
Histone Modifying Enzymes in Gynaecological Cancers
by
 , ,
, ,
Priya Ramarao-Milne
1,2, 
Olga Kondrashova
1,
Sinead Barry
3,4,
John D. Hooper
4,
Jason S. Lee
2,5,6,*,† and
Nicola Waddell
1,2,† 1
Medical Genomics Group, QIMR Berghofer Medical Research Institute, Brisbane, QLD 4006, Australia
2
Faculty of Medicine, The University of Queensland, Brisbane, QLD 4006, Australia
3
Department of Gynaecological Oncology, Mater Hospital Brisbane, Brisbane, QLD 4101, Australia
4
Mater Research Institute, The University of Queensland, Translational Research Institute, Woolloongabba, QLD 4102, Australia
5
Epigenetics and Disease Laboratory, QIMR Berghofer Medical Research Institute, Brisbane, QLD 4006, Australia
6
School of Biomedical Sciences, Queensland University of Technology, Brisbane, QLD 4000, Australia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2021, 13(4), 816; https://doi.org/10.3390/cancers13040816
Submission received: 19 January 2021
/
Revised: 10 February 2021
/
Accepted: 11 February 2021
/
Published: 16 February 2021
(This article belongs to the Special Issue Epigenomic Studies of Gynecological Cancer)
Abstract
:Simple Summary
Epigenetics is a process that allows genetic control, without the involvement of sequence changes to DNA or genes. In cancer, epigenetics is a key event in tumour development that can alter the expression of cancer driver genes and result in genomic instability. Due to the critical role of epigenetics in malignant transformation, therapies that target these processes have been developed to treat cancer. Here, we provide a summary of the epigenetic changes that have been described in a variety of gynaecological cancers. We then highlight how these changes are being targeted in preclinical models and clinical trials for gynaecological cancers.
Abstract
Genetic and epigenetic factors contribute to the development of cancer. Epigenetic dysregulation is common in gynaecological cancers and includes altered methylation at CpG islands in gene promoter regions, global demethylation that leads to genome instability and histone modifications. Histones are a major determinant of chromosomal conformation and stability, and unlike DNA methylation, which is generally associated with gene silencing, are amenable to post-translational modifications that induce facultative chromatin regions, or condensed transcriptionally silent regions that decondense resulting in global alteration of gene expression. In comparison, other components, crucial to the manipulation of chromatin dynamics, such as histone modifying enzymes, are not as well-studied. Inhibitors targeting DNA modifying enzymes, particularly histone modifying enzymes represent a potential cancer treatment. Due to the ability of epigenetic therapies to target multiple pathways simultaneously, tumours with complex mutational landscapes affected by multiple driver mutations may be most amenable to this type of inhibitor. Interrogation of the actionable landscape of different gynaecological cancer types has revealed that some patients have biomarkers which indicate potential sensitivity to epigenetic inhibitors. In this review we describe the role of epigenetics in gynaecological cancers and highlight how it may exploited for treatment.
1. Background
Cancer is a multifaceted group of diseases that develop due to an interplay between genetic and epigenetic factors. Somatic mutations alone do not account for the tumourigenic characteristics of cancer cells; and epigenetic deregulation of oncogenes and tumour suppressors is another important mechanism contributing to carcinogenesis [1,2]. There is a great interest in epigenetic drug targets in cancer [3,4], an area actively pursued in gynaecological cancers. Epigenetic inhibitors or “epidrugs” are being studied for their potential anti-tumour activity, and may be beneficial for cancers with global dysregulation of gene expression. The potential of these inhibitors to target reversible modifications to the genome, coupled with their ability to influence the expression of multiple genes concomitantly, make them attractive as novel anticancer compounds, through re-expression of tumour suppressor genes. This review will outline and discuss studies pertaining to epigenetic factors with a focus on histone modifying enzymes in common gynaecological cancers, specifically, ovarian, endometrial and cervical cancers. The mechanisms by which epigenetics contribute to tumorigenesis and the evidence that implicates epigenetic enzymes, in particular histone modifying enzymes, as treatment targets will be discussed.
1.1. DNA Methylation
DNA methylation, which has a key role in cancer [1,2], refers to the addition of methyl groups to DNA residues by groups of enzymes known as DNA methyltransferases (DNMTs) or demethylases. In particular, methylation of cytosine residues occurring in CpG dinucleotides can be methylated to form 5-methylcytosines. CpG sites are clustered within the genome to form CpG islands, with approximately 70% of genes in the human genome containing a CpG island within their promoter region. Global hypomethylation changes within the genome are associated with genomic instability, while hypermethylation occurs frequently at gene promoter regions. There is extensive evidence that hypermethylation of CpG sites in promoter regions is a crucial mechanism for tumour suppressor gene and microRNA deactivation [5,6], and can account for discrepancies in cases whereby gene mutations do not correlate with respective mRNA levels.
1.2. Histone Modifications
In addition to CpG methylation of DNA, proteins within nucleosome subunits are also subject to covalent modifications. Nucleosomes are comprised of a core set of histones that exist as four dimers of histone H2A, H2B, H3 and H4. Histones are the major determinants of chromosomal conformation and stability, and are amenable to post-translational modifications. These modifications can induce changes in chromatin regions that may result in alteration of gene expression. Histone marks that signify active promoters generally include H3 lysine 4 trimethylation (H3K4me3) [7], while hallmarks of histones flanking inactive promoters are H3 lysine 27 trimethylation (H3K27me3) [8], as well as H3 lysine 9 trimethylation (H3K9me3) [9]. Additionally, there is a tight association between enhancers and regions consisting of H3 lysine 4 monomethylation (H3K4me1) [9]. Global overrepresentation of repressive histone marks is commonly found in cancers compared to normal tissue, and can result in the silencing of important tumour suppressor genes [10].
1.3. Enzymes Involved in Epigenetic Regulation
Histone modifications are catalysed by histone modifying enzymes known as writers, erasers or readers. Writers are enzymes which catalyse addition of post-translational modifications onto histone tails, such as lysine methyltransferases (KMTs), histone acetyltransferases (HATs) and ubiquitin ligases. Erasers, such as histone deacetylases (HDACs) that are subdivided into four classes [11], as well as lysine demethylases (KDMs) and deubiquitinating enzymes remove post-translational modifications (Figure 1). Readers contain motifs that recognise post-translational modifications, recruiting co-factors, which modulate transcription. Together, these enzymes can modulate transcription, using a combination of histone marks to elicit responses by controlling the addition and removal of a variety of modifications, such as methylation, acetylation, ubiquitination, phosphorylation and sumoylation [8]. The same modification in a different position in the amino acid tail can recruit completely different chromatin remodelling complexes, and exhibit a total shift in function [12]. Collectively, varying combinations of covalent modifications comprise the “histone code” – a complex set of signals which recruit enzymes and alter the structure of chromatin, prompting conformational changes in nucleosomes [12]. The complexity of the histone code, in combination with other epigenetic factors allow for fine control of chromatin dynamics and therefore gene activation and repression. There is evidence that histone modifying enzymes are involved in tumorigenic behaviour of cancers including gynaecological cancers [13] (Table 1).
While histone modifications can act alone to recruit co-factors, there is significant crosstalk between different types of epigenetic modifications. This involves cooperation between the enzymes that catalyse these modifications, such as DNMTs that transfer methyl groups onto CpG sites within promoters of genes to elicit gene silencing, and histone modifying enzymes that transfer covalent modifications, such as methyl and acetyl groups onto amino acid tails of histones. For example, DNMT1, which transfers methyl groups onto CpG islands, interacts with KMTs, contributing to H3K9 and H3K27 methylation [33], as well as several HDACs [33]. In addition, EHMT2 (commonly known as G9a) and EZH2, which are the methyltransferases of H3K9me2 and H3K27me3, respectively, also interact with each other [34]. G9a can modulate recruitment of the Polycomb Repressive Group 2 (PRC2) complex, which contains EZH2 as the enzymatic domain, to catalyse H3K27me3. Together, G9a and EZH2 co-repress target genes, although the finer mechanisms remain unclear [34]. The activity of both enzymes increases in hypoxic tumour microenvironments and is linked to the silencing of tumour suppressor genes and survival in breast cancer [35], while EZH2 transcription is enhanced by HIF-1α to promote more aggressive phenotypes [36]. As such, chromatin remodelling enzymes may form a network of interactions which are involved in multiple cellular processes within a variety of cancers.
2. Epigenetic Modifiers in Ovarian Cancer
Ovarian cancer has a poor prognosis, with a 5-year survival rate of 40–50% [37]. It is a heterogeneous disease comprised of multiple subtypes, the most common being epithelial ovarian cancer (EOC), which accounts for > 90% of all cases. The vast majority of cases are an aggressive and rapidly progressive subtype, high grade serous ovarian carcinoma (HGSOC) [38]. Other rarer subtypes include low-grade serous, endometrioid, mucinous and clear cell carcinomas [38]. Gene expression subtypes of EOC have been described with clinical significance [39], which have also been replicated using methylation profiling [40]. The genomic landscape of HGSOC has been profiled by TCGA [41] and ICGC [42] revealing that 95% harbour TP53 somatic mutations. Other somatic genomic features include widespread genomic rearrangements including CCNE1 amplifications in around 20% of cases [43], and recurrent gene breakage of RB1, NF1, RAD51B and PTEN [42]. Other key HGSOC genes are involved in homologous recombination (HR) repair, with up to 18% of patients carrying a germline BRCA1 and BRCA2 mutation [44], an additional 8% of cases containing somatic BRCA1/2 mutations [41,45], and around 10–15% of cases have promoter hypermethylation of BRCA1, which leads to BRCA1 silencing [46,47]. Other aberrations in HR genes, include mutations in genes such as PALB2, BRIP1 and RAD51C [48], and methylation of RAD51C [49].
Drug development for ovarian cancer has generally lagged behind advances for other solid malignancies. However, recently PARP inhibition [50] is beginning to transform the treatment landscape for HGSOC cases. Multiple Phase III trials have supported the use of PARP inhibitors [51], especially those with germline or somatic BRCA1/2 variants or those with a defective HR repair [52], and these agents are now mainstays for recurrent disease with significant potential, also as maintenance therapies after primary treatments. Targeted therapies such as erlotinib for non-small cell lung cancer [53] and Herceptin have become entrenched into treatment regimens for breast cancer [54], but attempts to recapitulate such success in gynaecological cancers has been ineffective thus far. A significant impediment to the development of targeted therapies in ovarian cancer is the presence of extensive intra- and inter-tumoural heterogeneity, and apart from HR deficiency, the lack of clinically actionable mutations. Therefore, targeting epigenetic enzymes in cancers that exhibit extensive dysregulation of gene expression may be a suitable alternative.
2.1. Histone Methyltransferases in Ovarian Cancer
Extensive evidence implicates KMTs in cancer. EZH2 is involved in ovarian cancer although multifaceted roles likely exist, so whether it is acting as a tumour suppressor or oncogene is unclear and may be context dependent. For example, inhibition of EZH2 promotes epithelial-to-mesenchymal transition in ovarian cancer cells [14]. However, knockdown of EZH2 levels also correlate with an induction of apoptosis in epithelial ovarian cancer cells, and regression of tumour xenografts [55]. Similarly inhibition of KDM6B, an H3K27me3 demethylase, may also induce apoptosis in ovarian cancer cells [16]. The presence or absence of EZH2 has potential treatment implications. Knockdown of EZH2 was able to re-sensitise the A2780-DDP cell line that is resistant to platinum through G2/M cycle arrest [56], which may have important implications for patients with platinum-resistant ovarian cancer. While overexpression of EZH2 inhibits phosphorylation of the BRCA1 protein at serine 1423, reducing its function [57], whether this would result in HR deficiency and subsequent response to PARP inhibition is not known. Co-inhibition of EZH2 and DNMT1 in a tumour may have the potential to reprogram the immune microenvironment and increase the efficacy of PD-L1 checkpoint blockade therapy [58]. This suggests a role in modulating immune responses, and that combined methyltransferase and immune checkpoint inhibition may enhance anti-tumour activity.
G9a is a methyltransferase that can catalyse mono- and di-methylation of H3K9. Elevated expression of G9a has been reported in a cohort of ovarian cancer patients, and was significantly associated with poor prognosis [17]. G9a has also been associated with metastases, as omental, peritoneal and lymph node metastases showed significantly elevated levels of G9a compared to matched primary tumours [17] and overexpression of G9a increased migration of cells in in-vitro scratch wound assays [17]. Another methyltransferase with an emerging role in cancer progression is DOT1L, which mediates catalysis of mono-, di- and tri-methylation of H3K79. Most research has investigated its role in mixed lineage leukaemia [59,60,61]. However, DOT1L also modulates ERα target genes [62], inducing chemoresistance [63] with its elevated expression predicting poor prognosis in ovarian cancer patients [64].
The histone methyltransferase SMYD2 (known as KMT3C), in addition to catalysing H3K4me3, also methylates PARP1 at lysine 528 which increases the activity [65]. PARP1 is involved in DNA damage repair [66], and its elevated activity is a contributor to survival and chemoresistance [67]. Increased staining of SMYD2 has been observed in ovarian and cervical cancers, however its involvement in ovarian cancer in terms of histone methyltransferase activity, or its ability to methylate non-histone proteins such as PARP1, remains unclear [68]. Furthermore, whether tumours with a high level of SMYD2 would respond to PARP inhibition is not known.
2.2. Histone Demethylases in Ovarian Cancer
Members of the KDM family also have known roles in cancer. KDM1A overexpression has been observed in a variety of different malignancies, including prostate [69] and bladder [70] cancers and it may have a role in promoting tumorigenesis and metastasis. Activation of the PI3K pathway in several cancers is well known and is involved in the proliferation and migration of ovarian cancer cells. The ligand upstream of the P13K pathway, EGF, upregulates KDM1A, which results in decreased global H3K4me2 levels [18]. In support of a role for KDM1A in cell migration, its overexpression in several ovarian cancer cell lines increases cell motility [18]. While inhibition of KDM1A activity with tranylcypromine decreases migration of the SKOV3 ovarian cancer cell line, and restores global expression of H3K4me2 [18].
A key hallmark of HGSOC is loss of p53 and widespread chromosomal instability due to genomic rearrangements. Overexpression of the demethylase KDM4A induces specific acquisition of copy gains at 1q12, 1q21, and Xq13.1 [25], suggesting that this enzyme and histone modifying enzymes may be contributing to genomic instability. Another KDM, KDM3A, is overexpressed in various ovarian cancer tissues, and its expression was significantly elevated in three cisplatin resistant ovarian cancer cell lines, compared to normal ovarian tissue [26]. Knockdown of KDM3A reduced proliferation in vitro and tumour growth in vivo [26], and restored cisplatin sensitivity in resistant cell lines via apoptosis induction [26]. Given the role of KMTs and KDMs in ovarian cancer and their potential treatment implications, further investigation is required to clarify the mechanisms involved.
2.3. HDACs in Ovarian Cancer
Normal ovarian surface epithelium exhibits weak nuclear expression of HDAC1, HDAC2 and HDAC3 [71], whereas elevated levels are reported in a variety of ovarian tumour types [72,73]. High expression of HDAC1 has been linked to poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas [74]. There is mounting evidence supporting the inhibition of HDACs in cancer treatment. Drug-induced inactivation or gene silencing of HDAC1 suppressed ovarian cancer cell growth [75], and inhibition of HDAC1/2/3 using the inhibitor Panobinostat, slowed the growth of ovarian cancer in a xenograft mouse model [76]. Inhibition of HDAC2 in a chemosensitive ovarian cancer cell line, PEO1, increased the efficacy of carboplatin treatment and increased γH2AX foci, and caused downregulation of phosphorylated BRCA1 [77].
Chemoresistant relapses are a frequent occurrence in patients with HGSOC. HDACs have been implicated in chemoresistance and their inhibition can prolong chemosensitivity or induce sensitivity in inherently chemoresistant tumours. Treatment with DNMT inhibitors and HDAC inhibitors (HDACis) has been shown to re-sensitise the cells to platinum treatment [78]. Chemotherapy resistance is also associated with the presence of cancer stem cells (CSCs), which are thought to exist as a pluripotent fraction within a tumour harbouring multiple pro-survival characteristics that allow them to evade chemotherapy-induced death [79]. As such, there is a particular interest in eliminating this population of cells, in order to maintain, or induce chemosensitivity. HDAC1 and HDAC7 are involved in the generation and maintenance of these CSC populations in breast and ovarian cancer cell lines [80]. Encouragingly, treatment with a HDAC1 inhibitor dampened the CSC phenotype, reducing their tumorigenic characteristics [80]. HDACis can also affect DNMT1 levels, a DNA methyltransferase, and abolish the activity of DNMT1 by inhibition of HDAC2, thus depleting CpG island methylation at the promoters of tumour suppressor genes [81]. However such an effect could have significant negative results for patients that are HR deficient, as the loss of methylation from BRCA1 or RAD51C gene promoter, would restore HR proficiency, rendering patients insensitive to PARP inhibition [82]. Therefore, the molecular makeup of the tumour should be considered prior to treatment selection.
3. Epigenetic Modifiers in Endometrial Cancer
Endometrial cancer is the fifth most prevalent cancer in women in the developed world [38]. While the overall survival for endometrial cancer is around 85%, the prognosis for locally advanced and metastatic disease is considerably worse, with the five-year survival rate dropping to 25% [37]. Endometrial cancer is characterised by four molecular subtypes with distinct treatment and survival outcomes. These subtypes were first described by the TCGA [83] and have since been validated by multiple groups [84,85]. These subtypes include TP53-mutant (also known as copy number high), mismatch repair deficient (MMRd; or microsatellite unstable), POLE-mutant and TP53 wild-type (or copy number low). MMRd endometrial cancer is often caused by germline mutations in MMR genes, including MSH2, MLH1 and MSH6, collectively recognised as Lynch syndrome [86]. TP53-mutant and MMRd molecular subtypes are known as the more aggressive subtypes associated with worse survival [85], so there is a strong clinical need for effective treatments, particularly for these subtypes and advanced stage disease.
3.1. Histone Methyltransferases in Endometrial Cancer
Endometrial cancer cells have higher levels of G9a localised to the nuclei compared to normal tissues, which exhibit only weak nuclear staining [15]. Stronger G9a staining was correlated with depth of myometrial invasion [15], which is a strong predictor of poor prognosis and survival. Increased EZH2 expression correlates with significantly lower survival rates in endometrial cancer [87]. Similar to ovarian cancer, G9a knockdown significantly attenuated migration of endometrial cancer cell lines [15]. Intriguingly, it also showed significantly diminished occupancy of DNMT1 at the promoter of the E-cadherin gene, CDH1, coupled with increased protein levels of E-cadherin protein, thus providing a potential mechanism by which G9a regulates gene expression and directly contributes to tumorigenic behaviour [15]. This may suggest that DNA methylation inhibition alone in this context would be ineffective at re-expressing E-cadherin, and removal of repressive H3K9me/H3K9me2 must also occur, highlighting the crosstalk of and complexity of epigenetic factors associated with the tumorigenic behaviour of cancer cells.
3.2. Histone Demethylases in Endometrial Cancer
Expression of KDM4B and KDM4A is higher in endometrial cancer tissue compared to normal endometrium tissue [28]. KDM4B is a histone demethylase which recognises and removes methyl marks from H3K9me2 and H3K9me3 to subsequently activate transcription. One study exploring the role of androgen receptor (AC) in endometrial cancer suggested that KDM4B together with AR can activate the well-recognised oncogene, MYC, by removing H3K9me3 marks in endometrial cancer cells with high basal levels of AR [28]. This effect was not observed in endometrial cancer cell lines with low levels of AR; instead, another histone demethylase, KDM4A, reduced levels of H3K4me3 methylation, an activation mark, at the promoter of the tumour suppressor gene p27kip1 [28]. These results suggested that both KDM4A and KDM4B together with AR have a role in endometrial cancer development and progression.
Depending on the context, KDM1A can act as a co-repressor or co-activator, catalysing either H3K9me1 or H3K4me2 [88]. This is achieved by exchanging transcriptional modules which enables the switch in activity. Association of KDM1A with the CoREST complex permits it to catalyse H3K4me2 demethylation [89], however, KDM1A is also responsible for the regulation of AR target genes. A study showed that inhibition of KDM1A via HCI2509 increased levels of H3K4me2 and H3K9me1, as well as increased global H3K27me3, proposing potential involvement in crosstalk with EZH2 and its modifications [19].
Taken together this shows that histone demethylases from the same family can potentially target different histone marks, resulting in oncogenic activation or suppressive mechanisms.
3.3. HDACs in Endometrial Cancer
Similar to ovarian carcinomas, high-grade endometrial carcinomas express high levels of HDAC1, HDAC2 and HDAC3 [74], while less aggressive subtypes show lower levels of HDAC expression. In agreement with this, samples with high expression of HDACs showed a higher proliferating capacity in endometrial and ovarian cancer [74], consistent with the observation that HDACis can induce apoptosis and cell cycle arrest in vitro.
4. Epigenetic Modifiers in Cervical Cancer
With the introduction of a vaccine, cervical cancer is now a largely preventable disease, however it remains one of the most commonly diagnosed cancers in developing countries [90]. The main risk factor for cervical cancer is infection with variants of the human papilloma virus (HPV), which underlie the cause of almost all cervical cancer cases [91]. HPV infection alone is not sufficient for the development of cervical cancer; another event such as activation of oncogenes or deactivation of tumour suppressor genes is required to initiate carcinogenesis [92]. HPV contains an 8-kb circular genome which encodes for a number of proteins including viral “oncoproteins” E6 and E7 [91] which are responsible for the repression of host tumour suppressor genes TP53 and RB1. Increased viral E7 inhibits binding of HDACs to hypoxia inducible factor 1 (HIF-1), thus activating transcription of pro-angiogenic genes downstream of HIF-1 [93]. Upon integration of the virus into the host genome, regulators of E6 and E7 by HPV proteins E1 and E2, are disrupted and there is a loss of transcriptional control of E6 and E7 [94]. Epigenetics also plays an important role in the modulation of these proteins, and may contribute to cervical cancer pathogenesis [95]. A screen to identify E2 binding partners found that it binds EP400, a component of the histone acetyltransferase complex NuA4/TIP60, as well as KDM5C [20]. EP400 also acts as a transcriptional corepressor, and KDM5C possesses demethylase activity against the active promoter marks, H34me2 and H3K4me3. This has led to the speculation that the transcriptional repressor activity of EP400 and KDM5C may aid in E2 mediated repression of the E6 and E7 oncoproteins [20].
5. Epigenetic Treatments in Gynaecological Cancers
Research into epigenetic regulation of cancers has facilitated the development of multiple inhibitors targeting DNA modifying enzymes. The development of DNA methyltransferase inhibitors and HDAC inhibitors (Figure 1), has fuelled the application of epigenetic treatments in gynaecological cancers. There have been a variety of preclinical studies testing epigenetic inhibitors in models of gynaecological cancers, some of which are highlighted in this review and Table 2, but this is not an exhaustive list as only representative examples of recent studies have been included.
5.1. HDAC Inhibitors
Of all histone modifying enzymes, HDACs have been most extensively studied for their potential as cancer therapies. An array of HDACis are currently being tested within a variety of cancer types, which has facilitated the refinement of these agents. Several HDACis are FDA approved and have shown potential in haematological cancers, however less so as a single agent in solid tumours, where they produce at most modest decreases in tumour growth. Instead, inhibiting histone modifying enzymes could show more promise as drug resistance modulators, as they may be able to induce chemosensitivity or drastically lower chemotherapy doses. Here, the impact of the development of these inhibitors is discussed, with an emphasis on gynaecological cancers.
A variety of epigenetic inhibitors in particular HDAC inhibitors are currently or have been trialled within Phase I or II trials for gynaecological cancers, a summary of trials included on ClinicalTrials.gov (accessed on 18 January 2021) is provided in Table 3.
Although HDAC inhibitors have shown promise in T cell lymphomas [106], as well as other haematological malignancies [107], their efficacy in gynaecological cancers has been more limited. In 2008, a Phase II trial from the Gynecologic Oncology Group (GOG) showed that monotherapy of an oral dose of Vorinostat in patients with recurrent epithelial ovarian cancer, who were resistant to platinum based therapy, failed to produce encouraging results [108]. Vorinostat was also trialled as a combination treatment with carboplatin and gemcitabine in women with recurrent, platinum sensitive, fallopian tube or peritoneal cancer [109]. Although patients did demonstrate a response via the RECIST criteria, the study had to be terminated in its early stages due to toxicity. Despite many adjustments in dosing schedules, patients exhibited extensive haematologic toxicities with the incidence of Grade 4 thrombocytopaenia increasing to 23% with the addition of Vorinostat, from the 5% expected with gemcitabine and carboplatin alone. It would be interesting to determine if there is value of utilising Vorinostat in between chemotherapeutic regimens, or at a maintenance dose to achieve stable disease, or potentially with other agents which do not exacerbate side effects.
Panobinostat is another HDACi that inhibits Class I, II and IV HDAC enzymes. A Phase III trial of Panobinostat in combination with bortezomib and dexamethasone (the PANORAMA 1 trial) in patients with relapsed multiple myeloma yielded a modest survival benefit of approximately 3 months median progression free of survival [110]. However, the side effect profile showed high level toxicity in patients receiving this treatment, potentially limiting its use in the clinic [111]. Even so, several in vitro studies have continued to explore the use of Panobinostat in gynaecological cancers. In HR-proficient ovarian cancer cell lines, Panobinostat downregulated DNA damage repair genes and induced sensitivity to the PARP inhibitor olaparib [112]. This potential synergistic effect of HDACis warrants further investigation to determine if HR proficient tumours can be sensitised to PARP inhibition. Interestingly, Panobinostat may also be able to impart re-sensitisation of platinum-based compounds as demonstrated in cisplatin resistant ovarian cancer cell lines [113], possibly by rewiring these cells to a BRCA-deficiency like phenotype.
Another HDACi that has been trialled for treatment of ovarian cancer is Belinostat (Table 3). A Phase II trial of Belinostat alone in micropapillary/borderline and epithelial ovarian cancer found that it was generally well-tolerated with no Grade 4 toxicity events [114]. However there was only a modest progression-free survival improvement in the micropapillary/borderline cancer patients [114]. Belinostat was also tested in combination with carboplatin and paclitaxel [115], but the study was halted after the first stage due to little activity in the platinum-resistant ovarian cancers.
In the cervical cancer cell lines, SiHa and HeLa, the HDACi Panobinostat induces production of reactive oxygen species (ROS) and synergises strongly with topoisomerase inhibitors [116]. Specifically, Panobinostat induces cell cycle arrest and increases the percentage of cells in G1 cell cycle by affecting mitochondrial membrane potential, and increasing ROS. An increase in p21 was observed, consistent with the inhibition of CDK, illustrating the ability of Panobinostat to induce cell cycle arrest in the two tested cell lines. Significant downregulation of Bcl-xL, a component of mitochondrial anti-apoptotic machinery, was observed, in addition subsequent release of cytochrome c, a known precedent to apoptosis.
Despite in vitro data supporting a role for HDACis in gynaecological cancers, the unfavourable side effect profile and lack of clear efficacy hampers their use in the clinic [111]. Additionally, most of the HDACis tested are pan-HDAC inhibitors which may have many off-target effects and may be contributing to the adverse side effects observed. Therefore, further study to develop more specific inhibitors with on-target activity may be warranted.
5.2. Histone Methyltransferase and Demethylase Inhibitors
While HDACis are being extensively tested in clinical trials, progress in the development of histone methyltransferase inhibitors has been slower. Nonetheless, inhibitors targeting EZH2, KDM1A and DOT1L are being developed, while only inhibitors of EZH2 are currently in clinical trials for patients with gynaecological malignancies.
Through high-throughput drug screening and optimisation, small molecule inhibitors to EZH2 have been developed [117], many of which are competitive for the methyl co-factor S-adenosyl-L-methionine (SAM) required for the enzymatic activity of EZH2. For example, GSK126 was administered to patients with diffuse large B cell lymphoma (DLBCL) in a clinical trial (NCT02082977) [118], although this study was terminated in 2017 due to lack of clinical response. However, a new EZH2 inhibitor with greater oral bioavailability and specificity, tazemetostat was approved by the FDA for adult patients with relapsed or refractory follicular lymphoma whose tumors are positive for an EZH2 mutation who have received at least 2 prior systemic therapies, and for those patients with no satisfactory alternative treatment options (Study E7438-G000-101, NCT01897571 [119]).
In gynaecological cancers there are several ongoing trials for patients with small cell carcinoma of the ovary (NCT03874455), endometrial and ovarian carcinoma (NCT03348631). Other clinical trials include combining tazemetostat with immunotherapy in treating patients with locally advanced or metastatic urothelial carcinoma (NCT03854474). It would be interesting to see whether this newer compound will show greater efficacy.
While it is unlikely that histone modifiers will be introduced to chemo-naïve patients, or patients with chemoresistant disease as a first-line therapy, their use in addition to conventional chemotherapeutic regimes has shown some promise pre-clinically. Discrepancies between in vitro data and clinical data need to be addressed if histone methyltransferase inhibitors are to progress to the clinic for patients with gynaecological malignancies.
5.3. Targeting “Readers” of Histone Modifications
Another class of emerging anticancer agents are bromodomain and extra-terminal (BET) protein inhibitors that target the BET family proteins epigenetic “readers”, which recognise post-translational modifications such as those on histone residues. BRD4 is the most extensively studied member of the BET family of proteins; and binds acetylated lysines on histones. The relevance of BRD4 in cancer pertains to its occupancy at super enhancers of oncogenes, such as c-myc [120]. In ovarian cancer, preclinical models have suggested efficacy for BET inhibitors, and revealed a mechanism of action via disruption of Forkhead box protein M1 (FoxM1) pathway [32], one of the drivers of ovarian carcinoma. It has been subsequently shown in multiple studies that BET inhibitors can reduce HR activity and sensitize HR-proficient cell lines to PARP inhibitors [121,122,123,124,125]. This is significant as BET inhibitors may enable ovarian cancer patients that are HR-proficient access to PARP inhibition.
Despite promising pre-clinical studies, clinical trials involving BET inhibitors have yielded mixed results. Many of these studies have been conducted in non-gynaecological cancers. For example, a dose finding clinical trial administering OTX015 to glioblastoma patients (NCT02296476) was established based on promising anti-tumour effects in preclinical glioblastoma models [126]. However, the trial was terminated after one year due to a lack of clinical activity. Similarly, a Phase I trial of OTX015 inadvanced solid malignancies revealed partial responses in some patients, but most patients suffered adverse effects, including grade 3 and 4 thrombocytopaenia [127]. At present, there are several clinical trials listed using BET inhibitors for various malignancies, although 9 have either been withdrawn or terminated.
Several Phase I trials have been initiated with new BET inhibitors for a variety of tumour types including BMS-986158 for the treatment of recurrent or refractory solid tumors (CNS or Lymphoma) (NCT03936465 and NCT02419417), CPI-0610 for myelofibrosis (NCT02158858) and PLX51107 and azacitidine in treating patients with acute myeloid leukemia or myelodysplastic syndrome (NCT04022785). Two trials have entered Phase 2 examining the efficacy of the pan-BET inhibitor ZEN-3694 in combination with enzalutamide in metastatic castration-resistant prostate cancer patients (NCT04471974) or in combination with talazoparib in triple negative breast cancer (NCT03901469). In ovarian cancer, various pre-clinical studies have exhibited effective anti-tumour responses in vitro and in vivo [128,129,130], including strong synergistic effects when combined with PARP inhibitors [121,122,123,125,131]. Given the strong pre-clinical evidence for BET inhibitors in myc-addicted tumours, further study is warranted into this class of epigenetic inhibitor, and efforts should be focused into minimising the severe adverse effects encountered by patients undergoing BET inhibitor treatment.
5.4. Combination Therapy
The rationale for combination therapies in cancer treatment has been reviewed [132]. As alluded to throughout this review, combination therapy with various agents may augment the anticancer effect of epigenetic inhibitors [133,134]. For example, the combination of HDAC inhibitors and immunotherapy appears to be beneficial through transcriptional induction of endogenous retroviruses and testis antigens, resulting in a response called viral mimicry [133]. In vitro and in vivo xenograft studies in ovarian cancer cell lines and mouse models have shown that HDAC inhibitor treatment results in re-sensitisation of ovarian cancer cells to paclitaxel [135]. Additionally, the combination of PARP inhibitors with BET inhibitors resulted in increased DNA damage in ovarian cancer cells, potentially by blocking homologous recombination repair [136]. Moreover, epigenetic inhibitors can be combined with each other to induce cell death; for example, the combination of HDAC inhibition and demethylating agents such as 5-azacytidine and decitabine exerted synergistic effects in inhibiting proliferation of ovarian cancer cells [137]. The results from these studies illustrate the range of processes which can be modulated by epigenetic inhibition, and thus can be leveraged to enhance the cytotoxic activity of other agents. As such, more research into combination therapies with epigenetic inhibitors is well-warranted.
6. Concluding Remarks
Clearly, there is an urgent need for the development of novel therapies for metastatic disease in gynaecological cancers, alongside better strategies to achieve earlier diagnosis. Epigenetic enzymes play a vital role in carcinogenesis and may prove to be effective targets for cancer therapy. However, much progress needs to be made in order to ensure the right patients are targeted and the right combination of therapies is used to facilitate the translation of research findings into clinically relevant outcomes. There is evidence for the potential of targeting histone modifying enzymes in these aggressive cancers. However, rigorous preclinical studies, as well as careful drug development against robust biomarkers and well-designed clinical trials will be imperative if there is a chance for successful transition of inhibitors to histone modifying enzymes from the bench to the bedside.
Author Contributions
P.R.-M., O.K., J.S.L. and N.W. co-wrote the paper. S.B. and J.D.H. reviewed and edited the paper. All of the authors have read and approved the manuscript; and agree to be personally accountable for the author’s own contributions and for ensuring that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and documented in the literature.
Funding
N.W. is supported by a National Health and Medical Research Council of Australia (NHMRC) Senior Research Fellowship (APP1139071). J.S.L. is supported by the Ovarian Cancer Research Foundation Investigator-initiated Research Grant (GA-2019-18) and Donald and Joan Wilson Foundation.
Data Availability Statement
Data sharing not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
N.W. is a co-founder and Board member of genomiQa. O.K. has consulted for XING Technologies. The remaining authors have no competing interests. The conflicts had no role in this study or in the writing of the manuscript, or in the decision to publish the review.
References
- Chen, Q.W.; Zhu, X.Y.; Li, Y.Y.; Meng, Z.Q. Epigenetic regulation and cancer (review). Oncol. Rep. 2014, 31, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lujambio, A.; Esteller, M. CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle 2007, 6, 1455–1459. [Google Scholar] [CrossRef]
- Kim, T.H.; Barrera, L.O.; Zheng, M.; Qu, C.; Singer, M.A.; Richmond, T.A.; Wu, Y.; Green, R.D.; Ren, B. A high-resolution map of active promoters in the human genome. Nature 2005, 436, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, F.F. Epigenomics in cancer management. Cancer Manag. Res. 2010, 2, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J. Action at a distance: Epigenetic silencing of large chromosomal regions in carcinogenesis. Hum. Mol. Genet. 2007, 16, R88–R95. [Google Scholar] [CrossRef] [Green Version]
- Krusche, C.A.; Vloet, A.J.; Classen-Linke, I.; von Rango, U.; Beier, H.M.; Alfer, J. Class I histone deacetylase expression in the human cyclic endometrium and endometrial adenocarcinomas. Hum. Reprod. 2007, 22, 2956–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, H.; Zhao, J.; Vieth, E.; Nephew, K.P.; Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 2016, 7, 84453. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, S.M.; Chen, M.W.; Chen, C.A.; Chien, M.H.; Hua, K.T.; Hsiao, M.; Kuo, M.L.; Wei, L.H. The H3K9 Methyltransferase G9a Represses E-cadherin and is Associated with Myometrial Invasion in Endometrial Cancer. Ann. Surg. Oncol. 2015, 22 (Suppl 3), S1556–S1565. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, H.; Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Suzuki, S.; Seino, S.; Seino, M.; Ohta, T.; Nagase, S.; et al. GSKJ4, A Selective Jumonji H3K27 Demethylase Inhibitor, Effectively Targets Ovarian Cancer Stem Cells. Anticancer Res. 2015, 35, 6607–6614. [Google Scholar]
- Hua, K.-T.; Wang, M.-Y.; Chen, M.-W.; Wei, L.-H.; Chen, C.-K.; Ko, C.-H.; Jeng, Y.-M.; Sung, P.-L.; Jan, Y.-H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [Green Version]
- Shao, G.; Wang, J.; Li, Y.; Liu, X.; Xie, X.; Wan, X.; Yan, M.; Jin, J.; Lin, Q.; Zhu, H.; et al. Lysine-specific demethylase 1 mediates epidermal growth factor signaling to promote cell migration in ovarian cancer cells. Sci. Rep. 2015, 5, 15344. [Google Scholar] [CrossRef] [Green Version]
- Theisen, E.R.; Gajiwala, S.; Bearss, J.; Sorna, V.; Sharma, S.; Janat-Amsbury, M. Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. BMC Cancer 2014, 14, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.C.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.Z.; Luo, X.G.; Shen, J.; Zou, J.N.; Lu, Y.H.; Xi, T. Knockdown of SMYD3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008, 41, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Xu, B.; Jiang, D.; Huang, S.; Yu, H.; Wu, Z.; Wu, Q. Depletion of H3K79 methyltransferase Dot1L promotes cell invasion and cancer stem-like cell property in ovarian cancer. Am. J. Transl. Res. 2019, 11, 1145–1153. [Google Scholar] [PubMed]
- Dickson, K.A.; Cole, A.J.; Gill, A.J.; Clarkson, A.; Gard, G.B.; Chou, A.; Kennedy, C.J.; Henderson, B.R.; Fereday, S.; Traficante, N.; et al. The RING finger domain E3 ubiquitin ligases BRCA1 and the RNF20/RNF40 complex in global loss of the chromatin mark histone H2B monoubiquitination (H2Bub1) in cell line models and primary high-grade serous ovarian cancer. Hum. Mol. Genet. 2016, 25, 5460–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarcic, O.; Pateras, I.S.; Cooks, T.; Shema, E.; Kanterman, J.; Ashkenazi, H.; Boocholez, H.; Hubert, A.; Rotkopf, R.; Baniyash, M.; et al. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell Rep. 2016, 14, 1462–1476. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Manning, A.L.; Van Rechem, C.; Kim, J.; Ladd, B.; Cho, J.; Pineda, C.M.; Murphy, N.; Daniels, D.L.; Montagna, C.; et al. KDM4A Lysine Demethylase Induces Site-Specific Copy Gain and Rereplication of Regions Amplified in Tumors. Cell 2013, 154, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Ramadoss, S.; Sen, S.; Ramachandran, I.; Roy, S.; Chaudhuri, G.; Farias-Eisner, R. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 2016, 36, 6508. [Google Scholar] [CrossRef] [Green Version]
- Sone, K.; Piao, L.; Nakakido, M.; Ueda, K.; Jenuwein, T.; Nakamura, Y.; Hamamoto, R. Critical role of lysine 134 methylation on histone H2AX for γ-H2AX production and DNA repair. Nat. Commun. 2014, 5, 5691. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.-T.; Fan, Q.; Zhu, Z.; Kwan, S.-Y.; Chen, L.; Chen, J.-H.; Ying, Z.-L.; Zhou, Y.; Gu, W.; Wang, L.-H.; et al. KDM4B and KDM4A promote endometrial cancer progression by regulating androgen receptor, c-myc, and p27(kip1). Oncotarget 2015, 6, 31702–31720. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Zhang, J.; Cai, H.; Li, Y.; Zhang, Y.; Zhang, X.; Zhao, D.; Li, Z.; Ma, H.; Wang, J.; et al. HDAC as a therapeutic target for treatment of endometrial cancers. Curr. Pharm. Des. 2014, 20, 1847–1856. [Google Scholar] [CrossRef]
- Yano, M.; Yasuda, M.; Sakaki, M.; Nagata, K.; Fujino, T.; Arai, E.; Hasebe, T.; Miyazawa, M.; Miyazawa, M.; Ogane, N.; et al. Association of histone deacetylase expression with histology and prognosis of ovarian cancer. Oncol Lett 2018, 15, 3524–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Tian, G.; Murph, M.M. Molecular epigenetics in the management of ovarian cancer: Are we investigating a rational clinical promise? Front. Oncol. 2014, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ma, P.; Jing, Y.; Yan, Y.; Cai, M.-C.; Zhang, M.; Zhang, S.; Peng, H.; Ji, Z.-L.; Di, W.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating FoxM1. Theranostics 2016, 6, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Pontis, J.; Fritsch, L.; Robin, P.; Portoso, M.; Proux, C.; Margueron, R.; Ait-Si-Ali, S. The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 2014, 53, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casciello, F.; Al-Ejeh, F.; Kelly, G.; Brennan, D.J.; Ngiow, S.F.; Young, A.; Stoll, T.; Windloch, K.; Hill, M.M.; Smyth, M.J.; et al. G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 7077–7082. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA A Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Bodelon, C.; Killian, J.K.; Sampson, J.N.; Anderson, W.F.; Matsuno, R.; Brinton, L.A.; Lissowska, J.; Anglesio, M.S.; Bowtell, D.D.L.; Doherty, J.A.; et al. Molecular Classification of Epithelial Ovarian Cancer Based on Methylation Profiling: Evidence for Survival Heterogeneity. Clin. Cancer Res. 2019, 25, 5937–5946. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489. [Google Scholar] [CrossRef]
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19. [Google Scholar] [CrossRef]
- Saleemuddin, A.; Folkins, A.K.; Garrett, L.; Garber, J.; Muto, M.G.; Crum, C.P.; Tworoger, S. Risk factors for a serous cancer precursor ("p53 signature") in women with inherited BRCA mutations. Gynecol. Oncol. 2008, 111, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, J.F.; Gayther, S.A.; Russell, P.; Dearden, J.; Gore, M.; Blake, P.; Easton, D.; Ponder, B.A. Contribution of BRCA1 mutations to ovarian cancer. N. Engl. J. Med. 1997, 336, 1125–1130. [Google Scholar] [CrossRef]
- Ruscito, I.; Dimitrova, D.; Vasconcelos, I.; Gellhaus, K.; Schwachula, T.; Bellati, F.; Zeillinger, R.; Benedetti-Panici, P.; Vergote, I.; Mahner, S.; et al. BRCA1 gene promoter methylation status in high-grade serous ovarian cancer patients—A study of the tumour Bank ovarian cancer (TOC) and ovarian cancer diagnosis consortium (OVCAD). Eur. J. Cancer 2014, 50, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L.; Nemeth, E.; Tran, H.; Shvartsman, H.; Cass, I.; Narod, S.; Karlan, B.Y. BRCA1 promoter region hypermethylation in ovarian carcinoma: A population-based study. Cancer Res. 2000, 60, 5329–5333. [Google Scholar]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, R.W.; Coleman, R.L.; Brown, J.; Moore, K.N. Phase III trials in ovarian cancer: The evolving landscape of front line therapy. Gynecol. Oncol. 2019, 153, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Ruscito, I.; Bellati, F.; Ray-Coquard, I.; Mirza, M.R.; du Bois, A.; Gasparri, M.L.; Costanzi, F.; De Marco, M.P.; Nuti, M.; Caserta, D.; et al. Incorporating Parp-inhibitors in Primary and Recurrent Ovarian Cancer: A Meta-analysis of 12 phase II/III randomized controlled trials. Cancer Treat. Rev. 2020, 87, 102040. [Google Scholar] [CrossRef]
- Shash, E.; Peccatori, F.A.; Azim, H.A., Jr. Optimizing the use of epidermal growth factor receptor inhibitors in advanced non-small-lung cancer (NSCLC). J. Thorac. Dis. 2011, 3, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant Trastuzumab in HER2-Positive Breast Cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of Zeste Homolog 2 Promotes the Proliferation and Invasion of Epithelial Ovarian Cancer Cells. Mol. Cancer Res. 2010, 8, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zeng, X.; Chen, S.; Ding, L.; Zhong, J.; Zhao, J.C.; Wang, L.; Sarver, A.; Koller, A.; Zhi, J.; et al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J. 2013, 32, 1584–1597. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.Y.; Granowicz, E.M.; Maillard, I.; Thomas, D.; Hess, J.L. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 2011, 117, 4759–4768. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.J.; Wu, H.; Achille, N.J.; Reisenauer, M.R.; Chou, C.W.; Zeleznik-Le, N.J.; Hemenway, C.S.; Zhang, W. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 2010, 70, 10234–10242. [Google Scholar] [CrossRef] [Green Version]
- Nassa, G.; Salvati, A.; Tarallo, R.; Gigantino, V.; Alexandrova, E.; Memoli, D.; Sellitto, A.; Rizzo, F.; Malanga, D.; Mirante, T.; et al. Inhibition of histone methyltransferase DOT1L silences ERα gene and blocks proliferation of antiestrogen-resistant breast cancer cells. Sci. Adv. 2019, 5, eaav5590. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, X.-X.; Li, M.-C.; Cao, C.-H.; Wan, D.-Y.; Xi, B.-X.; Tan, J.-H.; Wang, J.; Yang, Z.-Y.; Feng, X.-X.; et al. C/EBPβ enhances platinum resistance of ovarian cancer cells by reprogramming H3K79 methylation. Nat. Commun. 2018, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, D.; Li, M.; Cao, C.; Wan, D.; Xi, B.; Li, W.; Tan, J.; Wang, J.; Wu, Z.; et al. Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer. J. Hematol. Oncol. 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Kang, D.; Suzuki, T.; Masuda, A.; Dohmae, N.; Nakamura, Y.; Hamamoto, R. The Histone Methyltransferase SMYD2 Methylates PARP1 and Promotes Poly(ADP-ribosyl)ation Activity in Cancer Cells. Neoplasia 2014, 16, 257–264.e252. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F.; et al. LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Hsieh, C.Y.; Huang, K.E.; Chang, C.; Kang, H.Y. Cryptotanshinone down-regulates androgen receptor signaling by modulating lysine-specific demethylase 1 function. Int. J. Cancer 2012, 131, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.L.; Pak, J.H.; Park, J.Y.; Choi, W.H.; Lee, J.Y.; Kim, J.H.; Nam, J.H. Expression profile of histone deacetylases 1, 2 and 3 in ovarian cancer tissues. J. Gynecol. Oncol. 2008, 19, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Denkert, C.; Noske, A.; Darb-Esfahani, S.; Dietel, M.; Kalloger, S.E.; Huntsman, D.G.; Köbel, M. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia 2008, 10, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Khabele, D.; Son, D.S.; Parl, A.K.; Goldberg, G.L.; Augenlicht, L.H.; Mariadason, J.M.; Rice, V.M. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: Implications for therapy. Cancer Biol. Ther. 2007, 6, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Garrett, L.A.; Growdon, W.B.; Rueda, B.R.; Foster, R. Influence of a novel histone deacetylase inhibitor panobinostat (LBH589) on the growth of ovarian cancer. J. Ovarian Res. 2016, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Langdon, S.P.; Tse, M.; Mullen, P.; Um, I.H.; Faratian, D.; Harrison, D.J. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 2016, 7, 4695–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 Modulate RGS10 Expression and Decrease Ovarian Cancer Chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, G.; Wolff, J.E.; Aldoss, I.; Hu, B.; Dinh, M.; Torres, A.; Chen, X.; Rizzieri, D.; Sood, A.; Odenike, O.; et al. First-in-human study of ABBV-075 (mivebresib), a pan-inhibitor of bromodomain and extra terminal (BET) proteins, in patients (pts) with relapsed/refractory (RR) acute myeloid leukemia (AML): Preliminary data. J. Clin. Oncol. 2018, 36, 7019. [Google Scholar] [CrossRef]
- Witt, A.E.; Lee, C.W.; Lee, T.I.; Azzam, D.J.; Wang, B.; Caslini, C.; Petrocca, F.; Grosso, J.; Jones, M.; Cohick, E.B.; et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene 2016, 36, 1707–1720. [Google Scholar] [CrossRef]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Res. 2011, 31, 2723–2732. [Google Scholar]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhouk, A.; McConechy, M.K.; Leung, S.; Yang, W.; Lum, A.; Senz, J.; Boyd, N.; Pike, J.; Anglesio, M.; Kwon, J.S.; et al. Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer. Cancer 2017, 123, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, Y.; Li, J.; Cragun, J.; Hatch, K.; Chambers, S.K.; Zheng, W. Lynch syndrome related endometrial cancer: Clinical significance beyond the endometrium. J. Hematol. Oncol. 2013, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.T.; Dovey, O.M.; Lezina, L.; Luo, J.L.; Gant, T.W.; Barlev, N.; Bradley, A.; Cowley, S.M. Lysine-Specific Demethylase 1 Regulates the Embryonic Transcriptome and CoREST Stability. Mol. Cell. Biol. 2010, 30, 4851–4863. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Franco, E.L.; Duarte-Franco, E.; Ferenczy, A. Cervical cancer: Epidemiology, prevention and the role of human papillomavirus infection. CMAJ 2001, 164, 1017–1025. [Google Scholar]
- Bodily, J.M.; Mehta, K.P.M.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1 mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.S.; Rajasekaran, N.; Ju, W.; Shin, Y.K. Human Papillomavirus: Current and Future RNAi Therapeutic Strategies for Cervical Cancer. J. Clin. Med. 2015, 4, 1126–1155. [Google Scholar] [CrossRef] [Green Version]
- Sartor, M.A.; Dolinoy, D.C.; Jones, T.R.; Colacino, J.A.; Prince, M.E.P.; Carey, T.E.; Rozek, L.S. Genome-wide methylation and expression differences in HPV(+) and HPV(−) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 2011, 6, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Salvati, A.; Gigantino, V.; Nassa, G.; Giurato, G.; Alexandrova, E.; Rizzo, F.; Tarallo, R.; Weisz, A. The Histone Methyltransferase DOT1L Is a Functional Component of Estrogen Receptor Alpha Signaling in Ovarian Cancer Cells. Cancers 2019, 11, 1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; Atiya, H.I.; Wang, Y.; Pisanic, T.R.; Wang, T.H.; Shih, I.M.; Foy, K.K.; Frisbie, L.; Buckanovich, R.J.; Chomiak, A.A.; et al. Epigenomic Reprogramming toward Mesenchymal-Epithelial Transition in Ovarian-Cancer-Associated Mesenchymal Stem Cells Drives Metastasis. Cell Rep. 2020, 33, 108473. [Google Scholar] [CrossRef] [PubMed]
- Oki, S.; Sone, K.; Oda, K.; Hamamoto, R.; Ikemura, M.; Maeda, D.; Takeuchi, M.; Tanikawa, M.; Mori-Uchino, M.; Nagasaka, K.; et al. Oncogenic histone methyltransferase EZH2: A novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 2017, 8, 40402–40411. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Wang, Z.; Liu, D. Interference with endogenous EZH2 reverses the chemotherapy drug resistance in cervical cancer cells partly by up-regulating Dicer expression. Tumour Biol. 2016, 37, 6359–6369. [Google Scholar] [CrossRef]
- Chen, R.J.; Shun, C.T.; Yen, M.L.; Chou, C.H.; Lin, M.C. Methyltransferase G9a promotes cervical cancer angiogenesis and decreases patient survival. Oncotarget 2017, 8, 62081–62098. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Shin, S.H.; Yoon, H.; Huh, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. FIH Is an Oxygen Sensor in Ovarian Cancer for G9a/GLP-Driven Epigenetic Regulation of Metastasis-Related Genes. Cancer Res. 2018, 78, 1184–1199. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Huang, L.; Wei, L.; Hou, Z.; Ye, W.; Huang, S. Chaetocin induces caspase-dependent apoptosis in ovarian cancer cells via the generation of reactive oxygen species. Oncol. Lett. 2019, 18, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Qiu, L.; Hong, Y.; Karnik, T.; Tadros, G.; Mau, B.; Ma, T.; Mu, Y.; New, J.; Louie, R.J.; et al. The histone demethylase KDM4B regulates peritoneal seeding of ovarian cancer. Oncogene 2017, 36, 2565–2576. [Google Scholar] [CrossRef] [Green Version]
- Soldi, R.; Ghosh Halder, T.; Weston, A.; Thode, T.; Drenner, K.; Lewis, R.; Kaadige, M.R.; Srivastava, S.; Daniel Ampanattu, S.; Rodriguez Del Villar, R.; et al. The novel reversible LSD1 inhibitor SP-2577 promotes anti-tumor immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) complex mutated ovarian cancer. PLoS ONE 2020, 15, e0235705. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Stimson, L.; Wood, V.; Khan, O.; Fotheringham, S.; La Thangue, N.B. HDAC inhibitor-based therapies and haematological malignancy. Ann. Oncol. 2009, 20, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P.; Gynecologic Oncology, G. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Matulonis, U.; Berlin, S.; Lee, H.; Whalen, C.; Obermayer, E.; Penson, R.; Liu, J.; Campos, S.; Krasner, C.; Horowitz, N. Phase I study of combination of vorinostat, carboplatin, and gemcitabine in women with recurrent, platinum-sensitive epithelial ovarian, fallopian tube, or peritoneal cancer. Cancer Chemother. Pharmacol. 2015, 76, 417–423. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Wilson, A.J.; Sarfo-Kantanka, K.; Barrack, T.; Steck, A.; Saskowski, J.; Crispens, M.A.; Khabele, D. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol. Oncol. 2016, 143, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues Moita, A.J.; Bandolik, J.J.; Hansen, F.K.; Kurz, T.; Hamacher, A.; Kassack, M.U. Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors. Int. J. Mol. Sci 2020, 21, 8300. [Google Scholar] [CrossRef]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.A.; Tsao, M.S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; Disilvestro, P.A.; Fader, A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Wasim, L.; Chopra, M. Panobinostat induces apoptosis via production of reactive oxygen species and synergizes with topoisomerase inhibitors in cervical cancer cells. Biomed. Pharmacother. 2016, 84, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, J.; Ding, H.; Chen, L.; Zhang, Y.; Liu, R.; Xu, P.; Du, D.; Lu, W.; Liu, J.; et al. Identification of novel EZH2 inhibitors through pharmacophore-based virtual screening and biological assays. Bioorganic Med. Chem. Lett. 2016, 26, 3813–3817. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [Green Version]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e408. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenguer-Daize, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Soria, J.C.; Stathis, A.; Delord, J.P.; Awada, A.; Peters, S.; Lewin, J.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. A phase Ib trial with MK-8628/OTX015, a small molecule inhibitor of bromodomain (BRD) and extra-terminal (BET) proteins, in patients with selected advanced solid tumors. Eur. J. Cancer 2016, 69, S2–S3. [Google Scholar] [CrossRef]
- Liu, A.; Fan, D.; Wang, Y. The BET bromodomain inhibitor i-BET151 impairs ovarian cancer metastasis and improves antitumor immunity. Cell Tissue Res. 2018, 374, 577–585. [Google Scholar] [CrossRef]
- Wilson, A.J.; Hoover, A.; Harris, W.; Liu, E.; Khabele, D.; Yull, F. Abstract 626: Bromodomain inhibition in ovarian cancer and the tumor microenvironment to improve PARP inhibitor response. Cancer Res. 2018, 78, 626. [Google Scholar] [CrossRef]
- Yin, M.; Guo, Y.; Hu, R.; Cai, W.L.; Li, Y.; Pei, S.; Sun, H.; Peng, C.; Li, J.; Ye, R.; et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 2020, 11, 1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, G.Y.L.; Shaw, R.; Schaub, F.X.; Stork, I.N.; Gurley, K.E.; Bridgwater, C.; Diaz, R.L.; Rosati, R.; Swan, H.A.; Ince, T.A.; et al. BET, SRC, and BCL2 family inhibitors are synergistic drug combinations with PARP inhibitors in ovarian cancer. EBioMedicine 2020, 60, 102988. [Google Scholar] [CrossRef]
- Boshuizen, J.; Peeper, D.S. Rational Cancer Treatment Combinations: An Urgent Clinical Need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS ONE 2011, 6, e29085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A.; Gynecologic Oncology, G. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.Y.; Liao, W.S.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Histone modifying enzymes and epigenetic drugs in gynaecological cancers to restore the balance of epigenetic factors. Left panel; HDACs, KDMs and KMTs are often over-expressed across many gynaecological cancers. Mechanisms include addition of methyl groups and/or removal of acetyl groups at key histone tail residues, resulting in the repression of key tumour suppressor genes. Right panel; Epigenetic intervention via small molecule inhibitors to epigenetic enzymes, or “epidrugs” induces changes in chromatin configuration, resulting in re-expression of tumour suppressor genes. Examples of HDAC inhibitor names are provided, question marks denote that KDM/KMT inhibitors are to be determined. Green wedges represent lysine demethylases (KDMs), yellow wedges represent KMTs, blue wedges represent HDACs, red diamonds represent methyl groups, green diamonds represent acetyl groups, alternating grey and orange circles represent amino acid residues that comprise histone tails, large purple ovals represent histone subunits, triangles with black, dark grey and light grey borders represent HDAC inhibitors, KDM inhibitors and KMT inhibitors respectively.
Figure 1.
Histone modifying enzymes and epigenetic drugs in gynaecological cancers to restore the balance of epigenetic factors. Left panel; HDACs, KDMs and KMTs are often over-expressed across many gynaecological cancers. Mechanisms include addition of methyl groups and/or removal of acetyl groups at key histone tail residues, resulting in the repression of key tumour suppressor genes. Right panel; Epigenetic intervention via small molecule inhibitors to epigenetic enzymes, or “epidrugs” induces changes in chromatin configuration, resulting in re-expression of tumour suppressor genes. Examples of HDAC inhibitor names are provided, question marks denote that KDM/KMT inhibitors are to be determined. Green wedges represent lysine demethylases (KDMs), yellow wedges represent KMTs, blue wedges represent HDACs, red diamonds represent methyl groups, green diamonds represent acetyl groups, alternating grey and orange circles represent amino acid residues that comprise histone tails, large purple ovals represent histone subunits, triangles with black, dark grey and light grey borders represent HDAC inhibitors, KDM inhibitors and KMT inhibitors respectively.


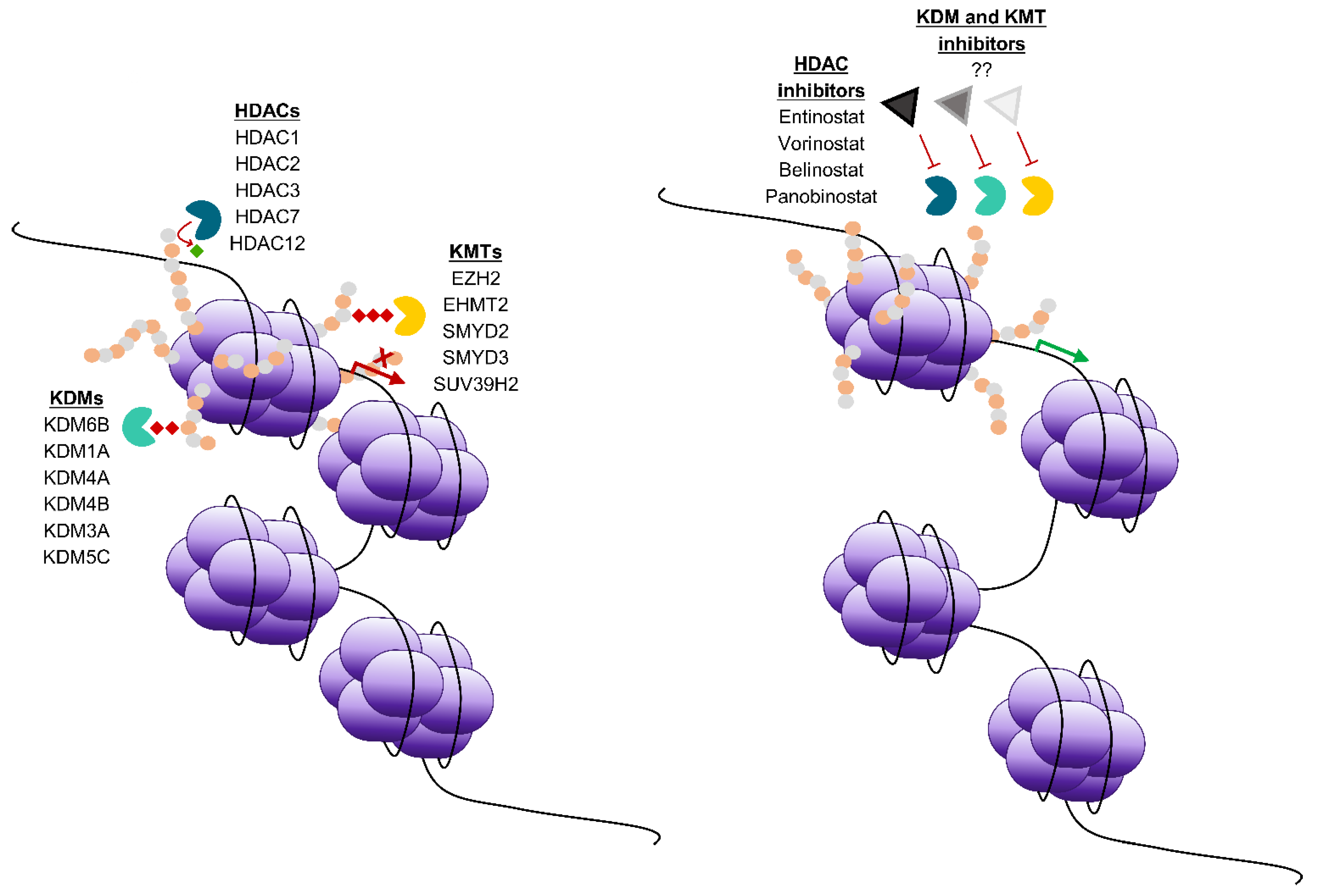{kind=link}
Table 1.
Histone modifying enzymes relevant to gynaecological cancers.
| Enzyme | Enzyme Type | Mechanism | Modification | Cancer | Expression | Reference(s) |
|---|---|---|---|---|---|---|
| EZH2 | Writer | Methyltransferase | H3K27me2, H3K27me3 | Ovarian, Endometrial | ↑ | [14,15] |
| KDM6B | Eraser | Demethylase | H3K27me2, H3K27me3 | Ovarian | ↑ | [16] |
| EHMT2 | Writer | Methyltransferase | H3K9me, H3K9me2 | Ovarian, Endometrial | ↑ | [15,17] |
| LSD1 | Eraser | Demethylase | H3K4me2, H3K4me | Ovarian, Endometrial | ↑ | [18] |
| LSD1 | Eraser | Demethylase | H3K9me | Endometrial | ↑ | [19] |
| EP400 | Writer | Acetyltransferase | HAc | Cervical | ↓ | [20] |
| KDM5C | Eraser | Demethylase | H3K4me, H3K4me2, H3K4me3 | Cervical | ↓ | [20] |
| SMYD2 | Writer | Methyltransferase | H3K4me3 | Ovarian | ↑ | [21] |
| SMYD3 | Writer | Methyltransferase | H3K4me3, H4K5me | Cervical | ↑ | [21] |
| DOT1L | Writer | Methyltransferase | H3K79me2 | Ovarian | ↑ | [22] |
| RNF20-RNF40 complex | Writer | Ubiquitinating enzyme | H2Bub | Ovarian | ↓ | [23,24] |
| KDM4A | Eraser | Demethylase | H3K9me3, H3K36 | Ovarian | ↑ | [25] |
| KDM3A | Eraser | Demethylase | H3K9me, H3K9me2 | Ovarian | ↑ | [26] |
| SUV39H2 | Writer | Methyltransferase | H3K9me3 | Cervical | ↑ | [27] |
| KDM4B | Eraser | Demethylase | H3K9me2, H3K9me3 | Endometrial | ↑ | [28] |
| HDAC1 | Eraser | Deacetylase | HAc | Ovarian, Endometrial | ↑ | [29,30] |
| HDAC2 | Eraser | Deacetylase | HAc | Ovarian, Endometrial | ↑ | [29,30] |
| HDAC3 | Eraser | Deacetylase | HAc | Ovarian, Endometrial | ↑ | [29,30] |
| HDAC7 | Eraser | Deacetylase | HAc | Ovarian | ↑ | [31] |
| HDAC12 | Eraser | Deacetylase | HAc | Ovarian | ↑ | [31] |
| BRD4 | Reader | BET protein | - | Ovarian | ↑ | [32] |
Up arrows (↑) represent increased expression in indicated cancer relative to normal tissue, down arrows (↓) represent decreased expression in indicated cancer relative to normal tissue.
Table 2.
Recent examples of preclinical studies that have tested epigenetic inhibitors in gynaecological cancers.
Table 2.
Recent examples of preclinical studies that have tested epigenetic inhibitors in gynaecological cancers.
| Enzyme | Epigenetic Inhibitor | Combination Agent(s) | Cancer Type | Model/Cell Lines Tested | Inhibitor Dose and Treatment Duration | Outcome | Reference(s) |
|---|---|---|---|---|---|---|---|
| KMT4/DOT1L | EPZ004777 | None | Ovarian | PEO1 and PEO4 cell lines | 0.1 µM, 72 h | Growth arrest | [96] |
| EZH2 | GSK126 | Cisplatin | Ovarian | OVCAR3 and CA-MSC orthotopic mouse model | Various concentrations | Cell viability of OVCAR3 cells unaffected, but decreased ability of OVCAR3 cells to metastasise | [97] |
| EZH2 | GSK126 | 5-AZA dC | Ovarian | NSG model | 30 mg/kg, 3 times a week for 2 weeks | Increased efficacy of adoptive T-cell therapy in vivo | [58] |
| EZH2 | GSK126 | None, Cisplatin or Doxorubicin | Endometrial | Various cancer cell lines | 0.025–20 µM, 8 days | Decreased cell proliferation and induction of apoptosis. Additive effects with cisplatin or doxorubicin | [98] |
| EZH2 | DZNep | None | Cervical | HeLa and HeLa/DDP cells | Various concentrations, 72 h | Reversal of cisplatin resistance observed in the HeLa/DDP cell line | [99] |
| G9a (EHMT2) | BIX01294 | None | Cervical | CaSki, HeLa and SiHa cell lines | 5 µM, 72 h | Cell migration and invasion attenuated in BIX01294-treated cells | [100] |
| G9a (EHMT2) | BIX01294 | None | Cervical | Subcutaneous SiHa cell line xenograft cervical cancer tumor model | 5 mg/kg and 10 mg/kg, 39 days | Xenograft tumour growth significantly attenuated from day 29 at a dose of 10 mg/kg compared to control | [100] |
| G9a (EHMT2) | UNC0638 | None | Ovarian | SKOV-3, ES-2, and PA-1 cell lines | 2 µM, 48 h | Increase in metastasis suppressor genes such as CDH10 | [101] |
| G9a (EHMT2) | UNC0638 | None | Ovarian | SKOV-3 cell line | 2 µM, 48 h | Decreased metastasis-related signaling | [17] |
| SUV39H1/SUV39H2 | Chaetocin | None | Ovarian | OVCAR3 cell line | IC50–60.66 nM, 24 h | Inhibited proliferation, induced ROS accumulation and resulted in caspase-induced cell death in OVCAR-3 cells | [102] |
| SUV39H1/SUV39H2 | Chaetocin | None | Cervical | HeLa and CaSki cell lines | 150 nM, 24 h | Restored the innate immune response to exogenous DNA | [103] |
| KDM6A/6B | GSK-J4 | None | Cervical | SiHa and HeLa cell lines | 25–100 µM, 72 h | Decreased cell viability in SiHa cell line, no effect in HeLa cell line | [104] |
| KDM6A/6B | GSK-J4 | None | Cervical | CaSki cell line | 0–30 µM, 72 h | Decreased cell viability | [104] |
| KDM6A/6B | GSK-J4 | None | Ovarian | A2780 cancer stem cell like cells | 0.5–10 µM, 72 h | Decreased cell viability | [16] |
| LSD1 | SP-2577 | None | Ovarian | SWI/SNF-mutated cell lines | 0.01–1.1 µM, 72 h | Affects cell viability, and induces expression of inflammatory cytokines in organoids | [105] |
| LSD1 | HCI2509 | None | Endometrial | AN3CA and KLE cell lines | IC50–500 nM, 96 h | Apoptotic cell death in cell lines, tumour regression in orthotopic xenografts | [19] |
| BRD4 | JQ1, I-BET151 | None | Ovarian | Various cell lines | 0.01–10 µM | Cell cycle arrest in all subtypes of ovarian cancer cell lines tested | [32] |
| BRD4 | JQ1 | None | Ovarian | OVCAR-3 cell line xenograft and patient-derived xenograft model | 50 mg/kg | Decreased tumour volume | [32] |
Table 3.
Clinical trials of HDAC inhibitors in gynaecological cancers.
| NCT Number | Trial | Inhibitor Name | Inhibitor Type | Combination Agent(s) | Epigenetic Target | Epigenetic Inhibitor Dose | Cancer Type * | Recruitment Status |
|---|---|---|---|---|---|---|---|---|
| NCT04651127 | Phase I/II | Chidamide | Class I HDAC inhibitor | Toripalimab | Class I HDACs | 30 mg/day orally, twice a week | Persistent, Recurrent, or Metastatic Cervical Cancer | Recruiting |
| NCT02728492 | Phase I | Quisinostat | HDAC inhibitor | Gemcitabine, Carboplatin, Paclitaxel | HDACs | 8 mg every other day | Non-small Cell Lung Cancer, Epithelial Ovarian Cancer | Completed |
| NCT02948075 | Phase II | Quisinostat | HDAC inhibitor | Carboplatin, Paclitaxel | HDACs | 12 mg every other day | Ovarian Cancer | Completed |
| NCT02915523 | Phase I/II | Entinostat | HDAC inhibitor | Avelumab | HDACs | 5 mg weekly, 3 months | Advanced Epithelial Ovarian Cancer | Unknown |
| NCT00772798 | Phase II | Vorinostat | HDAC inhibitor | Paclitaxel, Carboplatin | HDACs | 400 mg once daily orally, days 4–10 of a 25 day cycle | Recurrent Ovarian Cancer | Unknown |
| NCT03345485 | Phase I/II | Tinostamustine | alkylating HDAC inhibitor | - | HDACs | 60 mg/m2 up to 100 mg/m2, day 1 and 15 of a 28 day cycle | Advanced Solid Tumors | Recruiting |
| NCT00020579 | Phase I | Entinostat | HDAC inhibitor | - | HDACs | Dose escalation study | Advanced Solid Tumors or Lymphoma | Completed |
| NCT00421889 | Phase I/II | Belinostat | HDAC inhibitor | Carboplatin, Paclitaxel | HDACs | 1000 mg/m2, days 1–5 of a 21 day cycle | Ovarian Cancer in Need of Relapse Treatment | Completed |
| NCT00976183 | Phase I/II | Vorinostat | HDAC inhibitor | Carboplatin, Paclitaxel | HDACs | 200 mg once a day | Advanced Stage Ovarian Carcinoma | Terminated |
| NCT04315233 | Phase I | Belinostat | HDAC inhibitor | Ribociclib | HDACs | 600 mg/m2, days 1–5 of a 28 day cycle | Metastatic Triple Neg Breast Cancer & Recurrent Ovarian Cancer | Recruiting |
| NCT02601937 | Phase I | Tazemetostat | KMT inhibitor | - | EZH2 | Dose escalation study | Pediatric Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma | Recruiting |
| NCT00301756 | Phase II | Belinostat | HDAC inhibitor | - | HDACs | - | Recurrent or Persistent Ovarian Epithelial or Primary Peritoneal Cavity Cancer | Completed |
| NCT04703920 | Phase I | Belinostat | HDAC inhibitor | Talazoparib | HDACs | Up to 1000 mg/m2 IV once daily on days 1– 5 of a 21-day cycle | Metastatic Castration Resistant Prostate Cancer, and Metastatic Ovarian Cancer | Not yet recruiting |
| NCT03018249 | Phase I | Entinostat | HDAC inhibitor | Medroxyprogesterone Acetate | HDACs | - | Endometrial cancer | Active, not recruiting |
| NCT00132067 | Phase II | Vorinostat | HDAC inhibitor | - | HDACs | - | Recurrent or Persistent Ovarian Epithelial or Primary Peritoneal Cavity Cancer | Completed |
| NCT02661815 | Phase I | Ricolinostat | HDAC inhibitor | Paclitaxel | HDACs | 80 mg/m2 per week (3 out of 4 weeks). | Gynecologic cancers | Terminated |
| NCT04357873 | Phase II | Vorinostat | HDAC inhibitor | Pembrolizumab | HDACs | 400 mg once daily, until progression | metastatic squamous cell carcinoma (head and neck, lung, cervix, vulva, anus and penis) | Recruiting |
| NCT04498520 | Phase I | Abexinostat | HDAC inhibitor | Palbociclib, Fulvestrant | HDACs | - | Breast and Gynecologic cancers | Not yet recruiting |
| NCT00413322 | Phase I | Belinostat | HDAC inhibitor | 5-Fluorouracil | HDACs | 300, 600, or 1000 mg/m2 belinostat for 5 days every 21 days starting with cycle 1 | Advanced Solid Tumors | Completed |
* may include other cancers in addition to gynaecological cancers.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ramarao-Milne, P.; Kondrashova, O.; Barry, S.; Hooper, J.D.; Lee, J.S.; Waddell, N. Histone Modifying Enzymes in Gynaecological Cancers. Cancers 2021, 13, 816. https://doi.org/10.3390/cancers13040816
AMA Style
Ramarao-Milne P, Kondrashova O, Barry S, Hooper JD, Lee JS, Waddell N. Histone Modifying Enzymes in Gynaecological Cancers. Cancers. 2021; 13(4):816. https://doi.org/10.3390/cancers13040816
Chicago/Turabian StyleRamarao-Milne, Priya, Olga Kondrashova, Sinead Barry, John D. Hooper, Jason S. Lee, and Nicola Waddell. 2021. "Histone Modifying Enzymes in Gynaecological Cancers" Cancers 13, no. 4: 816. https://doi.org/10.3390/cancers13040816
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.