
Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Expression Analysis and Correlation Analysis
2.2. Cell Culture
2.3. qPCR
2.4. Drug Response Curves and Cell Proliferation
2.5. Clonogenic Assay
2.6. Western Blotting
2.7. Immunocytochemistry
2.8. Oxygen Consumption
2.9. CO2-Trapping
2.10. Ionizing Radiation
2.11. Statistics and Figures
3. Results
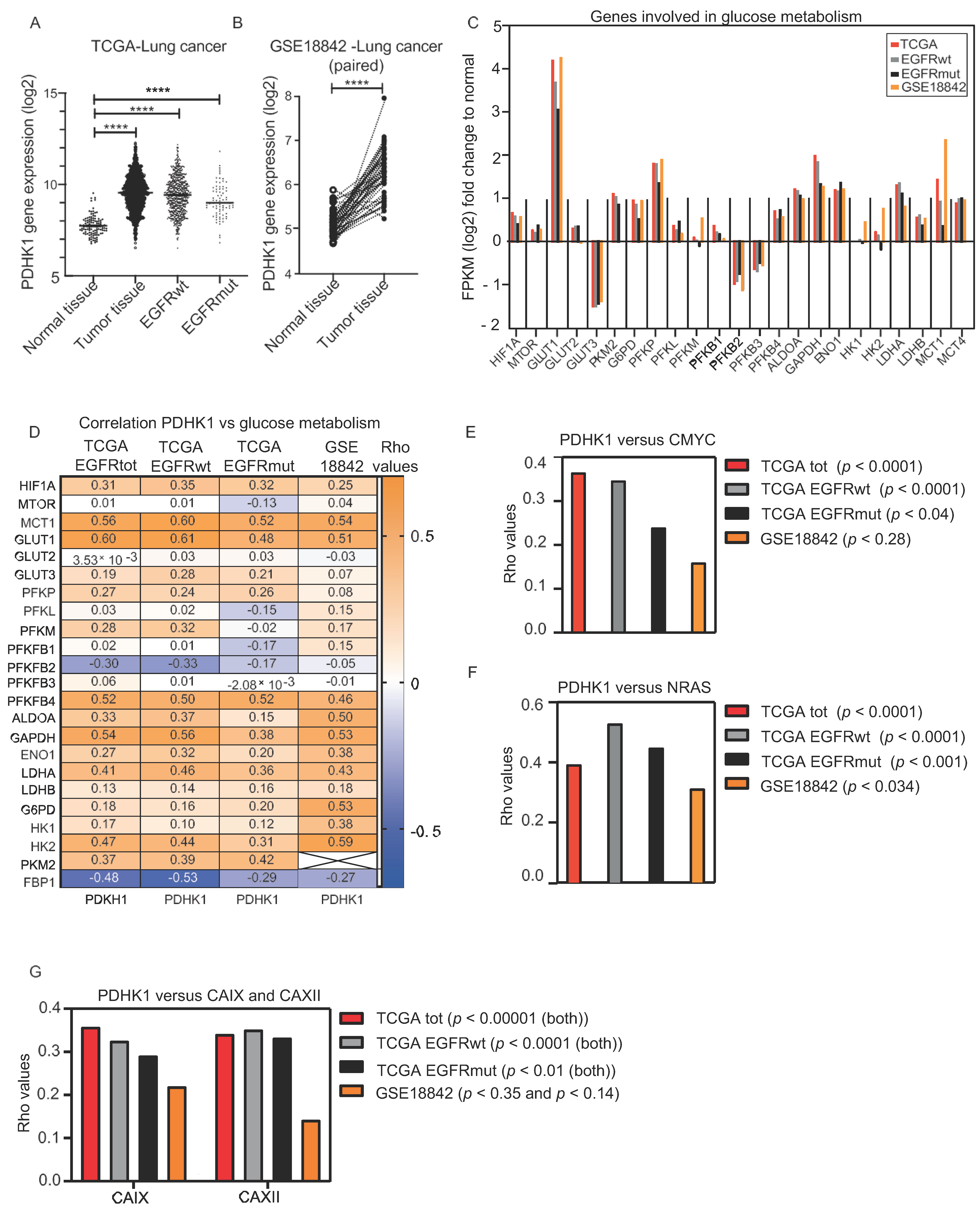
3.1. Increased PDHK1 Expression Is Associated with Altered Expression of Genes Involved in Glucose Metabolism in NSCLC Human Tumors
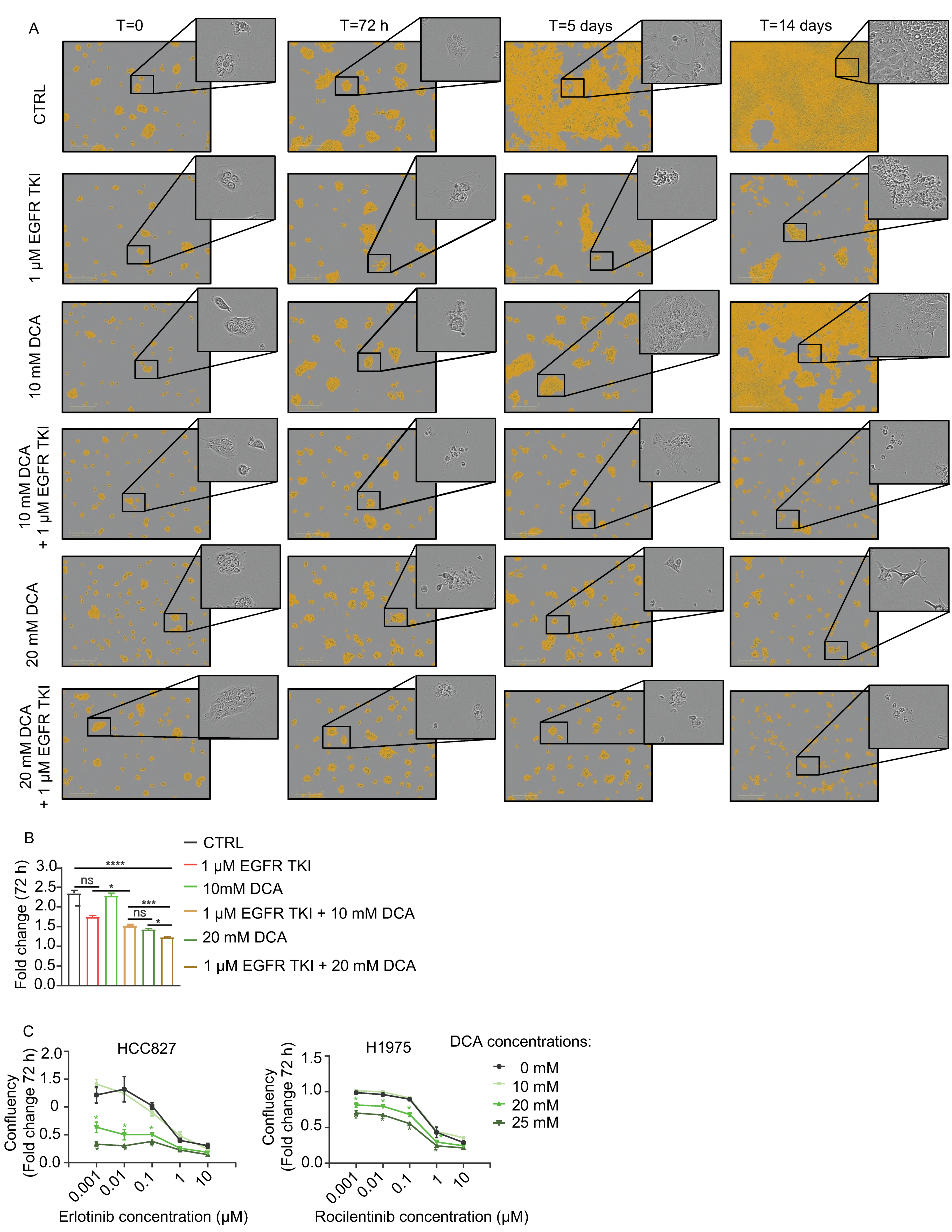
3.2. DCA Add to the Inhibitory Effect of Cell Growth by EGFR TKIs in NSCLC Cell Models
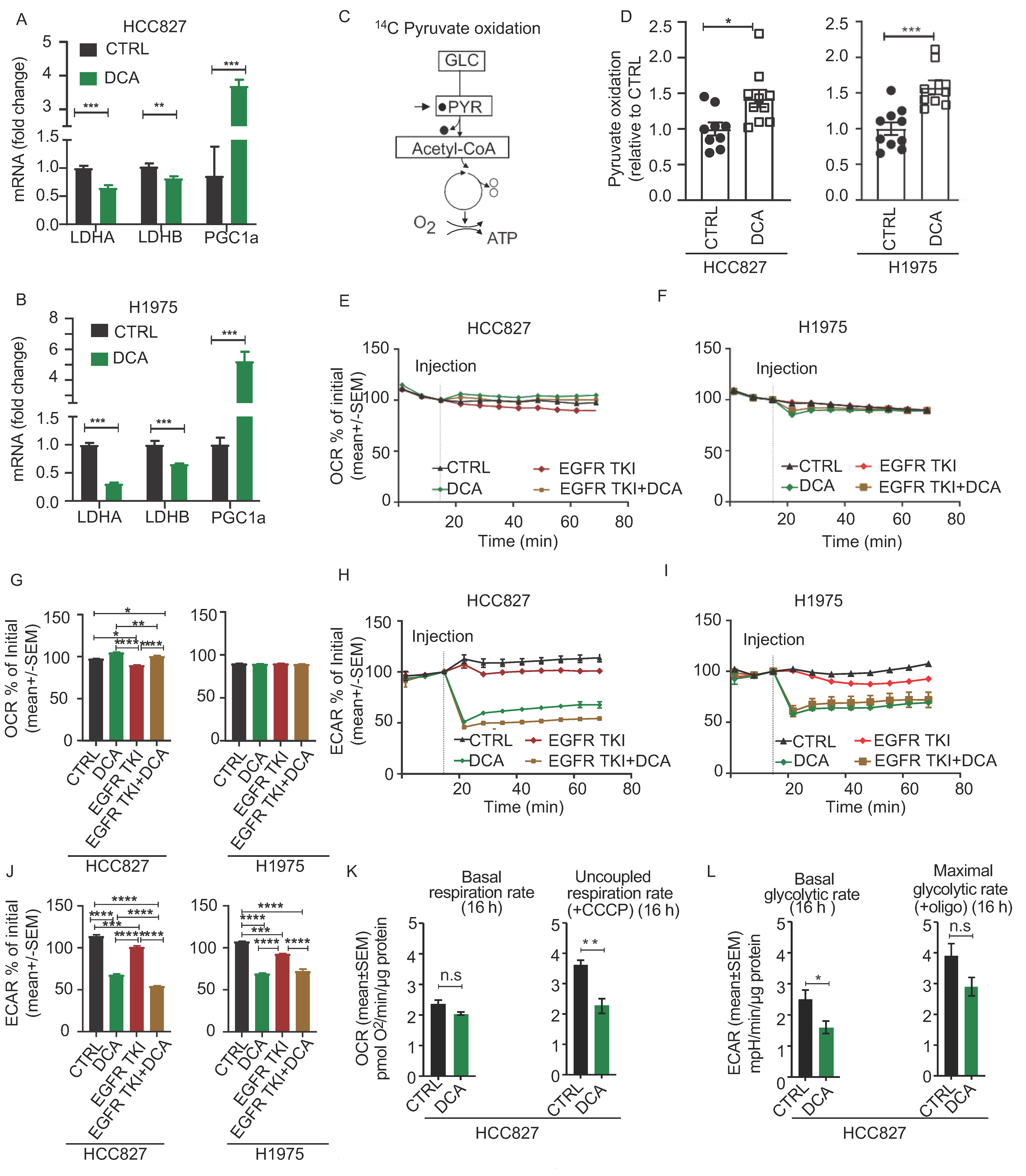
3.3. Acute Metabolic Effects of DCA in NSCLC Cell Models
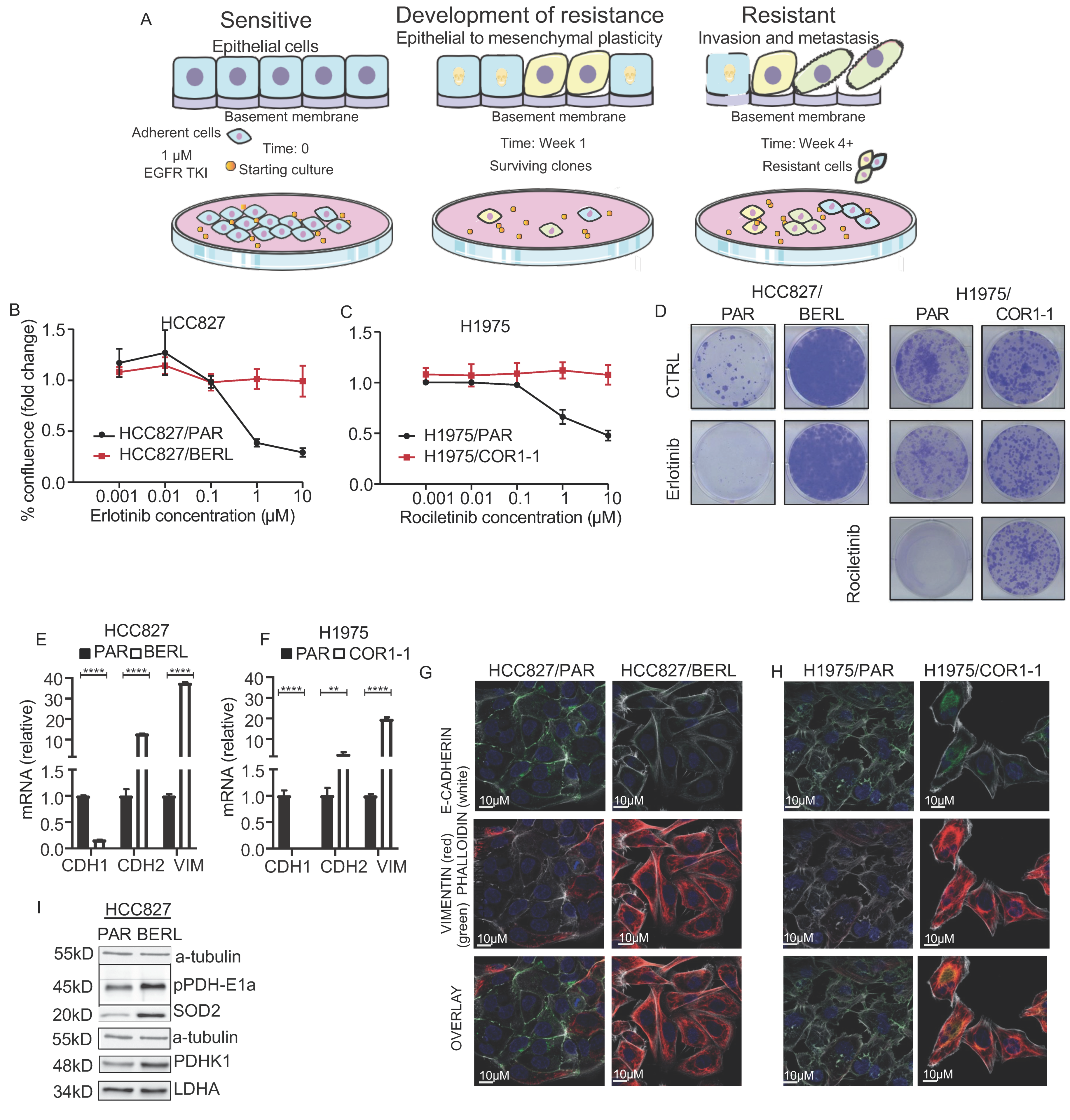
3.4. Development of EMT Mediated Resistance to EGFR TKI in NSCLC Cells
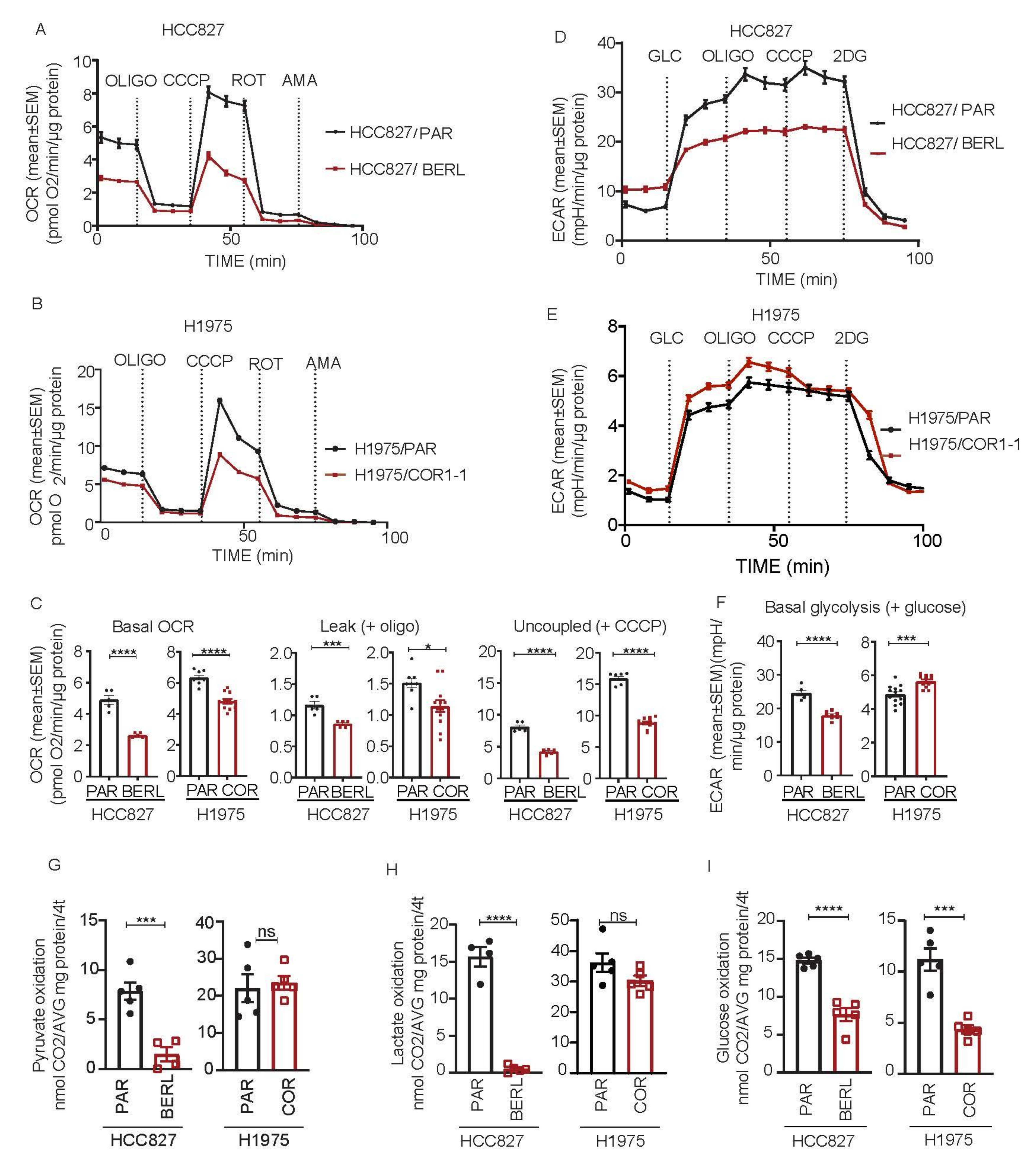
3.5. Mitochondrial Respiration Is Decreased in NSCLC in EGFR TKI Resistant Cell Models
3.6. DCA Disrupt the Metabolic Signature and Decrease Viability in NSCLC Cells Resistant to EGFR TKI Therapy
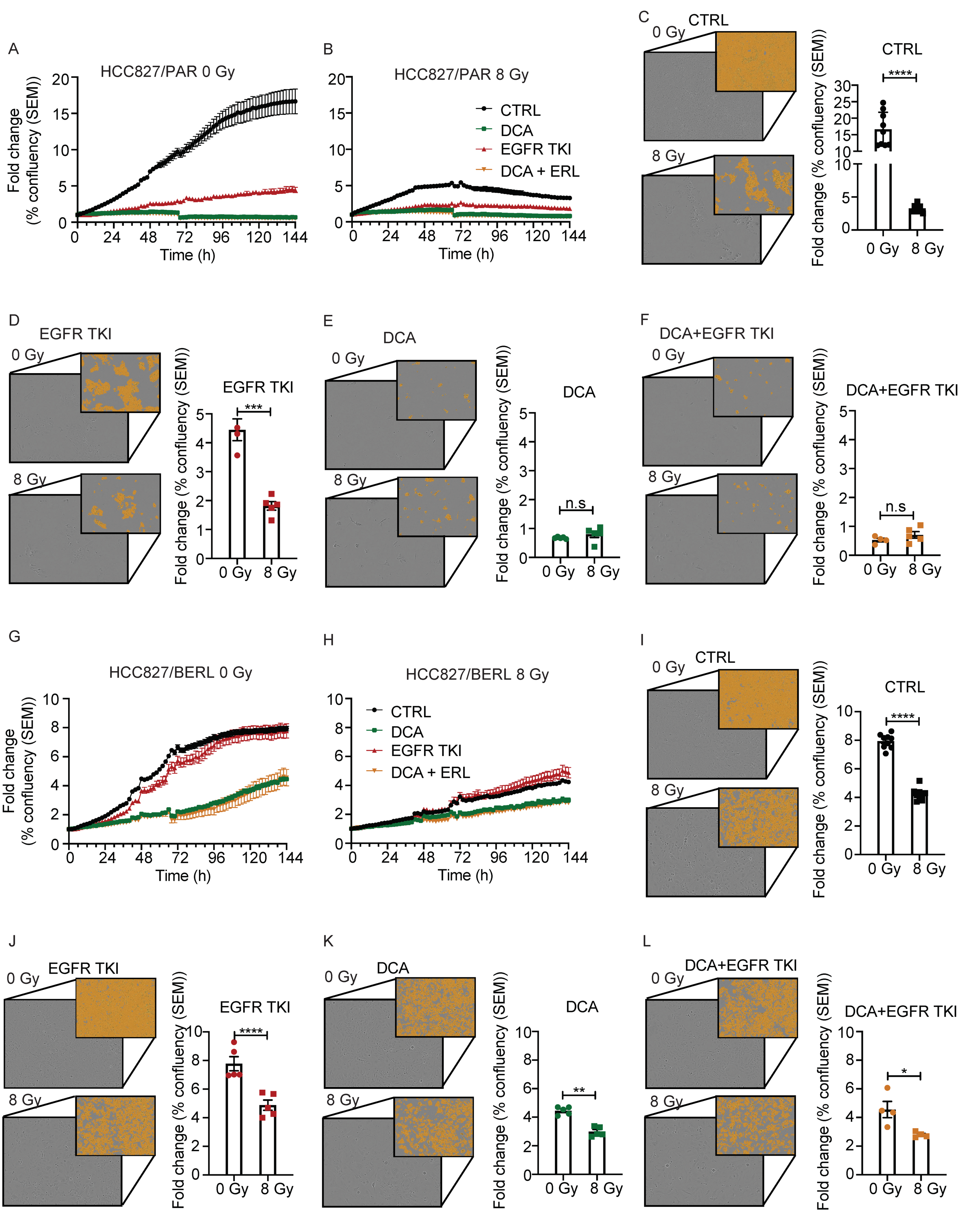
3.7. The Effect of Ionizing Radiation Is Increased in Combination with DCA and EGFR TKI in NSCLC Cells Resistant to EGFR TKI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crespi, B.; Summers, K. Evolutionary biology of cancer. Trends Ecol. Evol. 2005, 20, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 10. [Google Scholar] [CrossRef]
- Ferlay, J.; Laversanne, M.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow. International Agency for Research on Cancer: Lyon, France. Available online: https://gco.iarc.fr/tomorrow (accessed on 12 February 2021).
- Panani, A.; Roussos, C. Cytogenetic and molecular aspects of lung cancer. Cancer Lett. 2006, 239, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): The road to a success, paved with failures. Pharmacol. Ther. 2017, 174, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Asao, T.; Takahashi, F.; Takahashi, K. Resistance to molecularly targeted therapy in non-small-cell lung cancer. Respir. Investig. 2019, 57, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-S.; Gilligan, D.; Pacey, S.C. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Santos, G.D.C.; Shepherd, F.A.; Tsao, M.S. EGFR Mutations and Lung Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Ai, X.; Guo, X.; Wang, J.; Stancu, A.L.; Joslin, P.M.; Zhang, D.; Zhu, S. Targeted therapies for advanced non-small cell lung cancer. Oncotarget 2018, 9, 37589–37607. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [Green Version]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Okon, I.S.; Coughlan, K.A.; Zhang, M.; Wang, Q.; Zou, M.-H. Gefitinib-mediated Reactive Oxygen Species (ROS) Instigates Mitochondrial Dysfunction and Drug Resistance in Lung Cancer Cells. J. Biol. Chem. 2015, 290, 9101–9110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Huang, R.; Ding, J.; Ji, D.; Song, B.; Yuan, L.; Chang, H.; Chen, G. Multiple myeloma acquires resistance to EGFR inhibitor via induction of pentose phosphate pathway. Sci. Rep. 2015, 5, 9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On respiratory impariment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Shanghelin, Y.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Preter, G.; Neveu, M.-A.; Danhier, P.; Brisson, L.; Payen, V.L.; Porporato, P.E.; Jordan, B.F.; Sonveaux, P.; Gallez, B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 2015, 7, 2910–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.W.M.; Lane, A.N.; Higashi, R.M.; Farag, M.A.; Gao, H.; Bousamra, M.; Miller, D.M. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 2009, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, T.C.; Coelho, R.G.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Sola-Penna, M. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett. 2011, 585, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W.; Greene, Y.J. Dichloroacetate. Diabetes Care 1992, 15, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-W.; Lin, S.-C.; Chen, K.-F.; Lai, Y.-Y.; Tsai, S.-J. Induction of Pyruvate Dehydrogenase Kinase-3 by Hypoxia-inducible Factor-1 Promotes Metabolic Switch and Drug Resistance. J. Biol. Chem. 2008, 283, 28106–28114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holness, M.J.; Kraus, A.; Harris, R.A.; Sugden, M.C. Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 2000, 49, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Inskeep, K.; Bowker-Kinley, M.M.; Popov, K.M.; Harris, R.A. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999, 48, 1593–1599. [Google Scholar] [CrossRef]
- Pettersen, I.K.N.; Tusubira, D.; Ashrafi, H.; Dyrstad, S.E.; Hansen, L.; Liu, X.-Z.; Nilsson, L.I.H.; Løvsletten, N.G.; Berge, K.; Wergedahl, H.; et al. Upregulated PDK4 expression is a sensitive marker of increased fatty acid oxidation. Mitochondrion 2019, 49, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Cheng, Z.; Liu, Y.; Dai, Y.; Pang, Y.; Jiao, Y.; Ke, X.; Cui, W.; Zhang, Q.; Shi, J.; et al. Overexpression of PDK2 and PDK3 reflects poor prognosis in acute myeloid leukemia. Cancer Gene Ther. 2018, 27, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Lu, Y.; Xie, Y.; Qiu, S.; Li, X.; Fan, Z. Rational combination with PDK1 inhibition overcomes cetuximab resistance in head and neck squamous cell carcinoma. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of Metabolic Pathways between Cancer Cells and Stromal Cells in Colorectal Carcinomas: A Metabolic Survival Role for Tumor-Associated Stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Palencia, A.; Gomez-Morales, M.; Gomez-Capilla, J.A.; Pedraza, V.; Boyero, L.; Rosell, R.; Fárez-Vidal, M.E. Gene expression profiling reveals novel biomarkers in nonsmall cell lung cancer. Int. J. Cancer 2011, 129, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR that Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Dyrstad, S.E.; Tusubira, D.; Knappskog, S.; Tronstad, K.J.; Røsland, G.V. Introducing nano-scale quantitative polymerase chain reaction. Biochem. Biophys. Res. Commun. 2018, 506, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Røsland, G.V.; Dyrstad, S.E.; Tusubira, D.; Helwa, R.; Tan, T.Z.; Lotsberg, M.L.; Pettersen, I.K.N.; Berg, A.; Kindt, C.; Hoel, F.; et al. Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC. Cancer Metab. 2019, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Vanlinden, M.R.; Dölle, C.; Pettersen, I.K.N.; Kulikova, V.A.; Niere, M.; Agrimi, G.; Dyrstad, S.E.; Palmieri, F.; Nikiforov, A.A.; Tronstad, K.J.; et al. Subcellular Distribution of NAD+ between Cytosol and Mitochondria Determines the Metabolic Profile of Human Cells. J. Biol. Chem. 2015, 290, 27644–27659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensaas, A.J.; Rustan, A.C.; Lövstedt, K.; Kull, B.; Wikström, S.; Drevon, C.A.; Hallén, S. Cell-based multiwell assays for the detection of substrate accumulation and oxidation. J. Lipid Res. 2007, 48, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. The Interplay between MYC and HIF in the Warburg Effect. Oncog. Meet Metab. 2008, 10, 35–53. [Google Scholar] [CrossRef]
- Chesney, J.; Telang, S. Regulation of glycolytic and mitochondrial metabolism by ras. Curr. Pharm. Biotechnol. 2013, 14, 251–260. [Google Scholar] [CrossRef]
- Lotsberg, M.L.; Wnuk-Lipinska, K.; Terry, S.; Tan, T.Z.; Lu, N.; Trachsel-Moncho, L.; Røsland, G.V.; Siraji, M.I.; Hellesøy, M.; Rayford, A.; et al. AXL Targeting Abrogates Autophagic Flux and Induces Immunogenic Cell Death in Drug-Resistant Cancer Cells. J. Thorac. Oncol. 2020, 15, 973–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.J.; Yi, J.W.; Oh, S.W.; A Kim, Y.; Yi, K.H.; Kim, J.H.; Lee, K.E. Upregulation of SLC2 (GLUT) family genes is related to poor survival outcomes in papillary thyroid carcinoma: Analysis of data from The Cancer Genome Atlas. Surgery 2017, 161, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Jung, S.; Park, W.S.; Lee, J.-H.; Shin, R.; Heo, S.C.; Choe, E.K.; Lee, J.H.; Kim, K.; Chai, Y.J. Upregulation of SLC2A3 gene and prognosis in colorectal carcinoma: Analysis of TCGA data. BMC Cancer 2019, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.-E.; Kim, J.-Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Hyun, D.W.; Oh, S.-O. SLC2A2 (GLUT2) as a novel prognostic factor for hepatocellular carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilertsen, M.; Andersen, S.; Al-Saad, S.; Kiselev, Y.; Donnem, T.; Stenvold, H.; Pettersen, I.; Al-Shibli, K.; Richardsen, E.; Busund, L.-T.; et al. Monocarboxylate Transporters 1–4 in NSCLC: MCT1 Is an Independent Prognostic Marker for Survival. PLoS ONE 2014, 9, e105038. [Google Scholar] [CrossRef] [PubMed]
- Payen, V.L.; Hsu, M.Y.; Rädecke, K.S.; Wyart, E.; Vazeille, T.; Bouzin, C.; Porporato, P.E.; Sonveaux, P. Monocarboxylate Transporter MCT1 Promotes Tumor Metastasis Independently of Its Activity as a Lactate Transporter. Cancer Res. 2017, 77, 5591–5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Glare, E.M.; Divjak, M.; Bailey, M.J.; Walters, E.H. Beta-Actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels. Thorax 2002, 57, 765–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, H.J.M.; Fehrmann, R.S.N.; de Bont, E.S.J.M.; Hofstra, R.M.W.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.E.; van der Zee, A.G.J.; Meerman, G.J.T.; ter Elst, A. Evidence Based Selection of Housekeeping Genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, J.; Hu, J.; Li, D.; Wang, X.; Zhang, R.; Zhang, H.; Shi, M.; Chen, H. Cigarette smoke extract induces EGFR-TKI resistance via promoting EGFR signaling pathway and ROS generation in NSCLC cell lines. Lung Cancer 2017, 109, 109–116. [Google Scholar] [CrossRef]
- Chuang, M.-C.; Yang, Y.-H.; Tsai, Y.-H.; Hsieh, M.-J.; Lin, Y.-C.; Lin, C.-K.; Chen, P.-C.; Yang, T.-M. Survival benefit associated with metformin use in inoperable non-small cell lung cancer patients with diabetes: A population-based retrospective cohort study. PLoS ONE 2018, 13, e0191129. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.-B.; Li, T.-H.; Ren, Z.-P.; Liu, Y. Combination of 2-deoxy d-glucose and metformin for synergistic inhibition of non-small cell lung cancer: A reactive oxygen species and P-p38 mediated mechanism. Biomed. Pharmacother. 2016, 84, 1575–1584. [Google Scholar] [CrossRef]
- An, B.C.; Choi, Y.-D.; Oh, I.-J.; Kim, J.H.; Park, J.-I.; Lee, S.-W. GPx3-mediated redox signaling arrests the cell cycle and acts as a tumor suppressor in lung cancer cell lines. PLoS ONE 2018, 13, e0204170. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-like Breast Cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Van Poelje, P.D.; Potter, S.C.; Chandramouli, V.C.; Landau, B.R.; Dang, Q.; Erion, M.D. Inhibition of Fructose 1,6-Bisphosphatase Reduces Excessive Endogenous Glucose Production and Attenuates Hyperglycemia in Zucker Diabetic Fatty Rats. Diabetes 2006, 55, 1747–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, J.; Variend, S.; Howat, A.; Vawter, G. Investigation of Inborn Errors of Metabolism in Unexpected Infant Deaths. Lancet 1988, 332, 29–31. [Google Scholar] [CrossRef]
- Kitanovic, A.; Wölfl, S. Fructose-1,6-bisphosphatase mediates cellular responses to DNA damage and aging in Saccharomyces cerevisiae. Mutat. Res. Mol. Mech. Mutagen. 2006, 594, 135–147. [Google Scholar] [CrossRef]
- Son, B.; Lee, S.; Kim, H.; Kang, H.; Jeon, J.; Jo, S.; Seong, K.M.; Lee, S.-J.; Youn, H.; Youn, B. Decreased FBP1 expression rewires metabolic processes affecting aggressiveness of glioblastoma. Oncogene 2020, 39, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, E.J.; Moon, S.H.; Kim, S.; Hyun, S.H.; Cho, Y.S.; Choi, J.Y.; Kim, B.-T.; Lee, K.-H. Strong association of epidermal growth factor receptor status with breast cancer FDG uptake. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1438–1447. [Google Scholar] [CrossRef]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Jhung, S.; Sano, M.C.; Shungu, D.C.; Millar, W.S.; Hong, X.; Gooch, C.L.; Mao, X.; et al. Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurology 2006, 66, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Martyniuk, C.J.; James, M.O.; Calcutt, N.A. Dichloroacetate-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 211–238. [Google Scholar] [CrossRef]
- Holness, M.; Sugden, M. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Gore, D.C. In Vivo Metabolic Response of Glucose to Dichloroacetate in Humans. J. Surg. Res. 1996, 61, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Wells, P.G.; Moore, G.W.; Rabin, D.; Wilkinson, G.R.; Oates, J.A.; Stacpoole, P.W. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia 1980, 19, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhang, S.-L.; Hu, X.; Tam, K.Y. Inhibition of pyruvate dehydrogenase kinase 1 enhances the anti-cancer effect of EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Eur. J. Pharmacol. 2018, 838, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Yan, X.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; et al. Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells. Oncotarget 2016, 7, 59458–59470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneshmandi, S.; Wegiel, B.; Seth, P. Blockade of Lactate Dehydrogenase-A (LDH-A) Improves Efficacy of Anti-Programmed Cell Death-1 (PD-1) Therapy in Melanoma. Cancers 2019, 11, 450. [Google Scholar] [CrossRef]
- Hermans, D.; Gautam, S.; García-Cañaveras, J.C.; Gromer, D.; Mitra, S.; Spolski, R.; Li, P.; Christensen, S.; Nguyen, R.; Lin, J.-X.; et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8+ T cell stemness and antitumor immunity. Proc. Natl. Acad. Sci. USA 2020, 117, 6047–6055. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Yan, X.; Song, Q.; Wang, G.; Hu, Y.; Jiao, S.; Wang, J. Pretreatment lactate dehydrogenase may predict outcome of advanced non small-cell lung cancer patients treated with immune checkpoint inhibitors: A meta-analysis. Cancer Med. 2019, 8, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int. J. Cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, T.S.R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loriot, C.; Burnichon, N.; Gadessaud, N.; Vescovo, L.; Amar, L.; Libé, R.; Bertherat, J.; Plouin, P.-F.; Jeunemaitre, X.; Gimenez-Roqueplo, A.-P.; et al. Epithelial to Mesenchymal Transition Is Activated in Metastatic Pheochromocytomas and Paragangliomas Caused by SDHB Gene Mutations. J. Clin. Endocrinol. Metab. 2012, 97, e954–e962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, L.; Chai, W.; Zhao, T.; Jin, X.; Guo, X.; Han, L.; Yuan, C. Dichloroacetic acid upregulates apoptosis of ovarian cancer cells by regulating mitochondrial function. OncoTargets Ther. 2019, 12, 1729–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyrstad, S.E.; Lotsberg, M.L.; Tan, T.Z.; Pettersen, I.K.N.; Hjellbrekke, S.; Tusubira, D.; Engelsen, A.S.T.; Daubon, T.; Mourier, A.; Thiery, J.P.; et al. Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells. Cancers 2021, 13, 941. https://doi.org/10.3390/cancers13050941
Dyrstad SE, Lotsberg ML, Tan TZ, Pettersen IKN, Hjellbrekke S, Tusubira D, Engelsen AST, Daubon T, Mourier A, Thiery JP, et al. Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells. Cancers. 2021; 13(5):941. https://doi.org/10.3390/cancers13050941
Chicago/Turabian StyleDyrstad, Sissel E., Maria L. Lotsberg, Tuan Zea Tan, Ina K. N. Pettersen, Silje Hjellbrekke, Deusdedit Tusubira, Agnete S. T. Engelsen, Thomas Daubon, Arnaud Mourier, Jean Paul Thiery, and et al. 2021. "Blocking Aerobic Glycolysis by Targeting Pyruvate Dehydrogenase Kinase in Combination with EGFR TKI and Ionizing Radiation Increases Therapeutic Effect in Non-Small Cell Lung Cancer Cells" Cancers 13, no. 5: 941. https://doi.org/10.3390/cancers13050941