Int. J. Cancer 2014, 135, 2623–2632. [Google Scholar] [CrossRef]
- Garnelo, M.; Tan, A.; Her, Z.; Yeong, J.; Lim, C.J.; Chen, J.; Lim, K.H.; Weber, A.; Chow, P.; Chung, A.; et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017, 66, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Adams, W.C.; Chen, Y.-H.; Kratchmarov, R.; Yen, B.; Nish, S.A.; Lin, W.-H.W.; Rothman, N.J.; Luchsinger, L.L.; Klein, U.; Busslinger, M.; et al. Anabolism-Associated Mitochondrial Stasis Driving Lymphocyte Differentiation over Self-Renewal. Cell Rep. 2016, 17, 3142–3152. [Google Scholar] [CrossRef]
- Lin, W.-H.W.; Nish, S.A.; Yen, B.; Chen, Y.-H.; Adams, W.C.; Kratchmarov, R.; Rothman, N.J.; Bhandoola, A.; Xue, H.-H.; Reiner, S.L. CD8 + T Lymphocyte Self-Renewal during Effector Cell Determination. Cell Rep. 2016, 17, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [Green Version]
- Van Der Windt, G.J.W.; Everts, B.; Chang, C.-H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8+ T Cell Memory Development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T Cell Co-receptors for Cancer Therapy. Immunity 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Hoos, A. Development of immuno-oncology drugs-from CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, H.; Matsuda, T.; Takiguchi, M. Differentiation of Human CD8+T Cells from a Memory to Memory/Effector Phenotype. J. Immunol. 2002, 168, 5538–5550. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Sugiura, D.; Shimizu, K.; Maruhashi, T.; Watada, M.; Okazaki, I.-M.; Okazaki, T. PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front. Immunol. 2019, 10, 630. [Google Scholar] [CrossRef] [Green Version]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Cañas, M.; Melero, I.; Delgoffe, G.M. Metabolic Consequences of T-cell Costimulation in Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaseb, A.O.; Pestana, R.C.; Vence, L.M.; Blando, J.M.; Singh, S.; Ikoma, N.; Vauthey, J.-N.; Allison, J.P.; Sharma, P. Randomized, open-label, perioperative phase II study evaluating nivolumab alone versus nivolumab plus ipilimumab in patients with resectable HCC. J. Clin. Oncol. 2019, 37, 185. [Google Scholar] [CrossRef]
- Moreno-Cubero, E.; Larrubia, J.-R. Specific CD8+T cell response immunotherapy for hepatocellular carcinoma and viral hepatitis. World J. Gastroenterol. 2016, 22, 6469–6483. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Kaseb, A.O.; Vence, L.; Blando, J.; Yadav, S.S.; Ikoma, N.; Pestana, R.C.; Vauthey, J.N.; Allison, J.P.; Sharma, P. Immunologic Correlates of Pathologic Complete Response to Preoperative Immunotherapy in Hepatocellular Carcinoma. Cancer Immunol. Res. 2019, 7, 1390–1395. [Google Scholar] [CrossRef]
- Zhang, W.; Song, Z.; Xiao, J.; Liu, X.; Luo, Y.; Yang, Z.; Luo, R.; Li, A. Blocking the PD-1/PD-L1 axis in dendritic cell-stimulated Cytokine-Induced Killer Cells with pembrolizumab enhances their therapeutic effects against hepatocellular carcinoma. J. Cancer 2019, 10, 2578–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berretta, M.; Rinaldi, L.; Di Benedetto, F.; Lleshi, A.; De Re, V.; Facchini, G.; De Paoli, P.; Di Francia, R. Angiogenesis Inhibitors for the Treatment of Hepatocellular Carcinoma. Front. Pharmacol. 2016, 7, 428. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-Y.; Tan, C.-T.; Chen, C.-W.; Ou, D.-L.; Cheng, A.-L.; Hsu, C. Immunomodulatory Effects of Current Targeted Therapies on Hepatocellular Carcinoma: Implication for the Future of Immunotherapy. Semin. Liver Dis. 2018, 38, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Terme, M.; Colussi, O.; Marcheteau, E.; Tanchot, C.; Tartour, E.; Taieb, J. Modulation of Immunity by Antiangiogenic Molecules in Cancer. Clin. Dev. Immunol. 2012, 2012, 492920. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.B.; Meyronet, D.; Hervieu, V.; Frederick, M.J.; Bergsland, E.; Bergers, G. Intratumoral Myeloid Cells Regulate Responsiveness and Resistance to Antiangiogenic Therapy. Cell Rep. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Manning, E.A.; Ullman, J.G.; Leatherman, J.M.; Asquith, J.M.; Hansen, T.R.; Armstrong, T.D.; Hicklin, D.J.; Jaffee, E.M.; Emens, L.A. A Vascular Endothelial Growth Factor Receptor-2 Inhibitor Enhances Antitumor Immunity through an Immune-Based Mechanism. Clin. Cancer Res. 2007, 13, 3951–3959. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8+ T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef]
- Kimura, T.; Kato, Y.; Ozawa, Y.; Kodama, K.; Ito, J.; Ichikawa, K.; Yamada, K.; Hori, Y.; Tabata, K.; Takase, K.; et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018, 109, 3993–4002. [Google Scholar] [CrossRef]
- Ikeda, M.; Sung, M.W.; Kudo, M.; Kobayashi, M.; Baron, A.D.; Finn, R.S.; Kaneko, S.; Zhu, A.X.; Kubota, T.; Kraljevic, S.; et al. A phase 1b trial of lenvatinib (LEN) plus pembrolizumab (PEM) in patients (pts) with unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2018, 36, 4076. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Golan, T.; Dahan, L.; Fu, S.; Moreno, V.; Park, K.; Geva, R.; De Braud, F.; Wainberg, Z.A.; Reck, M.; et al. Ramucirumab and durvalumab for previously treated, advanced non–small-cell lung cancer, gastric/gastro-oesophageal junction adenocarcinoma, or hepatocellular carcinoma: An open-label, phase Ia/b study (JVDJ). Eur. J. Cancer 2020, 137, 272–284. [Google Scholar] [CrossRef]
- Finn, R.S.; Ducreux, M.; Qin, S.; Galle, P.R.; Zhu, A.X.; Ikeda, M.; Kim, T.-Y.; Xu, D.-Z.; Verret, W.; Liu, J.; et al. IMbrave150: A randomized phase III study of 1L atezolizumab plus bevacizumab vs sorafenib in locally advanced or metastatic hepatocellular carcinoma. J. Clin. Oncol. 2018, 36, TPS4141. [Google Scholar] [CrossRef]
- Farsaci, B.; Donahue, R.N.; Coplin, M.A.; Grenga, I.; Lepone, L.M.; Molinolo, A.A.; Hodge, J.W. Immune Consequences of Decreasing Tumor Vasculature with Antiangiogenic Tyrosine Kinase Inhibitors in Combination with Therapeutic Vaccines. Cancer Immunol. Res. 2014, 2, 1090–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprinzl, M.F.; Reisinger, F.; Puschnik, A.; Ringelhan, M.; Ackermann, K.; Hartmann, D.; Schiemann, M.; Weinmann, A.; Galle, P.R.; Schuchmann, M.; et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology 2013, 57, 2358–2368. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Qiu, M.; Wan, L.; Wang, G.; Huang, T.; Chen, Z.; Jiang, S.; Li, X.; Xie, L.; Cai, L. TGF-β1 Promotes Hepatocellular Carcinoma Invasion and Metastasis via ERK Pathway-Mediated FGFR4 Expression. Cell. Physiol. Biochem. 2018, 45, 1690–1699. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel III, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Faivre, S.; Santoro, A.; Kelley, R.K.; Gane, E.; Costentin, C.E.; Gueorguieva, I.; Smith, C.; Cleverly, A.; Lahn, M.M.; Raymond, E.; et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019, 39, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Morimoto, M.; Tajimi, M.; Inoue, K.; Benhadji, K.A.; Lahn, M.M.F.; Sakai, D. A phase 1b study of transforming growth factor-beta receptor I inhibitor galunisertib in combination with sorafenib in Japanese patients with unresectable hepatocellular carcinoma. Investig. New Drugs 2019, 37, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Lee, H.W.; Seong, J. Combination therapy with anti-T-cell immunoglobulin and mucin-domain containing molecule 3 and radiation improves antitumor efficacy in murine hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Sanmamed, M.F.; Rodriguez, I.; Palazon, A.; Martinez-Forero, I.; Labiano, S.; Hervas-Stubbs, S.; Sangro, B.; Ochoa, C.; Rouzaut, A.; et al. Combined Immunostimulatory Monoclonal Antibodies Extend Survival in an Aggressive Transgenic Hepatocellular Carcinoma Mouse Model. Clin. Cancer Res. 2013, 19, 6151–6162. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Wang, M.; Han, X.; Ren, J.; Huang, G.; Ju, S.; Zhang, Q. Combined use of microwave ablation and cell immunotherapy induces nonspecific immunity of hepatocellular carcinoma model mice. Cell Cycle 2020, 19, 3595–3607. [Google Scholar] [CrossRef]
- Qin, S.; Chen, Z.; Liu, Y.; Xiong, J.; Ren, Z.; Meng, Z.; Gu, S.; Wang, L.; Zou, J. A phase II study of anti–PD-1 antibody camrelizumab plus FOLFOX4 or GEMOX systemic chemotherapy as first-line therapy for advanced hepatocellular carcinoma or biliary tract cancer. J. Clin. Oncol. 2019, 37, 4074. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; Van Der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3–Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Du, X.; Wang, Z.; Ju, J.; Jia, M.; Huang, Q.; Xing, Q.; Xu, M.; Tan, Y.; Liu, M.; et al. Hyper-IL-15 suppresses metastatic and autochthonous liver cancer by promoting tumour-specific CD8+ T cell responses. J. Hepatol. 2014, 61, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.B.; Engels, B.; Schreiber, K.; Ciszewski, C.; Schietinger, A.; Jabri, B. IL-15 in tumor microenvironment causes rejection of large established tumors by T cells in a noncognate T cell receptor-dependent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 8158–8163. [Google Scholar] [CrossRef] [Green Version]
- Pilipow, K.; Roberto, A.; Roederer, M.; Waldmann, T.A.; Mavilio, D.; Lugli, E. IL15 and T-cell Stemness in T-cell–Based Cancer Immunotherapy. Cancer Res. 2015, 75, 5187–5193. [Google Scholar] [CrossRef] [Green Version]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dargel, C.; Bassani-Sternberg, M.; Hasreiter, J.; Zani, F.; Bockmann, J.-H.; Thiele, F.; Bohne, F.; Wisskirchen, K.; Wilde, S.; Sprinzl, M.F.; et al. T Cells Engineered to Express a T-Cell Receptor Specific for Glypican-3 to Recognize and Kill Hepatoma Cells In Vitro and in Mice. Gastroenterology 2015, 149, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Gehring, A.J.; Xue, S.-A.; Ho, Z.Z.; Teoh, D.; Ruedl, C.; Chia, A.; Koh, S.; Lim, S.G.; Maini, M.K.; Stauss, H.; et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011, 55, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Shi, D.; Gao, H.; Qi, X.; Jiang, H.; Zhang, Y.; Chi, J.; Ruan, H.; Wang, H.; Ru, Q.C.; et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). J. Clin. Oncol. 2017, 35, 3049. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, H.; Liu, L.; Cao, S.; Ren, B.; Zhang, N.; An, X.; Yu, J.; Li, H.; Ren, X. A randomized phase II study of autologous cytokine-induced killer cells in treatment of hepatocelluar carcinoma. J. Clin. Immunol. 2013, 34, 194–203. [Google Scholar] [CrossRef]
- Sawada, Y.; Yoshikawa, T.; Shimomura, M.; Iwama, T.; Endo, I.; Nakatsura, T. Programmed death-1 blockade enhances the antitumor effects of peptide vaccine-induced peptide-specific cytotoxic T lymphocytes. Int. J. Oncol. 2014, 46, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Multi Tyrosine Kinase Inhibitor | M1 TAM | M2 TAM | MDSC | CD8 | Treg | DC | NK |
|---|---|---|---|---|---|---|---|
| Sorafenib (low dose) | ↑↑ | ↓ | ↑↑ | ↓ | ↑ | ↑ | |
| Regorafenib | ↑ | ↓ | |||||
| Cabozantinib | ↑ | ↓ | ↑ | ↓ | |||
| Lenvatinib | ↓ | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Asensio, J.; Calvo, H.; Torralba, M.; Miquel, J.; Sanz-de-Villalobos, E.; Larrubia, J.-R. Anti-PD-1/PD-L1 Based Combination Immunotherapy to Boost Antigen-Specific CD8+ T Cell Response in Hepatocellular Carcinoma. Cancers 2021, 13, 1922. https://doi.org/10.3390/cancers13081922
Peña-Asensio J, Calvo H, Torralba M, Miquel J, Sanz-de-Villalobos E, Larrubia J-R. Anti-PD-1/PD-L1 Based Combination Immunotherapy to Boost Antigen-Specific CD8+ T Cell Response in Hepatocellular Carcinoma. Cancers. 2021; 13(8):1922. https://doi.org/10.3390/cancers13081922
Chicago/Turabian StylePeña-Asensio, Julia, Henar Calvo, Miguel Torralba, Joaquín Miquel, Eduardo Sanz-de-Villalobos, and Juan-Ramón Larrubia. 2021. "Anti-PD-1/PD-L1 Based Combination Immunotherapy to Boost Antigen-Specific CD8+ T Cell Response in Hepatocellular Carcinoma" Cancers 13, no. 8: 1922. https://doi.org/10.3390/cancers13081922