
Small Cell Lung Cancer: State of the Art of the Molecular and Genetic Landscape and Novel Perspective

 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Small Cell Lung Cancer (SCLC) General Considerations
2. Molecular Pathways Involved in SCLC Development and Progression
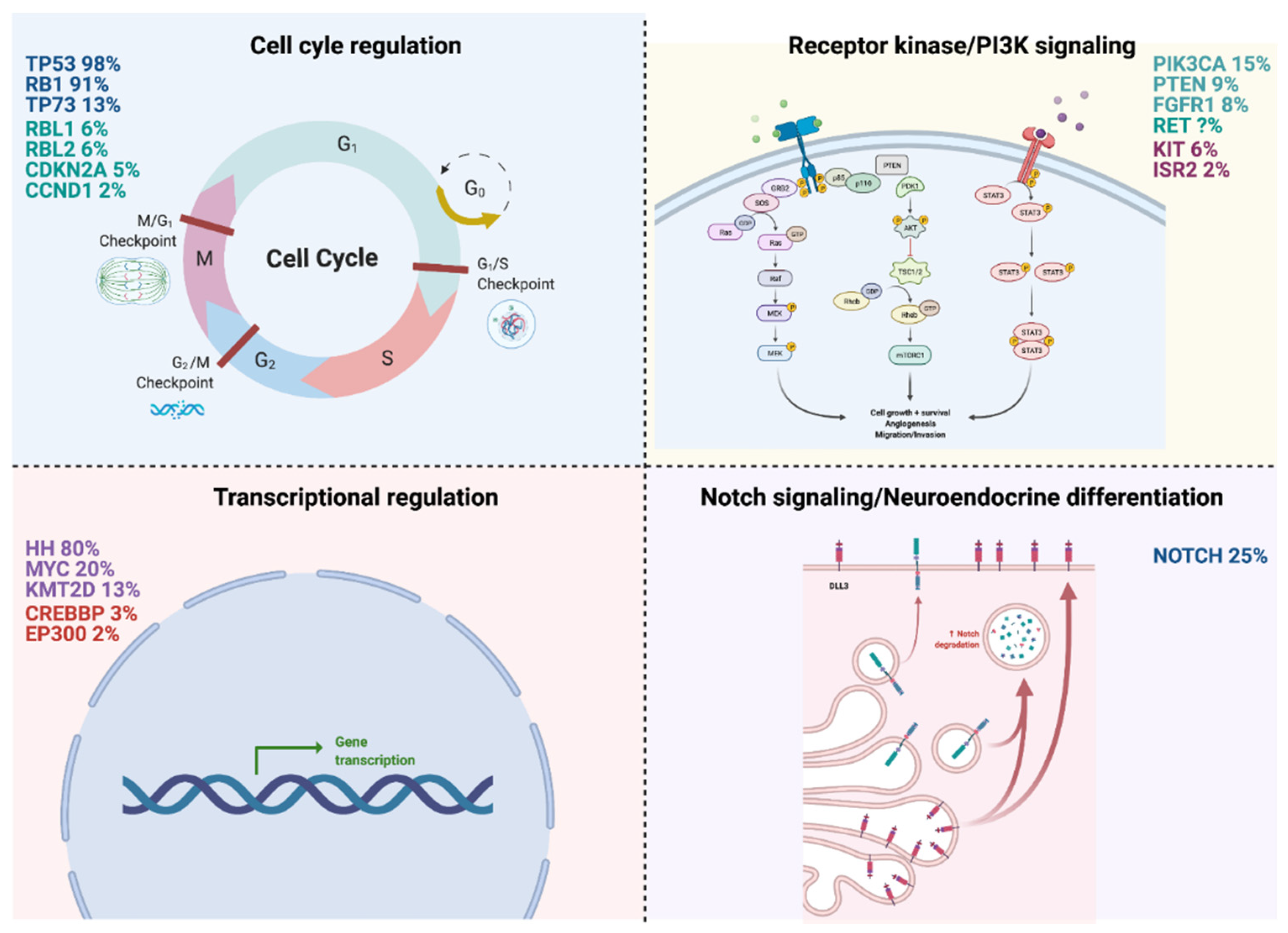
2.1. Cell Cycle Regulation
2.1.1. TP53/RB1 (98%/91%)
2.1.2. TP73 (13%)
2.2. Receptor Kinase/PI3K Signaling
2.2.1. PI3K3CA (15%)
2.2.2. PTEN (9%)
2.2.3. FGFR1 (8%)
2.2.4. RET
2.3. Transcriptional Regulation
2.3.1. Hedgehog Signaling Pathway (80%)
2.3.2. MYC (20%)
2.3.3. KMT2D (13%)
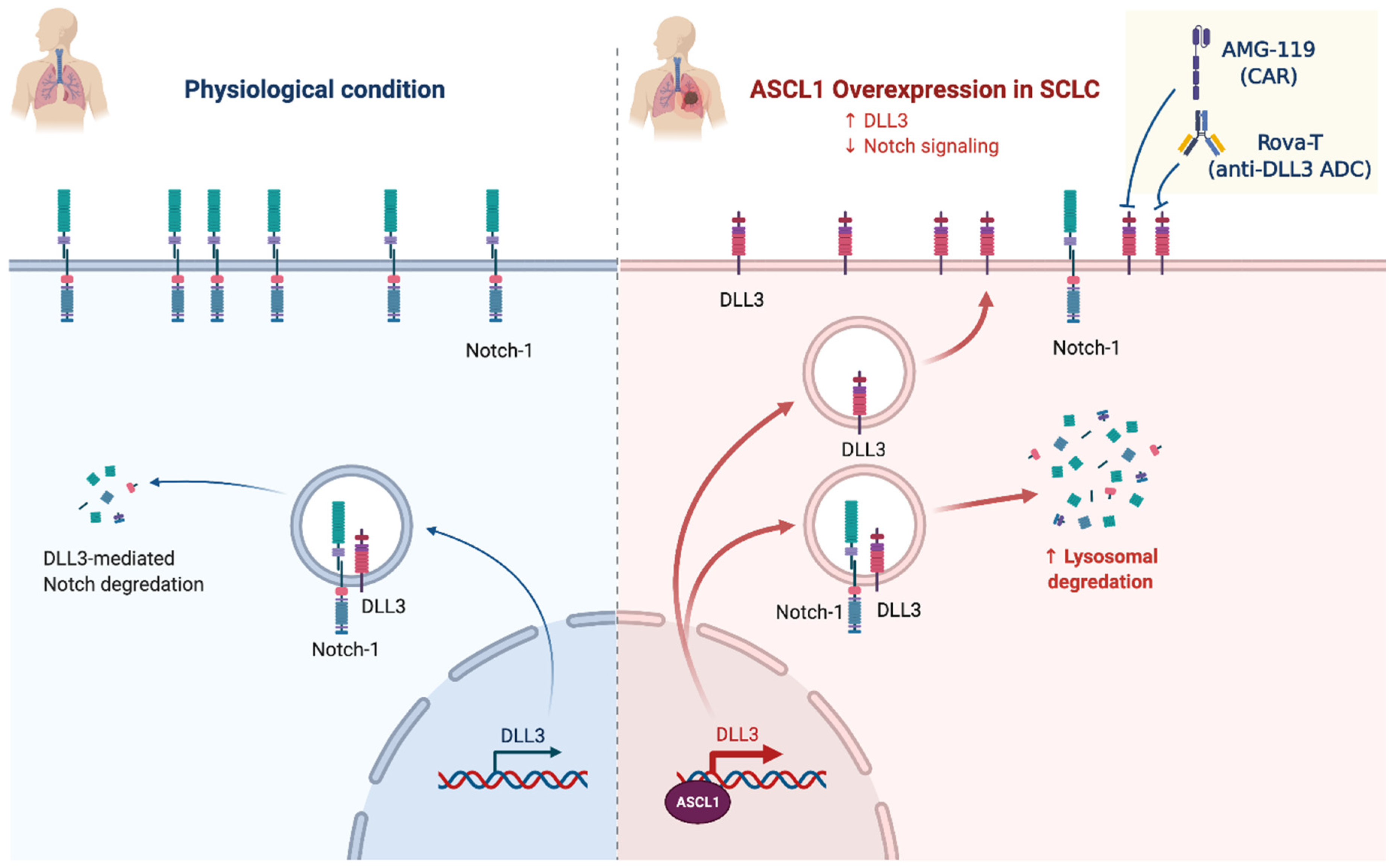
2.4. Notch Signaling/Neuroendocrine Differentiation
NOTCH (25%)
2.5. Epigenetic and Proteomic Changes
2.6. Transcriptional Addictions
3. Future Perspectives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bunn, P.A.; Minna, J.D.; Augustyn, A.; Gazdar, A.F.; Ouadah, Y.; Krasnow, M.A.; Berns, A.; Brambilla, E.; Rekhtman, N.; Massion, P.P.; et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J. Thorac. Oncol. 2016, 11, 453–474. [Google Scholar] [CrossRef] [Green Version]
- Lassen, U.; Osterlind, K.; Hansen, M.; Dombernowsky, P.; Bergman, B.; Hansen, H.H. Long-Term Survival in Small-Cell Lung Cancer: Posttreatment Characteristics in Patients Surviving 5 to 18+ Year—An Analysis of 1714 Consecutive Patients. J. Clin. Oncol. 1995, 13, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zimmermann, S.; Parikh, K.; Mansfield, A.S.; Adjei, A.A. Current Diagnosis and Management of Small-Cell Lung Cancer. Mayo Clin. Proc. 2019, 94, 1599–1622. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, A.; Lipka, M.B.; McColl, K.; Dabir, S.; Behtaj, M.; Kresak, A.; Miron, A.; Yang, M.; Sharma, N.; Fu, P.; et al. Clinical Correlation of Extensive-Stage Small-Cell Lung Cancer Genomics. Ann. Oncol. 2016, 27, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Sosa, A.E.; Rosell, R. Unraveling the Genomic Complexity of Small Cell Lung Cancer. Transl. Lung Cancer Res. 2016, 5, 363–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, J.T.; George, J.; Owonikoko, T.K.; Berns, A.; Brambilla, E.; Byers, L.A.; Carbone, D.; Chen, H.J.; Christensen, C.L.; Dive, C.; et al. New Approaches to SCLC Therapy: From the Laboratory to the Clinic. J. Thorac. Oncol. 2020, 15, 520–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, L.A.; Rudin, C.M. Small Cell Lung Cancer: Where Do We Go from Here? SCLC: Where Do We Go From Here? Cancer 2015, 121, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative Genome Analyses Identify Key Somatic Driver Mutations of Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive Genomic Analysis Identifies SOX2 as a Frequently Amplified Gene in Small-Cell Lung Cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Sun, Y.; Lei, Y.; Yang, K.; Tang, R. YAP1 Promotes Multidrug Resistance of Small Cell Lung Cancer by CD74-related Signaling Pathways. Cancer Med. 2020, 9, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Niu, H.; Nackaerts, K.; Csoszi, T.; Ostoros, G.; Mark, Z.; Baik, C.; Joy, A.A.; Chouaid, C.; Jaime, J.C.; et al. Randomized Phase II Study of Paclitaxel plus Alisertib versus Paclitaxel plus Placebo as Second-Line Therapy for SCLC: Primary and Correlative Biomarker Analyses. J. Thorac. Oncol. 2020, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information (NCBI). Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. 1988. Available online: https://www.Ncbi.Nlm.Nih.Gov/ (accessed on 14 February 2021).
- Yokouchi, H.; Nishihara, H.; Harada, T.; Yamazaki, S.; Kikuchi, H.; Oizumi, S.; Uramoto, H.; Tanaka, F.; Harada, M.; Akie, K.; et al. Detection of Somatic TP53 Mutation in Surgically Resected Small-Cell Lung Cancer by Targeted Exome Sequencing: Association with Longer Relapse-Free Survival. Heliyon 2020, 6, e04439. [Google Scholar] [CrossRef] [PubMed]
- Sonkin, D.; Vural, S.; Thomas, A.; Teicher, B.A. Neuroendocrine Negative SCLC Is Mostly RB1 WT and May Be Sensitive to CDK4/6 Inhibition. BioRxiv 2019, 516351. [Google Scholar] [CrossRef]
- Kim, K.-B.; Dunn, C.T.; Park, K.-S. Recent Progress in Mapping the Emerging Landscape of the Small-Cell Lung Cancer Genome. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Kokubu, A.; Tsuta, K.; Hirohashi, S. Oncogenic Mutation of PIK3CA in Small Cell Lung Carcinoma: A Potential Therapeutic Target Pathway for Chemotherapy-Resistant Lung Cancer. Cancer Lett. 2009, 283, 203–211. [Google Scholar] [CrossRef]
- Umemura, S.; Mimaki, S.; Makinoshima, H.; Tada, S.; Ishii, G.; Ohmatsu, H.; Niho, S.; Yoh, K.; Matsumoto, S.; Takahashi, A.; et al. Therapeutic Priority of the PI3K/AKT/MTOR Pathway in Small Cell Lung Cancers as Revealed by a Comprehensive Genomic Analysis. J. Thorac. Oncol. 2014, 9, 1324–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Feng, Z.; Han, J.; Cheng, W.; Su, B.; Mo, J.; Feng, X.; Feng, S.; Chen, G.; Huang, P.; et al. Antinociceptive Effects of Shenling Baizhu through PI3K-Akt-MTOR Signaling Pathway in a Mouse Model of Bone Metastasis with Small-Cell Lung Cancer. Evid. Based Complement. Altern. Med. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-Cell Lung Cancer. Nat. Rev. Dis. Primer 2021, 7, 3. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of Small Cell Lung Cancer by Somatic Inactivation of Both Trp53 and Rb1 in a Conditional Mouse Model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN Is a Potent Suppressor of Small Cell Lung Cancer. Mol. Cancer Res. 2014, 12, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferone, G.; Song, J.-Y.; Krijgsman, O.; van der Vliet, J.; Cozijnsen, M.; Semenova, E.A.; Adams, D.J.; Peeper, D.; Berns, A. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/P53 Mice. Cell Rep. 2020, 30, 3837–3850.e3. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Adjei, A.A. FGFR Signaling as a Target for Lung Cancer Therapy. J. Thorac. Oncol. 2016, 11, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.; McCusker, M.G.; Scilla, K.A.; Arensmeyer, K.E.; Mehra, R.; Adamo, V.; Rolfo, C. Immunotherapy in Lung Cancer: From a Minor God to the Olympus. In Immunotherapy; Naing, A., Hajjar, J., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1244, pp. 69–92. [Google Scholar] [CrossRef]
- Li, A.Y.; McCusker, M.G.; Russo, A.; Scilla, K.A.; Gittens, A.; Arensmeyer, K.; Mehra, R.; Adamo, V.; Rolfo, C. RET Fusions in Solid Tumors. Cancer Treat. Rev. 2019, 81, 101911. [Google Scholar] [CrossRef]
- Dabir, S.; Babakoohi, S.; Kluge, A.; Morrow, J.J.; Kresak, A.; Yang, M.; MacPherson, D.; Wildey, G.; Dowlati, A. RET Mutation and Expression in Small-Cell Lung Cancer. J. Thorac. Oncol. 2014, 9, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Lim, S.M.; Kim, M.-J.; Park, S.Y.; Kim, J.-H. Sonic Hedgehog Pathway as the Prognostic Marker in Patients with Extensive Stage Small Cell Lung Cancer. Yonsei Med. J. 2019, 60, 898. [Google Scholar] [CrossRef]
- Falkenstein, K.N.; Vokes, S.A. Transcriptional Regulation of Graded Hedgehog Signaling. Semin. Cell Dev. Biol. 2014, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.-S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A Crucial Requirement for Hedgehog Signaling in Small Cell Lung Cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, J.; Pedersen, M.W.; Pedersen, N.; Ensinger, C.; Tümer, Z.; Tommerup, N.; Poulsen, H.S.; Larsen, L.A. Hedgehog Signaling in Small-Cell Lung Cancer: Frequent in Vivo but a Rare Event in Vitro. Lung Cancer 2006, 52, 281–290. [Google Scholar] [CrossRef]
- Sos, M.L.; Dietlein, F.; Peifer, M.; Schottle, J.; Balke-Want, H.; Muller, C.; Koker, M.; Richters, A.; Heynck, S.; Malchers, F.; et al. A Framework for Identification of Actionable Cancer Genome Dependencies in Small Cell Lung Cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 17034–17039. [Google Scholar] [CrossRef] [Green Version]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalishazar, M.D.; Wait, S.J.; Huang, F.; Ireland, A.S.; Mukhopadhyay, A.; Lee, Y.; Schuman, S.S.; Guthrie, M.R.; Berrett, K.C.; Vahrenkamp, J.M.; et al. MYC-Driven Small-Cell Lung Cancer Is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin. Cancer Res. 2019, 25, 5107–5121. [Google Scholar] [CrossRef] [Green Version]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78.e12. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Yoo, S.; Kong, R.; Sato, T.; Sinha, A.; Karam, S.; Bao, L.; Fridrikh, M.; Emoto, K.; Nudelman, G.; et al. Prototypical Oncogene Family Myc Defines Unappreciated Distinct Lineage States of Small Cell Lung Cancer. Sci. Adv. 2021, 7, eabc2578. [Google Scholar] [CrossRef] [PubMed]
- Augert, A.; Zhang, Q.; Bates, B.; Cui, M.; Wang, X.; Wildey, G.; Dowlati, A.; MacPherson, D. Small Cell Lung Cancer Exhibits Frequent Inactivating Mutations in the Histone Methyltransferase KMT2D/MLL2: CALGB 151111 (Alliance). J. Thorac. Oncol. 2017, 12, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Gardner, E.E.; Poirier, J.T.; Rudin, C.M. Histone Code Aberrancies in Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 599–601. [Google Scholar] [CrossRef]
- Leonetti, A.; Facchinetti, F.; Minari, R.; Cortellini, A.; Rolfo, C.D.; Giovannetti, E.; Tiseo, M. Notch Pathway in Small-Cell Lung Cancer: From Preclinical Evidence to Therapeutic Challenges. Cell. Oncol. 2019, 42, 261–273. [Google Scholar] [CrossRef]
- Terragni, J.; Zhang, G.; Sun, Z.; Pradhan, S.; Song, L.; Crawford, G.E.; Lacey, M.; Ehrlich, M. Notch Signaling Genes: Myogenic DNA Hypomethylation and 5-Hydroxymethylcytosine. Epigenetics 2014, 9, 842–850. [Google Scholar] [CrossRef] [Green Version]
- Ardeshir-Larijani, F.; Wildey, G.; Fu, P.; Bhateja, P.; Dowlati, A. Frequency of NOTCH Pathway Mutation in Primary Tumor of SCLC Compared to Metastatic Biopsies and Association with Better Survival. J. Clin. Oncol. 2018, 36 (Suppl. 15), e20574. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Borges, M.W.; Ravi, R.K.; Arnold, D.R.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Notch Signaling Induces Cell Cycle Arrest in Small Cell Lung Cancer Cells. Cancer Res. 2001, 61, 3200–3205. [Google Scholar] [PubMed]
- Mohammad, H.P.; Smitheman, K.N.; Kamat, C.D.; Soong, D.; Federowicz, K.E.; Van Aller, G.S.; Schneck, J.L.; Carson, J.D.; Liu, Y.; Butticello, M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH Activation in Small Cell Lung Cancer through LSD1 Inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Pourat, J.; Abdel-Atti, D.; Carlin, S.D.; Piersigilli, A.; Bankovich, A.J.; Gardner, E.E.; Hamdy, O.; Isse, K.; Bheddah, S.; et al. Noninvasive Interrogation of DLL3 Expression in Metastatic Small Cell Lung Cancer. Cancer Res. 2017, 77, 3931–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.H.; Giffin, M.J.; Bailis, J.M.; Smit, M.-A.D.; Carbone, D.P.; He, K. DLL3: An Emerging Target in Small Cell Lung Cancer. J. Hematol. Oncol. 2019, 12, 61. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, N.; Song, W.; You, N.; Li, Q.; Sun, W.; Zhang, Y.; Wang, D.; Dou, K. The Significance of Notch1 Compared with Notch3 in High Metastasis and Poor Overall Survival in Hepatocellular Carcinoma. PLoS ONE 2013, 8, e57382. [Google Scholar] [CrossRef] [Green Version]
- Dupont, C.; Armant, D.; Brenner, C. Epigenetics: Definition, Mechanisms and Clinical Perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Krushkal, J.; Silvers, T.; Reinhold, W.C.; Sonkin, D.; Vural, S.; Connelly, J.; Varma, S.; Meltzer, P.S.; Kunkel, M.; Rapisarda, A.; et al. Epigenome-Wide DNA Methylation Analysis of Small Cell Lung Cancer Cell Lines Suggests Potential Chemotherapy Targets. Clin. Epigenetics 2020, 12, 93. [Google Scholar] [CrossRef]
- Stewart, P.A.; Welsh, E.A.; Slebos, R.J.C.; Fang, B.; Izumi, V.; Chambers, M.; Zhang, G.; Cen, L.; Pettersson, F.; Zhang, Y.; et al. Proteogenomic Landscape of Squamous Cell Lung Cancer. Nat. Commun. 2019, 10, 3578. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as Second-Line Treatment for Patients with Small-Cell Lung Cancer: A Single-Arm, Open-Label, Phase 2 Basket Trial. Lancet Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef]
- Russo, A.; De Miguel Perez, D.; Gunasekaran, M.; Scilla, K.; Lapidus, R.; Cooper, B.; Mehra, R.; Adamo, V.; Malapelle, U.; Rolfo, C. Liquid Biopsy Tracking of Lung Tumor Evolutions over Time. Expert Rev. Mol. Diagn. 2019, 19, 1099–1108. [Google Scholar] [CrossRef]
- Nong, J.; Gong, Y.; Guan, Y.; Yi, X.; Yi, Y.; Chang, L.; Yang, L.; Lv, J.; Guo, Z.; Jia, H.; et al. Circulating Tumor DNA Analysis Depicts Subclonal Architecture and Genomic Evolution of Small Cell Lung Cancer. Nat. Commun. 2018, 9, 3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almodovar, K.; Iams, W.T.; Meador, C.B.; Zhao, Z.; York, S.; Horn, L.; Yan, Y.; Hernandez, J.; Chen, H.; Shyr, Y.; et al. Longitudinal Cell-Free DNA Analysis in Patients with Small Cell Lung Cancer Reveals Dynamic Insights into Treatment Efficacy and Disease Relapse. J. Thorac. Oncol. 2018, 13, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, L.; Rothwell, D.G.; Mesquita, B.; Smowton, C.; Leong, H.S.; Fernandez-Gutierrez, F.; Li, Y.; Burt, D.J.; Antonello, J.; Morrow, C.J.; et al. Molecular Analysis of Circulating Tumor Cells Identifies Distinct Copy-Number Profiles in Patients with Chemosensitive and Chemorefractory Small-Cell Lung Cancer. Nat. Med. 2017, 23, 114–119. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Z.; Wang, Q. Emerging Therapies for Small Cell Lung Cancer. J. Hematol. Oncol. 2019, 12, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saltos, A.; Shafique, M.; Chiappori, A. Update on the Biology, Management, and Treatment of Small Cell Lung Cancer (SCLC). Front. Oncol. 2020, 10, 1074. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denninghoff, V.; Russo, A.; de Miguel-Pérez, D.; Malapelle, U.; Benyounes, A.; Gittens, A.; Cardona, A.F.; Rolfo, C. Small Cell Lung Cancer: State of the Art of the Molecular and Genetic Landscape and Novel Perspective. Cancers 2021, 13, 1723. https://doi.org/10.3390/cancers13071723
Denninghoff V, Russo A, de Miguel-Pérez D, Malapelle U, Benyounes A, Gittens A, Cardona AF, Rolfo C. Small Cell Lung Cancer: State of the Art of the Molecular and Genetic Landscape and Novel Perspective. Cancers. 2021; 13(7):1723. https://doi.org/10.3390/cancers13071723
Chicago/Turabian StyleDenninghoff, Valeria, Alessandro Russo, Diego de Miguel-Pérez, Umberto Malapelle, Amin Benyounes, Allison Gittens, Andres Felipe Cardona, and Christian Rolfo. 2021. "Small Cell Lung Cancer: State of the Art of the Molecular and Genetic Landscape and Novel Perspective" Cancers 13, no. 7: 1723. https://doi.org/10.3390/cancers13071723