
The Mitochondrial Protease LonP1 Promotes Proteasome Inhibitor Resistance in Multiple Myeloma

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Mitochondrial Proteases in Multiple Myeloma
2.2. Regulation of LONP1 and OSGEPL1 Genes in Multiple Myeloma Cells



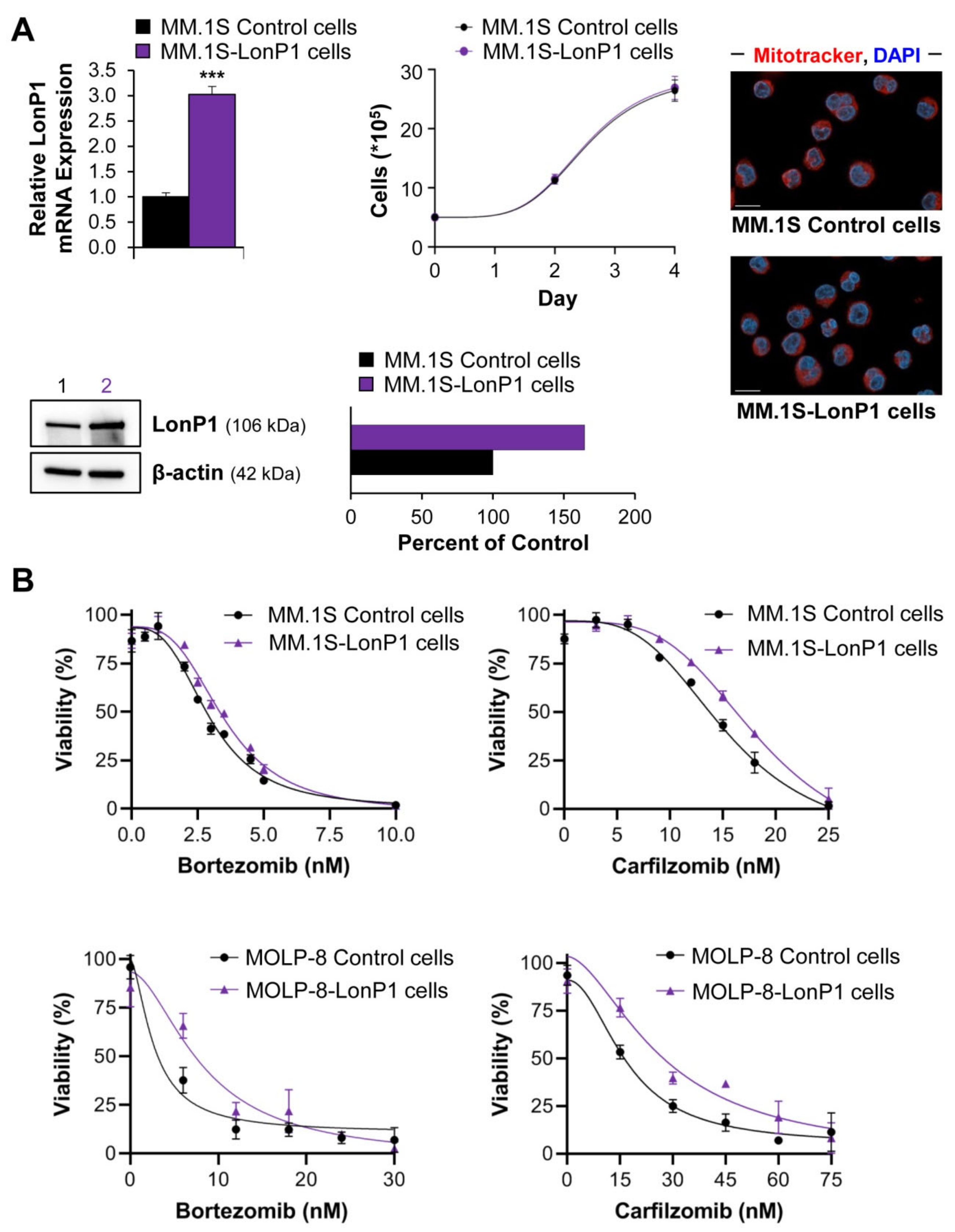
2.3. The Interplay of LonP1 and Proteasome Activities in Multiple Myeloma Cells
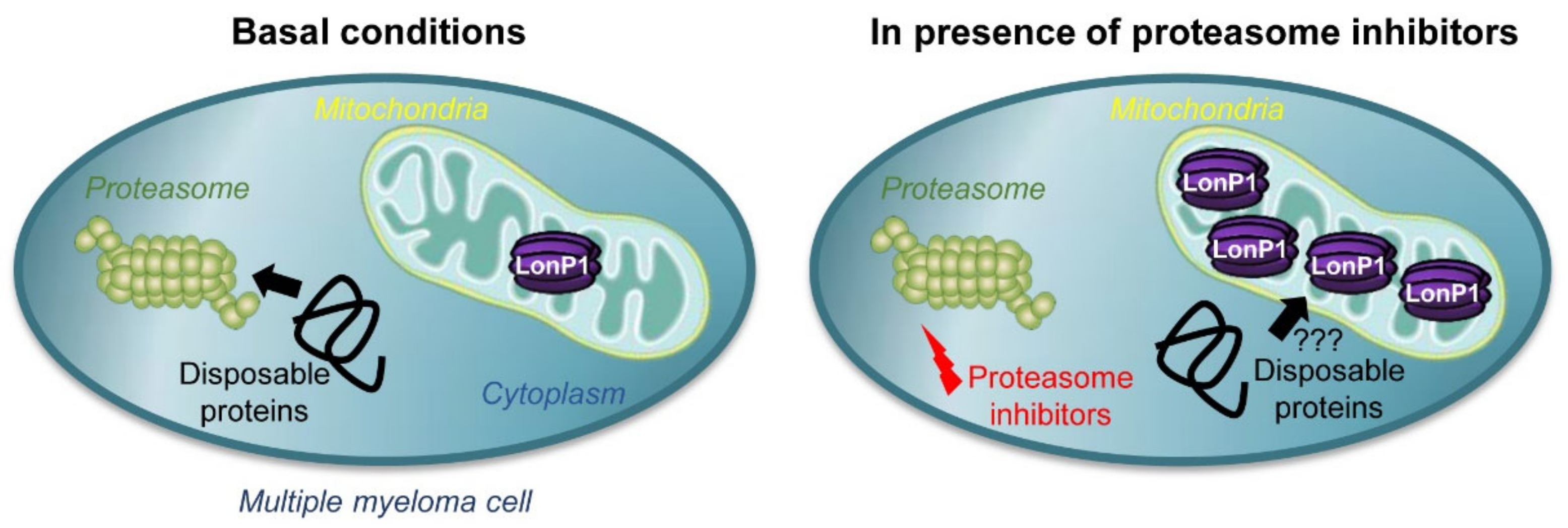
3. Discussion
4. Materials and Methods
4.1. Survival Analysis
4.2. Drug Treatments
4.3. Transcript Analysis
- β-actin (F-5’-CATGTACGTTGCTATCCAGGC; R-5′-CTCCTTAATGTCACGCACGAT);
- LONP1 (F-5′-ATGGAGGACGTCAAGAAACG; R-5′-GATCTCAGCCACGTCAGTCA);
- OSGEPL1 (F-5′-AAAACAGGTGGGATTGTTCCTC; R-5′-AGTAAGTGCATGAGCCTCCAT);
- CD40 (F-5′-ACTGAAACGGAATGCCTTCCT; R-5′-CCTCACTCGTACAGTGCCA);
- COX2 (F-5′-TAAGTGCGATTGTACCCGGAC; R-5′-TTTGTAGCCATAGTCAGCATTGT);
- ICAM-1 (F-5′-ATGCCCAGACATCTGTGTCC; R-5′-GGGGTCTCTATGCCCAACAA);
- IL-6 (F-5′-CCTGAACCTTCCAAAGATGGC; R-5′-TTCACCAGGCAAGTCTCCTCA);
- IRF3 (F-5′-AGAGGCTCGTGATGGTCAAG; R-5′-AGGTCCACAGTATTCTCCAGG).
4.4. Computational Modeling
4.5. Cytotoxicity Assays
4.6. Generation of Transgenic MM.1S and MOLP-8 Cells
4.7. Microscopy
4.8. Immunoblotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. SEER Cancer Statistics Review, 1975–2014, National Cancer Institute. Available online: https://seer.cancer.gov/csr/1975_2014/ (accessed on 12 May 2020).
- Ravi, P.; Kumar, S.K.; Cerhan, J.R.; Maurer, M.J.; Dingli, D.; Ansell, S.M.; Rajkumar, S.V. Defining cure in multiple myeloma: A comparative study of outcomes of young individuals with myeloma and curable hematologic malignancies. Blood Cancer J. 2018, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Chim, C.S.; Kumar, S.K.; Orlowski, R.Z.; Cook, G.; Richardson, P.G.; Gertz, M.A.; Giralt, S.; Mateos, M.V.; Leleu, X.; Anderson, K.C. Management of relapsed and refractory multiple myeloma: Novel agents, antibodies, immunotherapies and beyond. Leukemia 2018, 32, 252–262, Erratum in 2019, 33, 1058–1059. [Google Scholar] [CrossRef]
- Franke, N.E.; Niewerth, D.; Assaraf, Y.G.; van Meerloo, J.; Vojtekova, K.; van Zantwijk, C.H.; Zweegman, S.; Chan, E.T.; Kirk, C.J.; Geerke, D.P.; et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 2012, 26, 757–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerlikaya, A.; Okur, E. An investigation of the mechanisms underlying the proteasome inhibitor bortezomib resistance in PC3 prostate cancer cell line. Cytotechnology 2020, 72, 121–130. [Google Scholar] [CrossRef]
- Lü, S.; Wang, J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark. Res. 2013, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor- adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Kim, H.K.; Lee, S.R.; Jeong, S.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Besse, L.; Besse, A.; Mendez-Lopez, M.; Vasickova, K.; Sedlackova, M.; Vanhara, P.; Kraus, M.; Bader, J.; Ferreira, R.B.; Castellano, R.K.; et al. A metabolic switch in proteasome inhibitor-resistant multiple myeloma ensures higher mitochondrial metabolism, protein folding and sphingomyelin synthesis. Haematologica 2019, 104, e415–e419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell. Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Wada, N.; Kawano, Y.; Okuno, Y.; Kikukawa, Y.; Endo, S.; Nishimura, N.; Ueno, N.; Mitsuya, H.; Hata, H. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp. Hematol. Oncol. 2015, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Wirawan, E.; Lamkanfi, M.; Festjens, N.; Verspurten, J.; Saelens, X.; Vanden Berghe, T.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi cleaves RIP1 during apoptosis of Ba/F3 cells induced by growth factor withdrawal. Cell Res. 2010, 20, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef] [Green Version]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef]
- Granot, Z.; Kobiler, O.; Melamed-Book, N.; Eimerl, S.; Bahat, A.; Lu, B.; Braun, S.; Maurizi, M.R.; Suzuki, C.K.; Oppenheim, A.B.; et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: The unexpected effect of proteasome inhibitors. Mol. Endocrinol. 2007, 21, 2164–2177. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Langer, T.; López-Otín, C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, G.; Mitsiades, C.; Bryant, B.; Zhan, F.; Chng, W.J.; Roels, S.; Koenig, E.; Fergus, A.; Huang, Y.; Richardson, P.; et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood 2007, 109, 3177–3188. [Google Scholar] [CrossRef]
- Giusti, K. Company Profile: Multiple Myeloma Research Foundation. Per. Med. 2012, 9, 333–336. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Csizmadia, V.; Hales, P.; Tsu, C.; Ma, J.; Chen, J.; Shah, P.; Fleming, P.; Senn, J.J.; Kadambi, V.J.; Dick, L.; et al. Proteasome inhibitors bortezomib and carfilzomib used for the treatment of multiple myeloma do not inhibit the serine protease HtrA2/Omi. Toxicol. Res (Cambridge) 2016, 5, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Su, S.C.; Lin, C.C.; Tai, H.C.; Chang, M.Y.; Ho, M.R.; Babu, C.S.; Liao, J.H.; Wu, S.H.; Chang, Y.C.; Lim, C.; et al. Structural basis for the magnesium-dependent activation and hexamerization of the Lon AAA+ protease. Structure 2016, 24, 676–686. [Google Scholar] [CrossRef] [Green Version]
- Mia, S.; Watson, E.R.; Novick, S.J.; Griffin, P.; Wiseman, R.L.; Lander, G.C. Structures of the human LONP1 protease reveal regulatory steps involved in protease activation. BioRxiv 2020. [Google Scholar] [CrossRef]
- Huber, E.M.; Heinemeyer, W.; Groll, M. Bortezomib-resistant mutant proteasomes: Structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015, 23, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Goard, C.A.; Schimmer, A.D. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene 2014, 33, 2690–2699. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Gibellini, L.; Nasi, M.; De Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S.; Morse, K.; Ray, D.; Tompkins, A.; Young, S.M.; Hilchey, S.; Salim, S.; Konopleva, M.; Andreeff, M.; Phipps, R.; et al. The triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid and its derivatives elicit human lymphoid cell apoptosis through a novel pathway involving the unregulated mitochondrial permeability transition pore. Cancer Res. 2007, 67, 1793–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, S.H.; Venkatesh, S.; Li, M.; Lee, J.; Lu, B.; Hilchey, S.P.; Morse, K.M.; Metcalfe, H.M.; Skalska, J.; Andreeff, M.; et al. The mitochondrial ATP-dependent Lon protease: A novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 2012, 119, 3321–3329, Erratum in 2014, 124, 1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwart, A.; Arts, J.H.; ten Berge, W.F.; Appelman, L.M. Alternative acute inhalation toxicity testing by determination of the concentration-time-mortality relationship: Experimental comparison with standard LC50 testing. Regul. Toxicol. Pharmacol. 1992, 15, 278–290. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681, Erratum in 2007, 59, 124. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, Y.; Chen, Y. Predicting drug combination index and simulating the network-regulation dynamics by mathematical modeling of drug-targeted EGFR-ERK signaling pathway. Sci. Rep. 2017, 7, 40752. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me selectively attenuates CA1 neuronal death induced by status epilepticus via facilitating mitochondrial fission independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [Green Version]
- Stadheim, T.A.; Suh, N.; Ganju, N.; Sporn, M.B.; Eastman, A. The novel triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) potently enhances apoptosis induced by tumor necrosis factor in human leukemia cells. J. Biol. Chem. 2002, 277, 16448–16455. [Google Scholar] [CrossRef] [Green Version]
- Shishodia, S.; Sethi, G.; Konopleva, M.; Andreeff, M.; Aggarwal, B.B. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin. Cancer Res. 2006, 12, 1828–1838. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [Green Version]
- Groen, K.; van de Donk, N.; Stege, C.; Zweegman, S.; Nijhof, I.S. Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag. Res. 2019, 11, 2663–2675. [Google Scholar] [CrossRef] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellapragada, S.V.; Ifeorah, C.; Fletcher, L.; Rivero, G.; Yasin, Z.; Krieger, J.; Mulchandani, A.; Catic, A.; Mims, M.P. Differential response to carfilzomib based on initial therapy with bortezomib in multiple myeloma. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Shin, C.S.; Meng, S.; Garbis, S.D.; Moradian, A.; Taylor, R.W.; Sweredoski, M.J.; Lomenick, B.; Chan, D.C. LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat. Commun. 2021, 12, 265. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Liu, Y.; Xu, S.; Lu, B.; Cossarizza, A. Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell. Mol. Life Sci. 2015, 72, 4807–4824. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.A.; Iakova, P.; Maneix, L.; Shih, F.Y.; Cho, H.E.; Sahin, E.; Catic, A. The ubiquitin ligase Cullin-1 associates with chromatin and regulates transcription of specific c-MYC target genes. Sci. Rep. 2020, 10, 13942. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maneix, L.; Sweeney, M.A.; Lee, S.; Iakova, P.; Moree, S.E.; Sahin, E.; Lulla, P.; Yellapragada, S.V.; Tsai, F.T.F.; Catic, A. The Mitochondrial Protease LonP1 Promotes Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers 2021, 13, 843. https://doi.org/10.3390/cancers13040843
Maneix L, Sweeney MA, Lee S, Iakova P, Moree SE, Sahin E, Lulla P, Yellapragada SV, Tsai FTF, Catic A. The Mitochondrial Protease LonP1 Promotes Proteasome Inhibitor Resistance in Multiple Myeloma. Cancers. 2021; 13(4):843. https://doi.org/10.3390/cancers13040843
Chicago/Turabian StyleManeix, Laure, Melanie A. Sweeney, Sukyeong Lee, Polina Iakova, Shannon E. Moree, Ergun Sahin, Premal Lulla, Sarvari V. Yellapragada, Francis T. F. Tsai, and Andre Catic. 2021. "The Mitochondrial Protease LonP1 Promotes Proteasome Inhibitor Resistance in Multiple Myeloma" Cancers 13, no. 4: 843. https://doi.org/10.3390/cancers13040843