
Are We Benign? What Can Wnt Signaling Pathway and Epithelial to Mesenchymal Transition Tell Us about Intracranial Meningioma Progression


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Collection
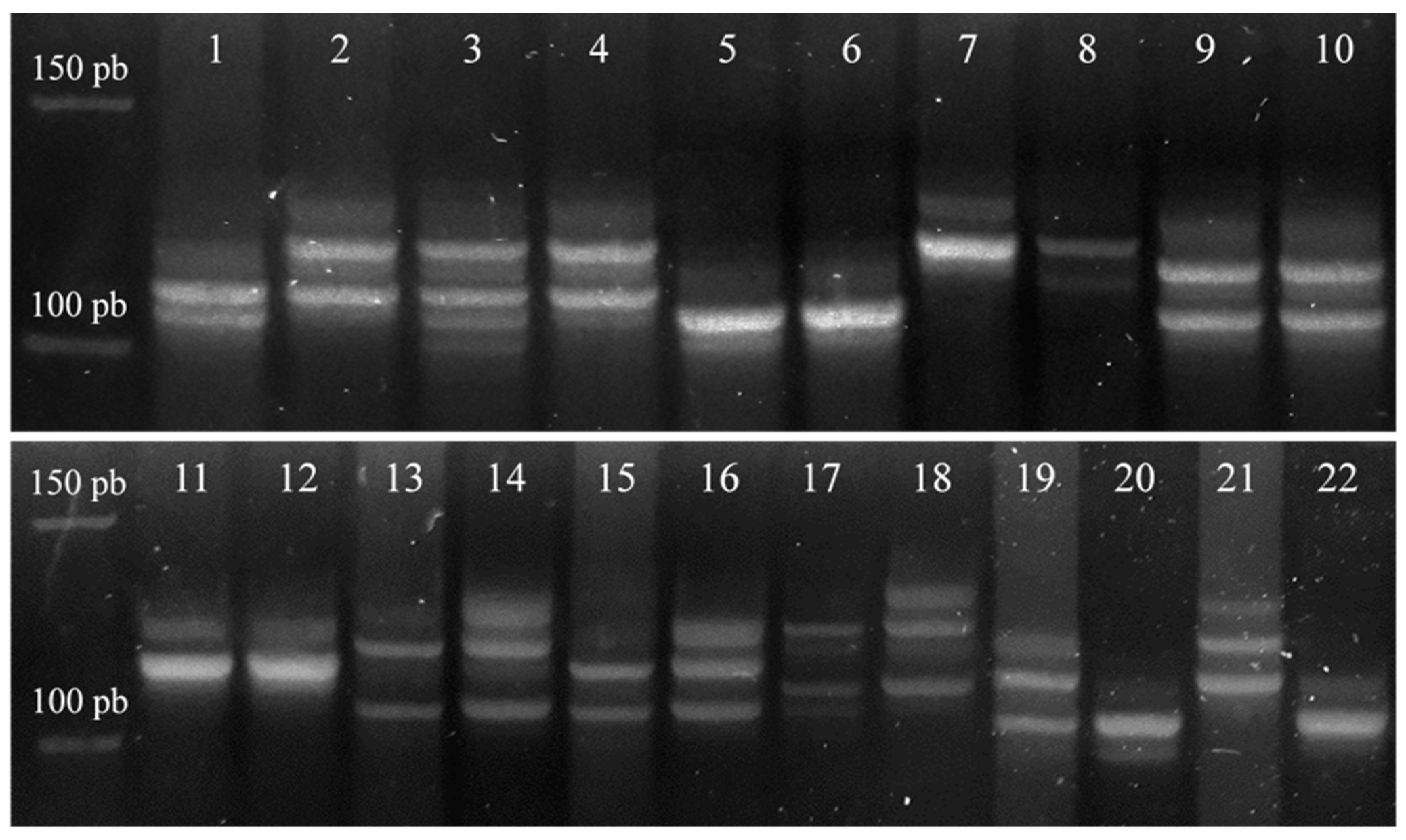
2.2. DNA Extraction and Analysis
2.3. Protein Localization and Expression
2.4. Statistical Analysis
3. Results
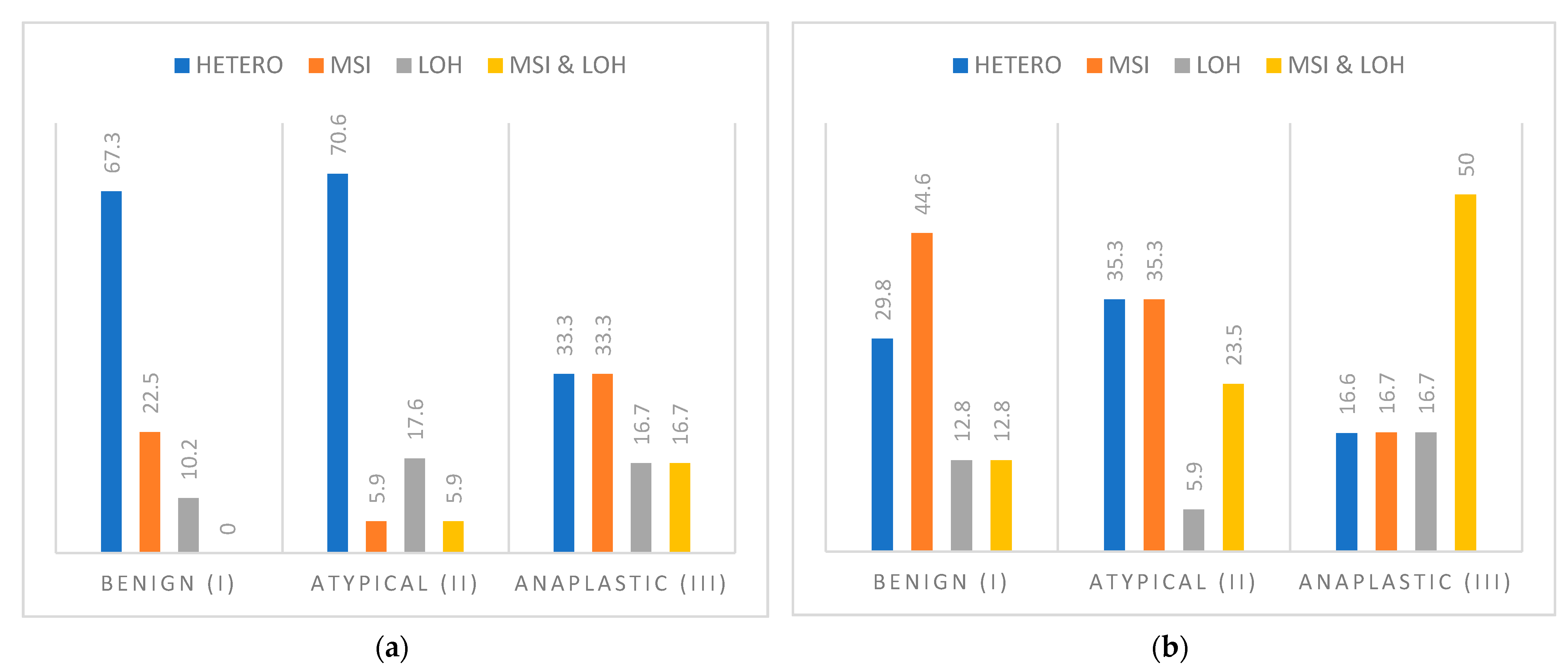
3.1. Loss of Heterozygosity and Microsatellite Instability of CDH1 and CDH2 Gene
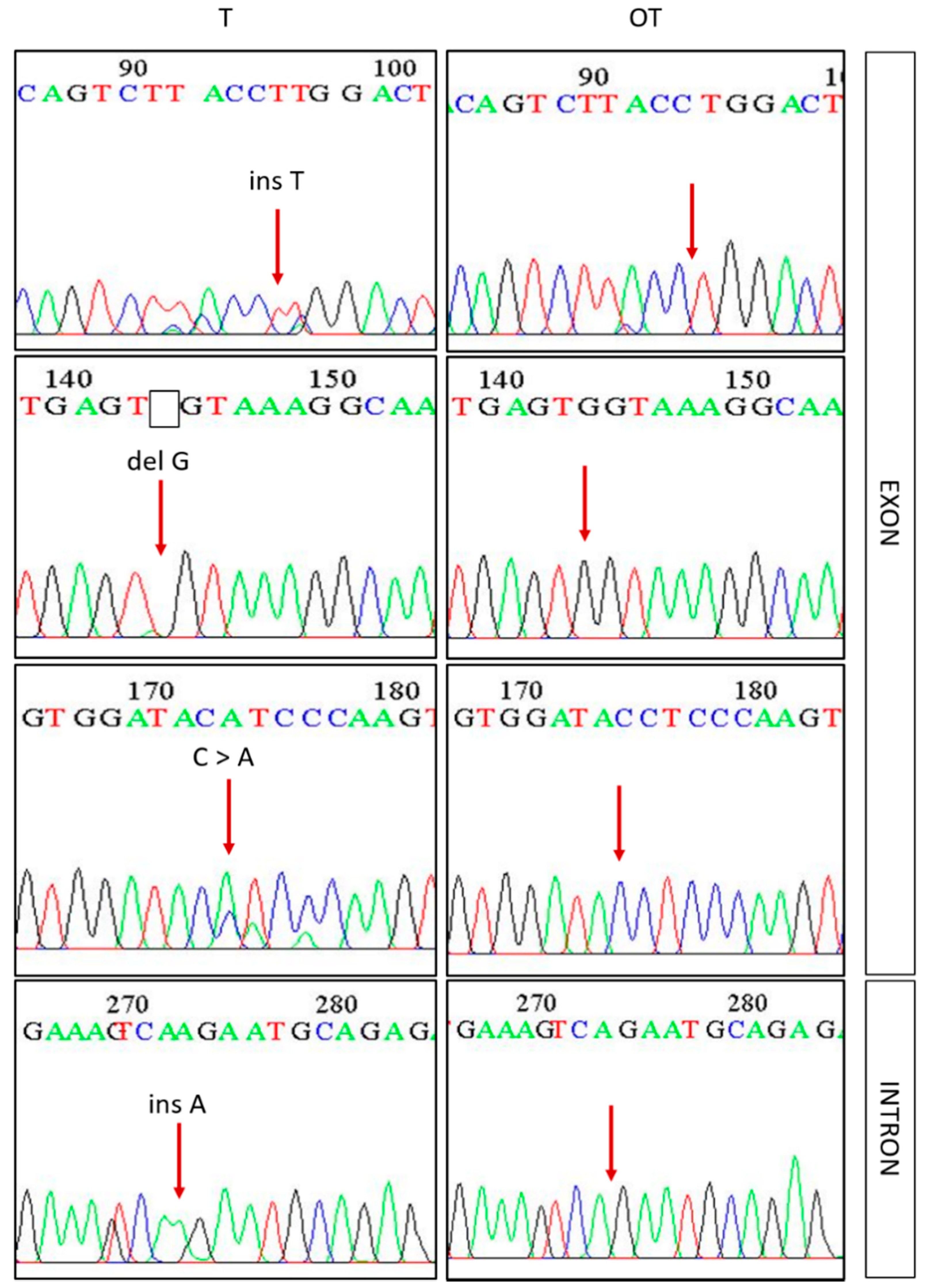
3.2. Mutations in Exon 3 β-Catenin (CTNNB1) and Prediction of Their Effects
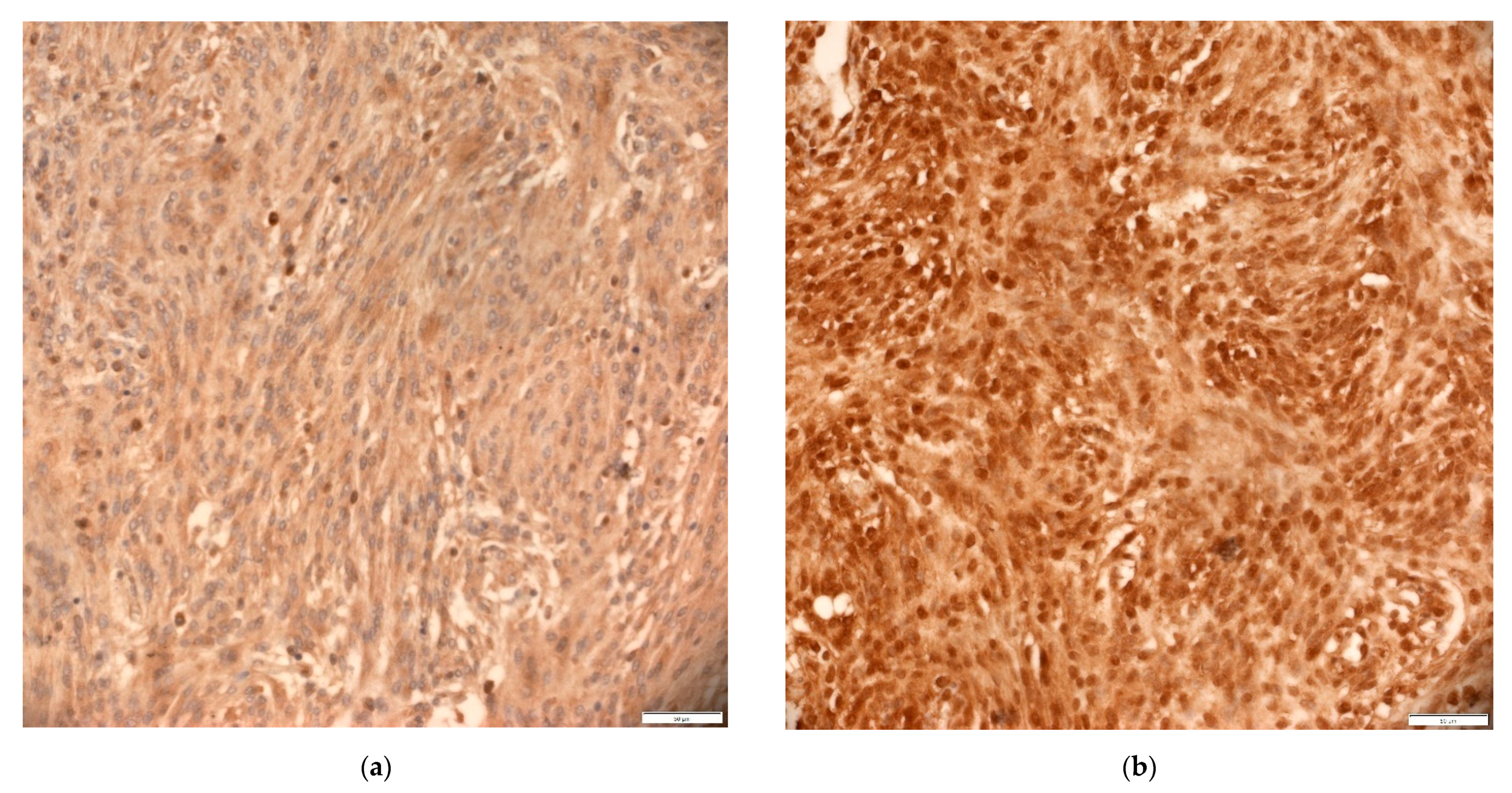
3.3. Expression of EMT Markers—E-Cadherin and N-Cadherin
3.4. Expression of EMT Transcription Factors—TWIST1, SNAIL and SLUG
3.5. Expression of Total and Non-Phosphorylated β-Catenin
3.6. Comparison of E-, N-Cadherin, β-Catenin, TWIST1, SNAIL and SLUG Expression
4. Discussion
4.1. The Role of E- and N-Cadherin in Intracranial Meningioma
4.2. The Role of SNAIL, SLUG and TWIST1 Transcription Factors in Intracranial Meningioma
4.3. Role of β-Catenin Phosphorylation Status and Wnt Signaling Pathway in Intracranial Meningioma
4.4. Interaction of Wnt Signaling Pathway and EMT in Intracranial Meningioma
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buttrick, S.; Shah, A.H.; Komotar, R.J.; Ivan, M.E. Management of Atypical and Anaplastic Meningiomas. Neurosurg. Clin. N. Am. 2016, 27, 239–247. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 WHO Classification of Tumors of the Central Nervous System; WHO: Geneva, Switzerland, 2016; Volume 131, ISBN 9789283244929. [Google Scholar]
- Buerki, R.A.; Horbinski, C.M.; Kruser, T.; Horowitz, P.M.; James, C.D.; Lukas, R.V. An overview of meningiomas. Futur. Oncol. 2018, 14, 2161–2177. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Çelen, I.; Ross, K.E.; Arighi, C.N.; Wu, C.H. Bioinformatics knowledge map for analysis of beta-catenin function in cancer. PLoS ONE 2015, 10, e0141773. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial–mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Wallesch, M.; Pachow, D.; Blücher, C.; Firsching, R.; Warnke, J.P.; Braunsdorf, W.E.K.; Kirches, E.; Mawrin, C. Altered expression of E-Cadherin-related transcription factors indicates partial epithelial-mesenchymal transition in aggressive meningiomas. J. Neurol. Sci. 2017, 380, 112–121. [Google Scholar] [CrossRef]
- Das, V.; Bhattacharya, S.; Chikkaputtaiah, C.; Hazra, S.; Pal, M. The basics of epithelial–mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J. Cell. Physiol. 2019, 234, 14535–14555. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Molecular Cloning—A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012. [Google Scholar]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrisi, M.; Kaleel, M.; Pollastri, G. Porter 5: State-of-the-art ab initio prediction of protein secondary structure in 3 and 8 classes. bioRxiv 2018, 289033. [Google Scholar] [CrossRef]
- Mazières, J.; Brugger, W.; Cappuzzo, F.; Middel, P.; Frosch, A.; Bara, I.; Klingelschmitt, G.; Klughammer, B. Evaluation of EGFR protein expression by immunohistochemistry using H-score and the magnification rule: Re-analysis of the SATURN study. Lung Cancer 2013, 82, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Latil, A.; Cussenot, O.; Fournier, G.; Driouch, K.; Lidereau, R. Loss of heterozygosity at chromosome 16q in prostate adenocarcinoma: Identification of three independent regions. Cancer Res. 1997, 57, 1058–1062. [Google Scholar]
- Berx, G.; Becker, K.F.; Höfler, H.; Van Roy, F. Mutations of the human E-cadherin (CDH1) gene. Hum. Mutat. 1998, 12, 226–237. [Google Scholar] [CrossRef]
- Cleton-Jansen, A.M. E-cadherin and loss of heterozygosity at chromosome 16 in breast cancinogenesis: Different genetic pathways in ductal and lobular breast cancer? Breast Cancer Res. 2002, 4, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Sarrió, D.; Moreno-Bueno, G.; Hardisson, D.; Sánchez-Estévez, C.; Guo, M.; Herman, J.G.; Gamallo, C.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int. J. Cancer 2003, 106, 208–215. [Google Scholar] [CrossRef]
- Green, A.R.; Krivinskas, S.; Young, P.; Rakha, E.A.; Paish, E.C.; Powe, D.G.; Ellis, I.O. Loss of expression of chromosome 16q genes DPEP1 and CTCF in lobular carcinoma in situ of the breast. Breast Cancer Res. Treat. 2009, 113, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Priya, T.P.; Kapoor, V.K.; Krishnani, N.; Agrawal, V.; Agrawal, S. Role of E-cadherin gene in gall bladder cancer and its precursor lesions. Virchows Arch. 2010, 456, 507–514. [Google Scholar] [CrossRef]
- Vladušić, T.; Hrašćan, R.; Vrhovac, I.; Krušlin, B.; Gamulin, M.; Grgić, M.; Pećina-Šlaus, N.; Franekić Čolić, J. Loss of heterozygosity of selected tumor suppressor genes in human testicular germ cell tumors. Pathol. Res. Pract. 2010, 206, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Nikuševa-Martić, T.; Beroš, V.; Pećina-Šlaus, N.; Pećina, H.I.; Bulić-Jakuš, F. Genetic changes of CDH1, APC, and CTNNB1 found in human brain tumors. Pathol. Res. Pract. 2007, 203, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Pećina-Šlaus, N.; Kafka, A.; Bukovac, A.; Vladušić, T.; Tomas, D.; Hrašćan, R. Genetic changes of MLH1 and MSH2 genes could explain constant findings on microsatellite instability in intracranial meningioma. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Kokkino, A.J.; Warnick, R.E.; Tew, J.M.; Von Deimling, A.; Menon, A.G. Role of genomic instability in meningioma progression. Genes Chromosom. Cancer 1996, 16, 265–269. [Google Scholar] [CrossRef]
- Wirtz, H.C.; Müller, W.; Noguchi, T.; Scheven, M.; Rüschoff, J.; Hommel, G.; Gabbert, H.E. Prognostic value and clinicopathological profile of microsatellite instability in gastric cancer. Clin. Cancer Res. 1998, 4, 1749–1754. [Google Scholar]
- Salvesen, H.B.; Macdonald, N.; Ryan, A.; Iversen, O.E.; Jacobs, I.J.; Akslen, L.A.; Das, S. Methylation of hMLH1 in a population-based series of endometrial carcinomas. Clin. Cancer Res. 2000, 6, 3607–3613. [Google Scholar] [PubMed]
- Pykett, M.J.; Murphy, M.; Harnish, P.R.; George, D.L. Identification of a Microsatellite Instability Phenotype in Meningiomas. Cancer Res. 1994, 54, 6340–6343. [Google Scholar] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Vladušić, T.; Pećina, H.I.; Hrašćan, R. AXIN1 Expression and Localization in Meningiomas and Association to Changes of APC and E-cadherin. Anticancer Res. 2016, 36, 4583–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- .Breivik, J.; Gaudernack, G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin. Cancer Biol. 1999, 9, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.M.; Novelli, M.R.; Bodmer, W.F. The mutation rate and cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 14800–14803. [Google Scholar] [CrossRef] [Green Version]
- Brewster, S.F.; Gingell, J.C.; Browne, S.; Brown, K.W. Loss of heterozygosity on chromosome 18q is associated with muscle-invasive transitional cell carcinoma of the bladder. Br. J. Cancer 1994, 70, 697–700. [Google Scholar] [CrossRef] [Green Version]
- Frank, C.J.; McClatchey, K.D.; Devaney, K.O.; Carey, T.E. Evidence that loss of chromosome 18q is associated with tumor progression. Cancer Res. 1997, 57, 824–827. [Google Scholar] [PubMed]
- Jones, J.W.; Raval, J.R.; Beals, T.F.; Worsham, M.J.; Van Dyke, D.L.; Esclamado, R.M.; Wolf, G.T.; Bradford, C.R.; Miller, T.; Carey, T.E. Frequent loss of heterozygosity on chromosome arm 18q in squamous cell carcinomas. Identification of 2 regions of loss—18q11.1-q12.3 and 18q21.1-q23. Arch Otolaryngol. Head Neck Surg. 2015, 123, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Druliner, B.R.; Ruan, X.; Sicotte, H.; O’Brien, D.; Liu, H.; Kocher, J.P.A.; Boardman, L. Early genetic aberrations in patients with sporadic colorectal cancer. Mol. Carcinog. 2018, 57, 114–124. [Google Scholar] [CrossRef]
- Takebayashi, S.; Hickson, A.; Ogawa, T.; Jung, K.Y.; Mineta, H.; Ueda, Y.; Grénman, R.; Fisher, S.G.; Carey, T.E. Loss of chromosome arm 18q with tumor progression in head and neck squamous cancer. Genes Chromosom. Cancer 2004, 41, 145–154. [Google Scholar] [CrossRef]
- Jen, J.; Kim, H.; Piantadosi, S.; Liu, Z.F.; Levitt, R.C.; Sistonen, P.; Kinzler, K.W.; Vogelstein, B.; Hamilton, S.R. Allelic Loss of Chromosome 18q and Prognosis in Colorectal Cancer. N. Engl. J. Med. 1994, 331, 931–933. [Google Scholar] [CrossRef]
- Sarli, L.; Bottarelli, L.; Bader, G.; Iusco, D.; Pizzi, S.; Costi, R.; D’Adda, T.; Bertolani, M.; Roncoroni, L.; Bordi, C. Association between recurrence of sporadic colorectal cancer, high level of microsatellite instability, and loss of heterozygosity at chromosome 18q. Dis. Colon Rectum 2004, 47, 1467–1482. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.; Biesma, H.D.; Cordes, M.; Smeets, D.; Neerincx, M.; Das, S.; Eijk, P.P.; Murphy, V.; Barat, A.; Bacon, O.; et al. Loss of chromosome 18q11.2-q12.1 is predictive for survival in patients with metastatic colorectal cancer treated with bevacizumab. J. Clin. Oncol. 2018, 36, 2052–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, H.C.; Derksen, P.W.B. Loss of E-cadherin-dependent cell–cell adhesion and the development and progression of cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Cao, Z.Q.; Wang, Z.; Leng, P. Aberrant N-cadherin expression in cancer. Biomed. Pharmacother. 2019, 118, 109320. [Google Scholar] [CrossRef]
- Zhao, P.; Zhu, T.; Tang, Q.; Liu, H.; Zhu, J.; Zhang, W. Immunohistochemical and genetic markers to distinguish hemangiopericytoma and meningioma. Int. J. Clin. Exp. Med. 2015, 8, 3291–3299. [Google Scholar]
- Ng, H.K.; Wong, A.T.C. Expression of epithelial and extracellular matrix protein markers in meningiomas. Histopathology 1993, 22, 113–126. [Google Scholar] [CrossRef]
- Sharma, S.; Ray, S.; Moiyadi, A.; Sridhar, E.; Srivastava, S. Quantitative proteomic analysis of meningiomas for the identification of surrogate protein markers. Sci. Rep. 2014, 4, 7140. [Google Scholar] [CrossRef] [Green Version]
- Ke, H.L.; Ke, R.H.; Li, B.; Wang, X.H.; Wang, Y.N.; Wang, X.Q. Association between laminin γ1 expression and meningioma grade, recurrence, and progression-free survival. Acta Neurochir. 2013, 155, 165–171. [Google Scholar] [CrossRef]
- Li, T.; Ren, J.; Ma, J.; Wu, J.; Zhang, R.; Yuan, H.; Han, X. LINC00702/miR-4652-3p/ZEB1 axis promotes the progression of malignant meningioma through activating Wnt/β-catenin pathway. Biomed. Pharmacother. 2019, 113. [Google Scholar] [CrossRef]
- Oh, K.Y.; Yoon, H.J.; Lee, J.I.; Ahn, S.H.; Hong, S.D. Twist and Snail expression in tumor and stromal cells of epithelial odontogenic tumors. J. Oral Pathol. Med. 2017, 46, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, A.M.; Mărgăritescu, C.; Taisescu, O.; Andreiana, B.C.; Florescu, M.M.; Ciurea, R.N. Immunoexpression of E-cadherin, snail and twist in colonic adenocarcinomas. Rom. J. Morphol. Embryol. 2019, 60, 531–536. [Google Scholar] [PubMed]
- Cho, Y.A.; Kim, E.K.; Cho, B.C.; Koh, Y.W.; Yoon, S.O. Twist and Snail/Slug expression in oropharyngeal squamous cell carcinoma in correlation with lymph node metastasis. Anticancer Res. 2019, 39, 6307–6316. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norozi, F.; Ahmadzadeh, A.; Shahjahani, M.; Shahrabi, S.; Saki, N. Twist as a new prognostic marker in hematological malignancies. Clin. Transl. Oncol. 2016, 18, 113–124. [Google Scholar] [CrossRef]
- De Morais, E.F.; Santos, H.B. de P.; Cavalcante, I.L.; Rabenhorst, S.H.B.; dos Santos, J.N.; Galvão, H.C.; de Almeida Freitas, R. Twist and E-cadherin deregulation might predict poor prognosis in lower lip squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 127, 318–329. [Google Scholar] [CrossRef]
- Wang, D.; Rai, B.; Qi, F.; Liu, T.; Wang, J.; Wang, X.; Ma, B. Influence of the twist gene on the invasion and metastasis of colon cancer. Oncol. Rep. 2018, 39, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Hu, P.; Shen, H.; Yu, J.; Liu, Q.; Du, J. Prognostic role of Twist or Snail in various carcinomas: A systematic review and meta-analysis. Eur. J. Clin. Invest. 2014, 44, 1072–1094. [Google Scholar] [CrossRef]
- Nordfors, K.; Haapasalo, J.; Mäkelä, K.; Granberg, K.J.; Nykter, M.; Korja, M.; Paavonen, T.; Haapasalo, H.; Soini, Y. Twist predicts poor outcome of patients with astrocytic glioma. J. Clin. Pathol. 2015, 68, 905–912. [Google Scholar] [CrossRef]
- Nagaishi, M.; Nobusawa, S.; Tanaka, Y.; Ikota, H.; Yokoo, H.; Nakazato, Y. Slug, Twist, and E-Cadherin as Immunohistochemical Biomarkers in Meningeal Tumors. PLoS ONE 2012, 7, e46053. [Google Scholar] [CrossRef] [Green Version]
- Galván, J.A.; González, M.V.; Crespo, G.; Folgueras, M.V.; Astudillo, A. Snail nuclear expression parallels higher malignancy potential in neuroendocrine lung tumors. Lung Cancer 2010, 69, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Huang, C.L.; Sonobe, M.; Kikuchi, R.; Ishikawa, M.; Imamura, N.; Kitamura, J.; Iwakiri, S.; Itoi, K.; Yasumizu, R.; et al. Snail expression is associated with a poor prognosis in malignant pleural mesotheliomas. Ann. Thorac. Surg. 2013, 95, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2018, 9, 5492–5508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Jeong, S. Mutation hotspots in the β-catenin gene: Lessons from the human cancer genome databases. Mol. Cells 2019, 42, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.I.; Hsu, M.Y.; Chou, C.H.; Wang, C.; Lo, Y.S.; Loh, J.K.; Howng, S.L.; Hong, Y.R. CTNNB1 (β-catenin) mutation is rare in brain tumours but involved as a sporadic event in a brain metastasis. Acta Neurochir. 2009, 151, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Nishida, Y.; Hamada, S.; Koike, H.; Ikuta, K.; Ota, T.; Ishiguro, N. Immunohistochemical staining with non-phospho β-catenin as a diagnostic and prognostic tool of COX-2 inhibitor therapy for patients with extra-peritoneal desmoid-type fibromatosis. Diagn. Pathol. 2017, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol. Pharmacol. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Shimada, S.; Ishizawa, K.; Hirose, T. Expression of E-cadherin and catenins in meningioma: Ubiquitous expression and its irrelevance to malignancy. Pathol. Int. 2005. [Google Scholar] [CrossRef]
- Brunner, E.C.; Romeike, F.M.; Jung, M.; Comtesse, N.; Meese, E. Altered expression of b-catenin/E-cadherin in meningiomas. Histopathology 2006, 49, 178–187. [Google Scholar] [CrossRef]
- Rutkowski, R.; Chrzanowski, R.; Trwoga, M.; Kochanowicz, J.; Turek, G.; Mariak, Z.; Reszeć, J. Expression of N-cadherin and β-catenin in human meningioma in correlation with peritumoral edema. Int. J. Neurosci. 2018, 128, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z. E-cadherin’s dark side: Possible role in tumor progression. Biochim. Biophys. Acta 2012, 1826, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-cadherin cell surface regulation in cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Natl. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | (Pre)Denaturation | Denaturation | Annealing | Extending | No. of Cycles |
|---|---|---|---|---|---|
| D16S752 (CDH1) | 96 °C/3 min | 96 °C/30 s | 55 °C/35 s | 72 °C/30 + 1 s | 35 |
| D16S3025 (CDH1) | 95 °C/10 min | 95 °C/45 s | 48 °C/30 s | 72 °C/1 min | 30 |
| D18S66 (CDH2) | 95 °C/5 min | 95 °C/35 s | 56 °C/35 s | 72 °C/30 + 1 s | 35 |
| D18S819 (CDH2) | 95 °C/5 min | 95 °C/35 s | 56 °C/35 s | 72 °C/30 + 1 s | 35 |
| CTNNB1 (exon 3) | 96 °C/3 min | 96 °C/30 s | 55 °C/35 s | 72 °C/30 + 1 s | 40 |
| Correlations | E-Cadherin | N-Cadherin | NON-P β-Catenin | β-Catenin | SNAIL & SLUG | TWIST1 | |
|---|---|---|---|---|---|---|---|
| E-cadherin | Pearson Correlation | 1 | 0.140 | 0.247 * | 0.373 ** | 0.227 | 0.005 |
| Sig. (2-tailed) | 0.240 | 0.037 | 0.001 | 0.056 | 0.970 | ||
| N | 72 | 72 | 72 | 72 | 72 | 72 | |
| N-cadherin | Pearson Correlation | 0.140 | 1 | 0.277 * | 0.305 ** | −0.008 | −0.035 |
| Sig. (2-tailed) | 0.240 | 0.018 | 0.009 | 0.948 | 0.769 | ||
| N | 72 | 72 | 72 | 72 | 72 | 72 | |
| NON-P β-catenin | Pearson Correlation | 0.247 * | 0.277 * | 1 | 0.628 ** | 0.279 * | 0.233 * |
| Sig. (2-tailed) | 0.037 | 0.018 | 0.000 | 0.017 | 0.048 | ||
| N | 72 | 72 | 72 | 72 | 72 | 72 | |
| β-catenin | Pearson Correlation | 0.373 ** | 0.305 ** | 0.628 ** | 1 | 0.204 | 0.252 * |
| Sig. (2-tailed) | 0.001 | 0.009 | 0.000 | 0.086 | 0.033 | ||
| N | 72 | 72 | 72 | 72 | 72 | 72 | |
| SNAIL & SLUG | Pearson Correlation | 0.227 | −0.008 | 0.279 * | 0.204 | 1 | 0.251 * |
| Sig. (2-tailed) | 0.056 | 0.948 | 0.017 | 0.086 | 0.033 | ||
| N | 72 | 72 | 72 | 72 | 72 | 72 | |
| TWIST1 | Pearson Correlation | 0.005 | −0.035 | 0.233 * | 0.252 * | 0.251 * | 1 |
| Sig. (2-tailed) | 0.970 | 0.769 | 0.048 | 0.033 | 0.033 | ||
| N | 72 | 72 | 72 | 72 | 72 | 72 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukovac, A.; Kafka, A.; Raguž, M.; Brlek, P.; Dragičević, K.; Müller, D.; Pećina-Šlaus, N. Are We Benign? What Can Wnt Signaling Pathway and Epithelial to Mesenchymal Transition Tell Us about Intracranial Meningioma Progression. Cancers 2021, 13, 1633. https://doi.org/10.3390/cancers13071633
Bukovac A, Kafka A, Raguž M, Brlek P, Dragičević K, Müller D, Pećina-Šlaus N. Are We Benign? What Can Wnt Signaling Pathway and Epithelial to Mesenchymal Transition Tell Us about Intracranial Meningioma Progression. Cancers. 2021; 13(7):1633. https://doi.org/10.3390/cancers13071633
Chicago/Turabian StyleBukovac, Anja, Anja Kafka, Marina Raguž, Petar Brlek, Katarina Dragičević, Danko Müller, and Nives Pećina-Šlaus. 2021. "Are We Benign? What Can Wnt Signaling Pathway and Epithelial to Mesenchymal Transition Tell Us about Intracranial Meningioma Progression" Cancers 13, no. 7: 1633. https://doi.org/10.3390/cancers13071633