
Clinical and Molecular Characteristics and Outcome of Cystic Partially Differentiated Nephroblastoma and Cystic Nephroma: A Narrative Review of the Literature

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
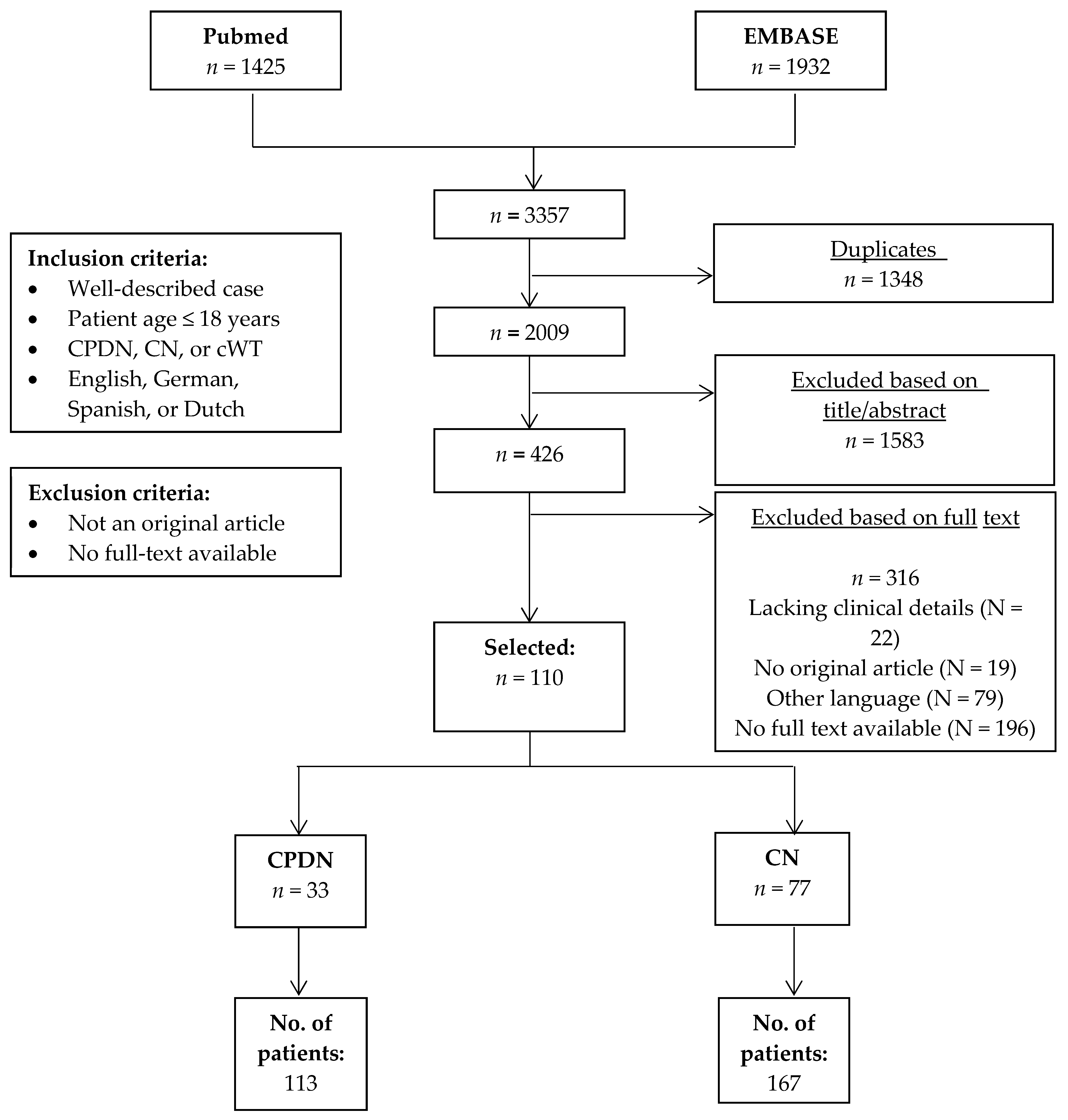
2. Methods
2.1. Literature Search
2.2. Classification of Tumors
3. Results
3.1. Cystic Partially Differentiated Nephroblastoma (CPDN)
3.1.1. Clinical Characteristics and Disease Stage
3.1.2. Treatment and Outcome
3.1.3. Molecular Testing and Genetic Predisposition

{kind=link}
| Reference | No. of Tested Patients | Karyotype | DICER1 Mutation | Other molecular/Germline Aberrations | |
|---|---|---|---|---|---|
| Somatic | Germline | ||||
| Charles et al. [10] | 1 | 52, XY, +2, +5, +7, +8, +17, +18. LOH p13b, p15T, and 16q in CPDN and nephrogenic rest (somatic) | NA | NA | - |
| Cummings et al. [9] | 1 | 46XY, no LOH 11p13, 11p15, 16q, or 7p (somatic) | NA | NA | - |
| De Chadarévian et al. [27] | 1 | 51XY, +7, +8, +12, +13, +17, one cell disomic for chromosome 7 (somatic) | NA | NA | - |
| Doros et al. [32] | 6 | Not performed | 0/6 | 0/6 | - |
| Kaneko et al. [29] | 2 | Case no. 1 (74 in article): 50, XY, +6, +7 + 12, + 13, + 18/51, + 17 Case no. 2 (384 in article): 50, X, -Y, +6, +12, +13, +der(?)t(1;)(q21;?), +der(?)t(1;?)(q21;?) (both somatic) | NA | NA | - |
| MdZin et al. [11] | 1 | Hyperdiploidy with presence of trisomy 12 (somatic) | NA | NA | - |
| Stout et al. [14] | 1 | Not performed | 0/1 | 0/1 | - |
| Timmons et al. [28] | 1 | Clonal hyperdiploid karyotype, mostly: 51, XY, +8, + 12, +17, + 19, +20. trisomy 8 was present in every hyperdiploid examined. Of the three hyperdiploid cells with 50 chromosomes, two lacked the third copy of chromosome 17 and one lacked the third copy of chromosome 12 (somatic) | NA | NA | - |
| Furukawa et al. [25] | 1 | Clonal hyperdiploid karyotype, mosaic trisomies occurring in 18 to 23% of somatic cells, in nephroblastoma cells mostly: 51, XX, +2, +7, +8, +10, +12. Trisomy 11, 18, 19, and 20 and monosomy 17 were also present in some aneuoploid cells (somatic and germline) | NA | NA | Mosaic variegated aneuploidy |
| Taskinen et al. [30] | 1 | Not performed | NA | NA | Mulibrey Nanism (germline TRIM37 mutation) |
| TOTAL | 16 | TOTAL | 0 | 0 | |
3.2. Cystic Nephroma (CN)
3.2.1. Clinical Characteristics and Disease Stage
3.2.2. Treatment and Outcome
3.2.3. Molecular Testing and Genetic Predisposition
3.3. Differential Diagnosis of CN and CPDN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- van den Heuvel-Eibrink, M.M.; Hol, J.A.; Pritchard-Jones, K.; van Tinteren, H.; Furtwangler, R.; Verschuur, A.C.; Vujanic, G.M.; Leuschner, I.; Brok, J.; Rübe, C.; et al. Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat. Rev. Urol. 2017, 14, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Hoek, J.; De Krijger, R.; Van De Ven, K.; Lequin, M.; Van Den Heuvel-Eibrink, M.M. Cystic nephroma, cystic partially differentiated nephroblastoma and cystic Wilms’ tumor in children: A spectrum with therapeutic dilemmas. Urol. Int. 2009, 82, 65–70. [Google Scholar] [CrossRef]
- Luithle, T.; Szavay, P.; Furtwängler, R.; Graf, N.; Fuchs, J. Treatment of Cystic Nephroma and Cystic Partially Differentiated Nephroblastoma-A Report From the SIOP/GPOH Study Group. J. Urol. 2007, 177, 294–296. [Google Scholar] [CrossRef]
- Cajaiba, M.M.; Khanna, G.; Smith, E.A.; Gellert, L.; Chi, Y.Y.; Mullen, E.A.; Hill, D.A.; Geller, J.I.; Dome, J.S.; Perlman, E.J. Pediatric cystic nephromas: Distinctive features and frequent DICER1 mutations. Hum. Pathol. 2016, 48, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujaniç, G.M.; Sandstedt, B.; Harms, D.; Kelsey, A.; Leuschner, I.; de Kraker, J. Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood. Med. Pediatr. Oncol. 2002, 38, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Andrews Jr, M.J.; Askin, F.B.; Fried, F.A.; McMillan, C.W.; Mandell, J. Cystic partially differentiated nephroblastoma and polycystic Wilms tumor: A spectrum of related clinical and pathologic entities. J. Urol. 1983, 129, 577–580. [Google Scholar] [CrossRef]
- Fowler, M. Differentiated nephroblastoma: Solid, cystic or mixed. J. Pathol. 1971, 105, 215–218. [Google Scholar] [CrossRef]
- Joshi, V.V.; Beckwith, J.B. Multilocular cyst of the kidney (cystic nephroma) and cystic, partially differentiated nephroblastoma. Terminology and criteria for diagnosis. Cancer 1989, 64, 466–479. [Google Scholar] [CrossRef]
- Cummings, M.; Brown, K.W. Low frequency of genetic lesions in Wilms tumors by representational difference analysis. Cancer Genet. Cytogenet. 2001, 127, 155–160. [Google Scholar] [CrossRef]
- Charles, A.K.; Brown, K.W.; Berry, P.J. Microdissecting the genetic events in nephrogenic rests and wilms’ tumor development. Am. J. Pathol. 1998, 153, 991–1000. [Google Scholar] [CrossRef]
- MdZin, R.; Murch, A.; Charles, A. Cytogenetic findings in Wilms’ tumour: A single institute study. Pathology 2010, 42, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Blakely, M.L.; Shamberger, R.C.; Norkool, P.; Beckwith, J.B.; Green, D.M.; Ritchey, M.L. Outcome of children with cystic partially differentiated nephroblastoma treated with or without chemotherapy. J. Pediatr. Surg. 2003, 38, 897–900. [Google Scholar] [CrossRef]
- Thambi Dorai, C.R.; Boucaut, H.A.P.; Le Quesne, G.W.; Toogood, I.J.R.; Bourne, A.J.; Byard, R.W. Unilateral cystic, partially-differentiated nephroblastoma with bilateral nephroblastomatosis. Pediatr. Surg. Int. 1994, 9, 137–140. [Google Scholar] [CrossRef]
- Stout, T.E.; Au, J.K.; Hicks, J.M.; Gargollo, P.C. A Case of Bilateral Cystic Partially Differentiated Nephroblastoma vs Cystic Wilms’ Tumor: Highlighting a Diagnostic Dilemma. Urology 2016, 92, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Kurian, J.J.; Jehangir, S.; Korula, A. Multiloculated Cystic Renal Tumors of Childhood: Has the Final Word Been Spoken. J. Indian Assoc. Pediatr. Surg. 2018, 23, 22–26. [Google Scholar] [CrossRef]
- Vujaniç, G.M.; Gessler, M.; Ooms, A.H.A.G.; Collini, P.; Coulomb-l’Hermine, A.; D’Hooghe, E.; de Krijger, R.R.; Perotti, D.; Pritchard-Jones, K.; Vokuhl, C.; et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat. Rev. Urol. 2018, 15, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odubanjo, M.O.; Ademuyiwa, A.O.; Daramola, A.O.; Orah, N.O.; Elebute, O.A.; Abdulkareem, F.B.; Akinda, O.R. Cystic poorly differentiated nephroblastoma: A case report and review of literature. Afr. J. Urol. 2014, 20, 144–148. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.M.; Viero, S.; Kim, P.C.; Grant, R.M. Stage III cystic partially differentiated nephroblastoma recurring after nephrectomy and chemotherapy. Pediatr. Blood Cancer 2008, 50, 129–131. [Google Scholar] [CrossRef]
- Saula, P.W.; Hadley, G.P. Pediatric non-wilms’ renal tumors: A third world experience. World J. Surg. 2012, 36, 565–572. [Google Scholar] [CrossRef]
- Babut, J.M.; Bawab, F.; Jouan, H.; Coeurdacier, P.; Treguier, C.; Fremond, B. Renal cystic tumours in children—A diagnostic challenge. Eur. J. Pediatr. Surg. 1993, 3, 157–160. [Google Scholar] [CrossRef]
- Patriarca, C.; Orazi, A.; Massimino, M.; Luksch, R. A cystic partially differentiated nephroblastoma producing α-fetoprotein. Am. J. Pediatr. Hematol. Oncol. 1992, 14, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M. Cystic partially differentiated nephroblastoma. J. Pathol. 1975, 115, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, L.P.V.; Carvalho, A.C.M.; Silva, I.M.; Martins, F.P.; Amaro, A.P.; Carvalho, E.M. Cystic partially differentiated nephroblastoma: A rare pediatric renal tumor—Case report. Radiol. Case Rep. 2020, 15, 1133–1137. [Google Scholar] [CrossRef]
- Dey, P.; Das, A.; Radhika, S. Fine needle aspiration cytology of cystic partially differentiated nephroblastoma: A case report. Acta Cytol. 1996, 40, 770–772. [Google Scholar] [CrossRef]
- Furukawa, T.; Azakami, S.; Kurosawa, H.; Ono, Y.; Ueda, Y.; Konno, Y. Cystic Partially Differentiated Nephroblastoma, Embryonal Rhabdomyosarcoma, and Multiple Congenital Anomalies Associated with Variegated Mosaic Aneuploidy and Premature Centromere Division: A Case Report. J. Pediatr. Hematol. Oncol. 2003, 25, 896–899. [Google Scholar] [CrossRef]
- Billiet, I.; Van Poppel, H.; Baert, L. Actual approach to cystic nephroma. Eur. Urol. 1988, 14, 280–286. [Google Scholar] [CrossRef] [PubMed]
- De Chadarévian, J.P.; Punnett, H.H.; Billmire, D.F.; Tomczak, E.Z. Hyperdiploidy and trisomy 12 in the cystic partially differentiated nephroblastoma. Hum. Pathol. 1996, 27, 980–981. [Google Scholar] [CrossRef]
- Timmons, C.F.; McGavran, L.; Unterkircher, L.; Beckwith, J.B.; Wilson, H.L. Hyperdiploidy including trisomy 8 in a cystic partially differentiated nephroblastoma. Cancer Genet. Cytogenet. 1989, 41, 79–85. [Google Scholar] [CrossRef]
- Kaneko, Y.; Homma, C.; Maseki, N.; Sakurai, M.; Hata, J. Correlation of chromosome abnormalities with histological and clinical features in Wilms’ and other childhood renal tumors. Cancer Res. 1991, 51, 5937–5942. [Google Scholar]
- Taskinen, S.; Lohi, J.; Kivisaari, R.; Fagerholm, R.; Rintala, R.; Taskinen, M. Segmental cystic kidney tumours in children. Scand. J. Urol. Nephrol. 2009, 43, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Kajii, T.; Ikeuchi, T.; Yang, Z.Q.; Nakamura, Y.; Tsuji, Y.; Yokomori, K.; Kawamura, M.; Fukuda, S.; Horita, S.; Asamoto, A. Cancer-prone syndrome of mosaic variegated aneuploidy and total premature chromatid separation: Report of five infants. Am. J. Med. Genet. 2001, 104, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doros, L.A.; Rossi, C.T.; Yang, J.; Field, A.; Williams, G.M.; Messinger, Y.; Cajaiba, M.M.; Perlman, E.J.; Schultz, K.A.; Cathro, H.P.; et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod. Pathol. 2014, 27, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, F.A.; McKenna, P.H. Partial nephrectomy in a metachronous multilocular cyst of the kidney (cystic nephroma). J. Urol. 1994, 151, 1358–1360. [Google Scholar] [CrossRef]
- Boybeyi, O.; Karnak, I.; Orhan, D.; Ciftci, A.O.; Tanyel, F.C.; Kale, G.; Şenocak, M.E. Cystic nephroma and localized renal cystic disease in children: Diagnostic clues and management. J. Pediatr. Surg. 2008, 43, 1985–1989. [Google Scholar] [CrossRef]
- Ishida, Y.; Kato, K.; Kigasawa, H.; Ohama, Y.; Ijiri, R.; Tanaka, Y. Synchronous occurrence of pleuropulmonary blastoma and cystic nephroma: Possible genetic link in cystic lesions of the lung and the kidney. Med. Pediatr. Oncol. 2000, 35, 85–87. [Google Scholar] [CrossRef]
- Vujaniç, G.M.; Jenney, M.E.; Adams, H.; Meyrick, S.M. Juxtaposed cystic nephroma and Wilms’ tumor. Pediatr. Dev. Pathol. 2000, 3, 91–94. [Google Scholar] [PubMed]
- Frazier, T.H. Multilocular cysts of the kidney. J. Urol. 1951, 65, 351–363. [Google Scholar] [CrossRef]
- Gallo, G.E.; Penchansky, L. Cystic nephroma. Cancer 1977, 39, 1322–1327. [Google Scholar] [CrossRef]
- Staicu, A.; Popa-Stanila, R.; Gheban, D.; Chiriac, L.; Turcu, F.R.; Caracostea, G.; Stamatian, F. Imagistic and histopathological description of a cystic nephroma during early second trimester of gestation. Case report. Med. Ultrason. 2016, 19, 327–329. [Google Scholar] [CrossRef] [Green Version]
- Bouron-Dal Soglio, D.; Harvey, I.; Yazbeck, S.; Rypens, F.; Oligny, L.L.; Fournet, J.C. An association of pleuropulmonary blastoma and cystic nephroma: Possible genetic association. Pediatr. Dev. Pathol. 2006, 9, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Lenz, M.P.; Warmann, S.W.; Scheel-Walter, H.G.; Schafer, J.; Wehrmann, M.; Hacker, H.W.; Fuchs, F. A complicated case of bilateral cystic nephroma in a 16-month-old boy. Pediatr. Surg. Int. 2005, 21, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.F.; Zanetti, G.; Rosito, P.; Capucci, M.C.; Federici, S.; Malossi, R.; Di Caro, A. Cystic nephroma in children. Report of a case. Tumori 1986, 72, 99–104. [Google Scholar] [CrossRef]
- Garrett, A.; Carty, H.; Pilling, D. Multilocular cystic nephroma: Report of three cases. Clin. Radiol. 1987, 38, 55–57. [Google Scholar] [CrossRef]
- Faria, P.A.; Zerbini, M.C. Dedifferentiated cystic nephroma with malignant mesenchymoma as the dedifferentiated component. Pediatr. Pathol. Lab. Med. J. Soc. Pediatr. Pathol. Affil. Int. Paediatr. Pathol. Assoc. 1996, 16, 1003–1011. [Google Scholar]
- Boggs, L.K.; Kimmelstiel, P. Benign multilocular cystic nephroma: Report of two cases of so-called multilocular cyst of the kidney. J. Urol. 1956, 76, 530–541. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Das, S.; Agarwal, A.; Ghosh, D.; Chaterjee, N.; Pal, M.S. Pleuropulmonary blastema with cystic nephroma—A rare presentation and surgical dilemma. Indian J. Pediatr. 2008, 75, 1266–1268. [Google Scholar] [CrossRef]
- Bal, N.; Kayaselcuk, F.; Polat, A.; Bolat, F.; Yilmaz, Z.; Tuncer, I. Familial cystic nephroma in two siblings with pleuropulmonary blastoma. Pathol. Oncol. Res. 2005, 11, 53–56. [Google Scholar] [CrossRef]
- de Kock, L.; Geoffrion, D.; Rivera, B.; Wagener, R.; Sabbaghian, N.; Bens, S.; Ellezam, B.; Bouron-Dal Soglio, D.; Ordóñez, J.; Sacharow, S.; et al. Multiple DICER1-related tumors in a child with a large interstitial 14q32 deletion. Genes Chromosomes Cancer 2018, 57, 223–230. [Google Scholar] [CrossRef]
- Saskin, A.; de Kock, L.; Sabbaghian, N.; Apellaniz-Ruiz, M.; Bozkurt, C.; Bouron-Dal Soglio, D.; Foulkes, W.D. A case of neuroblastoma in DICER1 syndrome: Chance finding or noncanonical causation? Pediatr. Blood Cancer 2018, 65, e26715. [Google Scholar] [CrossRef]
- Fernández-Martinez, L.; Villegas, J.A.; Santamaria, I.; Pitiot, A.S.; Alvarado, M.G.; Fernandez, S.; Torres, H.; Paredes, A.; Blay, P.; Balbín, M. Identification of somatic and germ-line DICER1 mutations in pleuropulmonary blastoma, cystic nephroma and rhabdomyosarcoma tumors within a DICER1 syndrome pedigree. BMC Cancer 2017, 17, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kock, L.; Druker, H.; Weber, E.; Hamel, N.; Traubici, J.; Malkin, D.; Arseneau, J.; Stewart, C.J.R.; Bouron-Dal Soglio, D.; Priest, J.R.; et al. Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum. Pathol. 2015, 46, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.K.; Cotter, M.B.; Pears, J.; McDermott, M.B.; Fabian, M.R.; Foulkes, W.D.; O’Sullivan, M.J. Tumor progression in DICER1-mutated cystic nephroma—witnessing the genesis of anaplastic sarcoma of the kidney. Hum. Pathol. 2016, 53, 114–120. [Google Scholar] [CrossRef]
- Apellaniz-Ruiz, M.; Cullinan, N.; Grant, R.; Marrano, P.; Priest, J.R.; Thorner, P.S.; Goudie, C.; Foulkes, W.D. DICER1 screening in 15 paediatric paratesticular sarcomas unveils an unusual DICER1-associated sarcoma. J. Pathol. Clin. Res. 2020, 6, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Faure, A.; Atkinson, J.; Bouty, A.; O’Brien, M.; Levard, G.; Hutson, J.; Heloury, Y. DICER1 pleuropulmonary blastoma familial tumour predisposition syndrome: What the paediatric urologist needs to know. J. Pediatr. Urol. 2016, 12, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.T.; Martinez-Rios, C.; de la Puente Gregorio, A.; Ahyad, R.A.; Villani, A.; Druker, H.; van Engelen, K.; Gallinger, B.; Aronoff, L.; Grant, R.; et al. Pediatric imaging in DICER1 syndrome. Pediatr Radiol. 2017, 47, 1292–1301. [Google Scholar] [CrossRef]
- Dural, O.; Kebudi, R.; Yavuz, E.; Yilmaz, I.; Buyukkapu Bay, S.; Schultz, K.A.P.; Hill, D.A. DICER1-Related Embryonal Rhabdomyosarcoma of the Uterine Corpus in a Prepubertal Girl. J. Pediatr. Adolesc. Gynecol. 2020, 33, 173–176. [Google Scholar] [CrossRef]
- Bardón-Cancho, E.J.; Haro-Diaz, A.; Alonso-Garcia-de la Rosa, F.J.; Huerta-Aragones, J.; Garcia-Morin, M.; Gonzalez-Martinez, F.; Garrido-Colino, C. DICER1 mutation and tumors associated with a familial tumor predisposition syndrome: Practical considerations. Fam. Cancer 2017, 16, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pawel, B.R.; Hill, D.A.; Epstein, J.I.; Argani, P. Pediatric Cystic Nephroma Is Morphologically, Immunohistochemically, and Genetically Distinct From Adult Cystic Nephroma. Am. J. Surg. Pathol. 2017, 41, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.K.; Sharma, P.D.; Mittal, A.; Sharma, A. Bilateral cystic nephroma with pleuropulmonary blastoma. BMJ Case Rep. 2011, 2011, bcr0520114171. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, I.S.; Fitzpatrick, M.; Brownlee, K.; Bhuskute, N.; Elliott, M.; Powis, M.; Ahmad, N.; Tyerman, K. Bilateral progressive cystic nephroma in a 9-month-old male infant requiring renal replacement therapy. Pediatr. Nephrol. 2010, 25, 1755–1758. [Google Scholar] [CrossRef] [PubMed]
- Nisen, P.D.; Rich, M.A.; Gloster, E.; Valderrama, E.; Saric, O.; Shende, A.; Lanzkowsky, P.; Alt, F.W. N-myc oncogene expression in histopathologically unrelated bilateral pediatric renal tumors. Cancer 1988, 61, 1821–1826. [Google Scholar] [CrossRef]
- Kousari, Y.M.; Khanna, G.; Hill, D.A.; Dehner, L.P. Case 211: Pleuropulmonary blastoma in association with cystic nephroma-DICER1 syndrome. Radiology 2014, 273, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Boman, F.; Hill, D.A.; Williams, G.M.; Chauvenet, A.; Fournet, J.C.; Bouron-dal Soglio, D.; Messinger, Y.; Priest, J.R. Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: A report from the International Pleuropulmonary Blastoma Registry. J. Pediatr. 2006, 149, 850–854. [Google Scholar] [CrossRef]
- Gimpel, C.; Avni, E.F.; Breysem, L.; Burgmaier, K.; Caroli, A.; Cetiner, M.; Haffner, D.; Hartung, E.A.; Franke, D.; König, J.; et al. Imaging of Kidney Cysts and Cystic Kidney Diseases in Children: An International Working Group Consensus Statement. Radiology 2019, 290, 769–782. [Google Scholar] [CrossRef] [Green Version]
- Breslow, N.; Olshan, A.; Beckwith, J.B.; Green, D.M. Epidemiology of Wilms tumor. Med. Pediatr. Oncol. 1993, 21, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.D.; Fitzgibbon, W.; Sas, K.; Stenbit, A.E.; Amria, M.; Houston, A.; Reichert, R.; Gilley, S.; Siegal, G.P.; Bissler, J.; et al. Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J. Am. Soc. Nephrol. 2011, 22, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.H.A.G.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.R.; et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, C.; Ozkaynak, M.F.; Tugal, O.; Jayabose, S. Hyperdiploidy in a case of favorable histology Wilms tumor. Cancer Genet. Cytogenet. 2001, 127, 89–90. [Google Scholar] [CrossRef]
- Jiménez, I.; Chicard, M.; Colmet-Daage, L.; Clément, N.; Danzon, A. Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. Int. J. Cancer 2019, 144, 68–79. [Google Scholar] [CrossRef] [Green Version]

| Tumor Type | No. of Patients | Sex (M/F/NA) | Median Age (Months) | Median Tumor Weight (Grams) | Median Tumor Volume (mL) | Stage | LN + Patients/Tested Patients | Relapse | Survival | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | NA | |||||||||
| CPDN | 113 | 73/38/2 | 12 (0.1–36) | 484 (170–3110) | 338 (122–405) | 62 | 1 | 9 | 0 | 5 | 37 | 0/52 | 4 | 107 |
| CN | 167 | 93/72/2 | 16 (0–198) | 540 (240–4000) | 292 (6.7–1490) | 51 | 0 | 1 | 0 | 9 | 106 | 0/24 | 1 | 161 |
| Tumor Type | No. of Patients | Pre-Op CT | Pre-Op RT | Surgery Type | Post-Op Treatment | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TN | NSS | NA | None | CT | RT | CT + RT | NA | No Post-Op Therapy | ||||
| CPDN | 113 | 20 | 0 | 94 | 9 | 9 | 1 | 40 | 0 | 7 | 15 | 52 |
| CN | 167 | 9 | 5 | 121 | 28 | 16 | 2 | 5 | 0 | 0 | 110 | 52 |
| # | Type | Age | Sex | Preop Tx | Initial Histology | Surgery | Postop Tx | Time to Relapse | Histology of Relapse | Relapse Treatment | Survival |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CPDN [8] # | 4m | M | None | >50% of the tissue in the septa contained immature elements. Possibly positive margins. | TN, possibly incomplete | None | 1 y/2 y *** | NA | Surgery | Yes |
| 2 | CPDN [18] # | 1y | M | None | Pre- and intraoperative cyst rupture, cystic tumor, septa contained tissue reminiscent of triphasic WT with nodules of blastema | TN, intraoperative tumor rupture | Chemo: Vincristine and actinomycin-D | 6 weeks (while on chemo) | WT | Chemo, surgery | Yes |
| 3 | CPDN bilateral [15] # | 9m | M | None | NA | Bilateral NSS | None | 6 months | Bilateral WT | Chemo, surgery | No * |
| 4 | CPDN [15] # | 3y | M | Chemo | NA | TN (positive margins) | Chemo | 5 months | NA | Chemo | No ** |
| 5 | CN [26] | 6m | F | None | Probable positive margins | NSS (probably positive margins) | None | 3 months | NA | NA | Yes |
| Reference | No. of Tested Patients | Karyotype (Tumor; Somatic) | DICER1-Mutation Positive | |
|---|---|---|---|---|
| Tumor | Germline | |||
| Saskin et al., 2017 [49] | 1 | Not performed | c.5113G > A, p.E1705K | Heterozygous c.4566_4570dupCTTTG p.V1524Afs * 38 |
| Fernández-Martinez et al., 2017 [50] | 1 | Not performed | c.5425G > A (p.G1809R) | Heterozygous c.5387C > T; p.Q1783 * |
| De Kock et al., 2015 [51] | 1 | Not performed | c.5439G > T p.(Glu1813Asp) | Heterozygous c.1196_1197dupAG p.(Trp400Serfs * 59) |
| De Kock et al., 2018 [48] | 1 | Normal (46, XY) | c.5425G > A, p.G1809R | Heterozygous 5.82 Mb deletion at the 14q32.13q32.2 region |
| Bardón-Cancho et al., 2017 [57] | 1 | Not performed | Not tested | Heterozygous c.3540C > G; p.Y1180 |
| Cajaiba et al., 2016 [4] | 16 | Not performed | 12 patients with LOF (nonsense or frameshift) and hotspot mutations 3 patients with hotspot mutations only | Not tested * |
| Faure et al., 2016 [54] | 2 | Not performed | Not tested | Patient 1 LOF mutation Patient 2 mutation (NOS) |
| Li et al., 2017 [58] | 7 | Not performed | Patient 1. c.5125G > A; p.D1709N Patient 2. c.5437G > A; pE1813K and c.3452_3453delTG; p.V1151Efs12 * Patient 3. c.5425G > A; G1809R Patient 4. c. 5125G > A; p.D1709N and c.958A>T; p.K320 *; Patient 5. c.5428G > C; p.D1810H and c. 3091C > T; p.Q1031 * Patient 6: c.5125G > A; p.D1709N and c.1177C > T; p.Q393 * | Not tested * |
| Bueno et al., 2017 [55] | 2 | Not performed | Not tested | Patient 1: Germline DICER1 variant (NOS) Patient 2: Germline DICER1 variant (NOS) |
| Wu et al., 2016 [52] | 1 | Not performed | c.5438A > G; p.E1813G | Heterozygous c.2450delC; p.P817LfsX15 |
| Apellaniz-Ruiz et al., 2020 [53] | 1 | Not performed | c.5428G > T, p.D1810Y | c.4308_4311del, p.A1436fs |
| Dural et al., 2020 [56] | 1 | Not performed | Not tested | Heterozygous c.1525C>T p.Arg509 * |
| TOTAL | 35 | TOTAL | 27 | 12 |
| Patient | Age (Months) | Gender | Tumor | DICER1 | Survival | |
|---|---|---|---|---|---|---|
| Somatic | Germline | |||||
| CN (bilateral) [15] | 4 | Male | ERMS urethra ### | NA | NA | Yes |
| CN [49] | 14 | Female | NBL *,## | + | + | Yes |
| CN [50] | 11 | Female | PPB ### | + | + | Yes |
| CN (bilateral) [59] | 30 | Male | PPB ## | NA | NA | NA |
| CN (bilateral) [60] | 9 | Male | PPB **,## | NA | NA | Yes |
| CN [30] | 18 | Female | PPB ## | NA | NA | Yes |
| CN [46] | 20 | Male | PPB ### | NA | NA | No |
| CN [51] | 144 | Female | ERMS ovary *,**,# Focal nodular liver hyperplasia #, Fibro-adenoma of the breast ### | + | + | NA |
| CN [47] | 54 | Female | PPB ### | NA | NA | No |
| CN [48] | 12 | Male | Malignant teratoid CBME, High-grade cerebral SCS, PPB ### | Somatic DICER1 mutations in the CBME and spindle-cell sarcoma | + | No |
| CN [61] | 30 | Male | NBL ## | NA | NA | Yes |
| CN [35] | 22 | Male | PPB ## | NA | NA | Yes |
| CN [40] | 32 | Male | PPB ## | NA | NA | Yes |
| CN [54] | 66 | Female | SLCT # | - | + | NA |
| CN [38] | 24 | Female | WT ## | NA | NA | Yes |
| CN [62] | 5 | Female | PPB ## | NA | NA | NA |
| CN [63] | 11 | Female | PPB | NA | NA | Yes |
| CN (bilateral) [63] | 10 | Female | PPB ** | NA | NA | Yes |
| CN [63] | 26 | Female | PPB | NA | NA | Yes |
| CN [55] | 13 | Male | Pineal cyst | - | + | NA |
| CN [55] | 78 | Female | ERMS ovary, fibroadenomas, thyroid cysts, exostoses | - | + | NA |
| CN [36] | 21 | Female | WT ## | NA | NA | Yes |
| CN [52] | 7 | Female | ASK ## | + (in CN and ASK) | + | Yes |
| CN [53] | 10 | Male | Paratesticular sarcoma ## | + (in CN and paratesticular sarcoma) | + | Yes |
| CN [56] | 12 | Female | ERMS uterus ### | - | + | Yes |
| CN [44] | 26 | Female | Anaplastic malignant mesenchymoma of the kidney ### | NA | NA | Yes |
| CPDN [8] | 30 | Female | WT ## | NA | Yes | |
| CPDN [25] | 2 | Female | RMS # | Multiple variegated aneuploidy | No | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Peer, S.E.; Pleijte, C.J.H.; de Krijger, R.R.; Jongmans, M.C.J.; Kuiper, R.P.; Lilien, M.R.; van Grotel, M.; Graf, N.; van den Heuvel-Eibrink, M.M.; Hol, J.A. Clinical and Molecular Characteristics and Outcome of Cystic Partially Differentiated Nephroblastoma and Cystic Nephroma: A Narrative Review of the Literature. Cancers 2021, 13, 997. https://doi.org/10.3390/cancers13050997
van Peer SE, Pleijte CJH, de Krijger RR, Jongmans MCJ, Kuiper RP, Lilien MR, van Grotel M, Graf N, van den Heuvel-Eibrink MM, Hol JA. Clinical and Molecular Characteristics and Outcome of Cystic Partially Differentiated Nephroblastoma and Cystic Nephroma: A Narrative Review of the Literature. Cancers. 2021; 13(5):997. https://doi.org/10.3390/cancers13050997
Chicago/Turabian Stylevan Peer, Sophie E., Corine J. H. Pleijte, Ronald R. de Krijger, Marjolijn C. J. Jongmans, Roland P. Kuiper, Marc R. Lilien, Martine van Grotel, Norbert Graf, Marry M. van den Heuvel-Eibrink, and Janna A. Hol. 2021. "Clinical and Molecular Characteristics and Outcome of Cystic Partially Differentiated Nephroblastoma and Cystic Nephroma: A Narrative Review of the Literature" Cancers 13, no. 5: 997. https://doi.org/10.3390/cancers13050997