
Dual-Target CAR-Ts with On- and Off-Tumour Activity May Override Immune Suppression in Solid Cancers: A Mathematical Proof of Concept

 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mathematical Models
2.1. Model of Solid Tumour Response to a CAR-T Cell Treatment in the Presence of Immune Suppression
2.2. Modelling CAR-T Cells Targetting On-Tumour and Off-Tumour Antigens
2.3. Parameter Estimation
3. Results (I): Therapy Outcomes under Immune Suppression Using CAR-T Cells with a Single CAR Group Targetting a Tumour Antigen
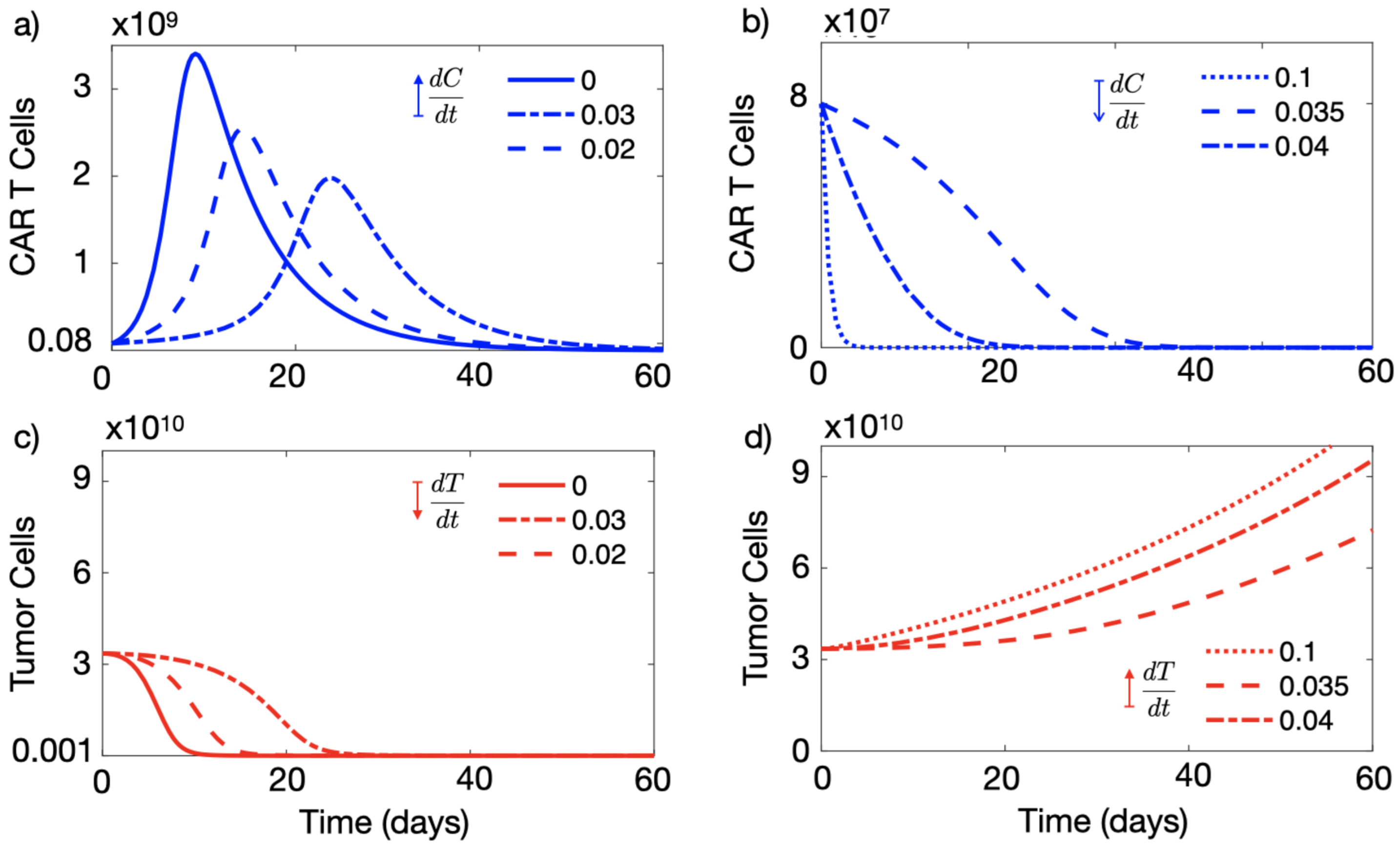
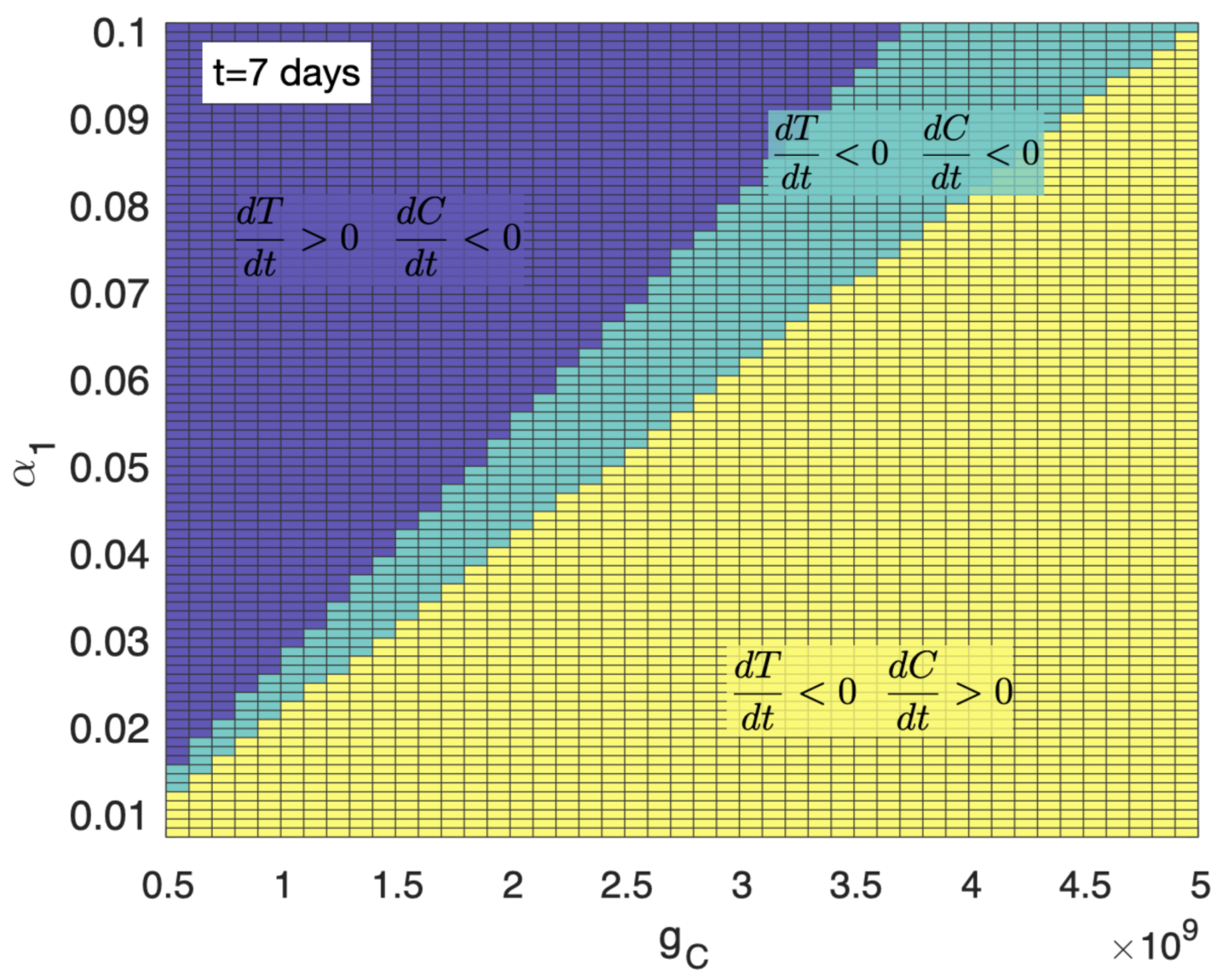
3.1. A High Level of Immune Suppression Prevents In-Patient Expansion of CAR-T Cells
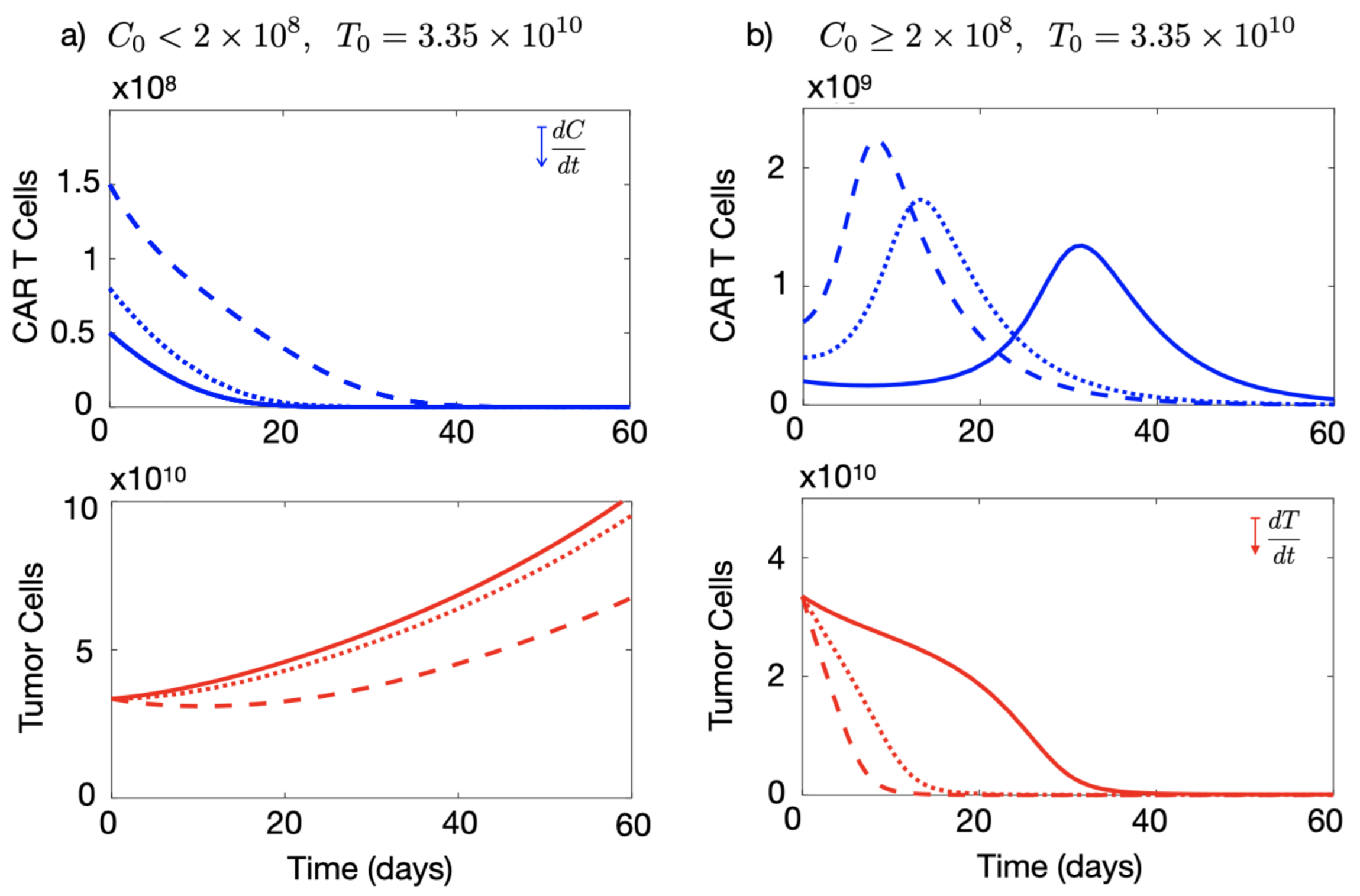
3.2. Initial Number of CAR-T Cells Injected Affects the Outcome of the Therapy
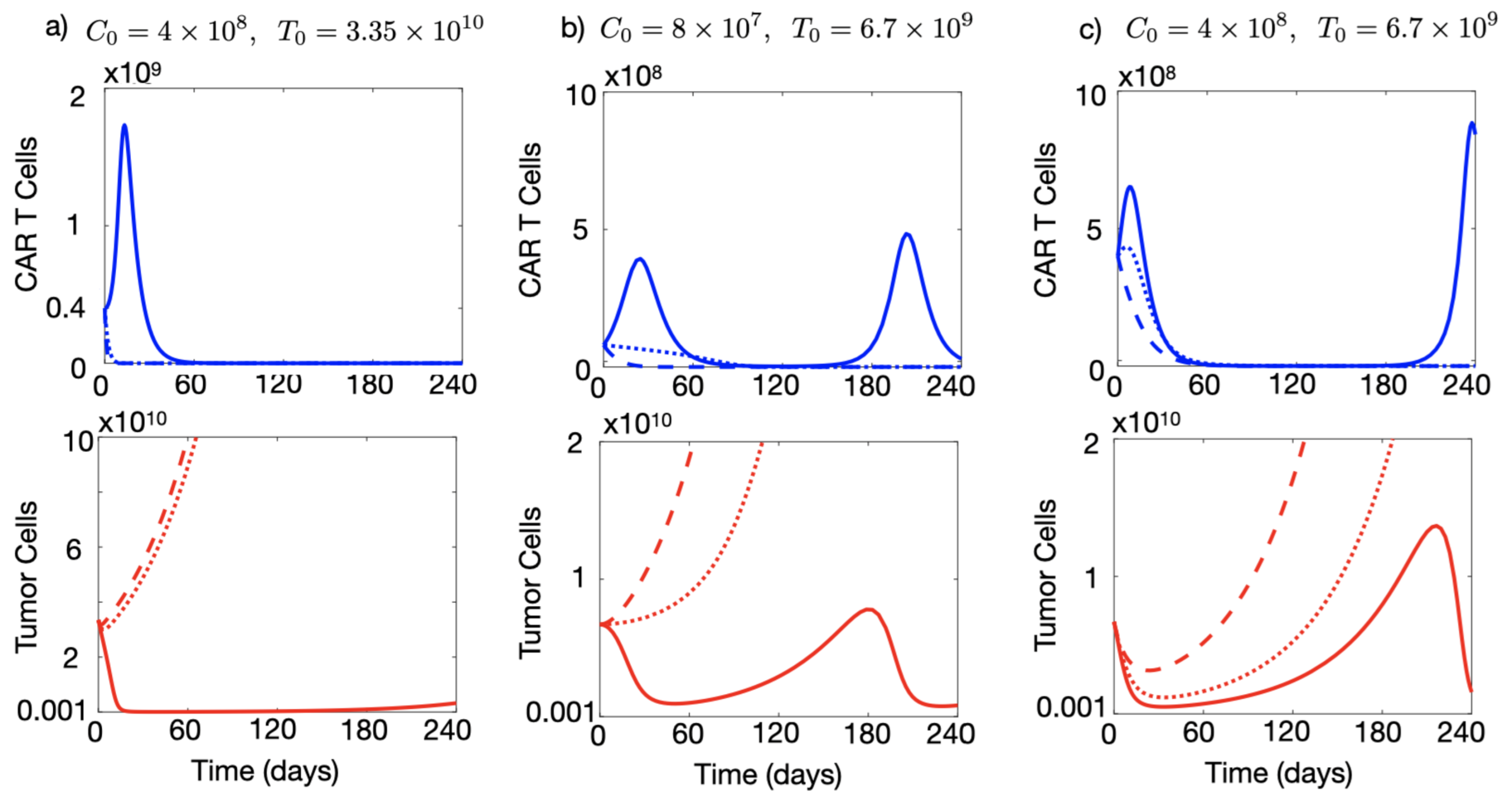
3.3. Injection of a Large Number of CAR-T Cells Could Allow for Cure or Prolonged Tumour Control in the Presence of Immune Suppression
3.4. A High Initial Tumour Load Favours CAR-T Cell Expansion
4. Results (II): Therapy Outcomes under Tumour Immune Suppression Using CAR-T Cells with Dual CAR Groups with On- and Off-Tumour Activity
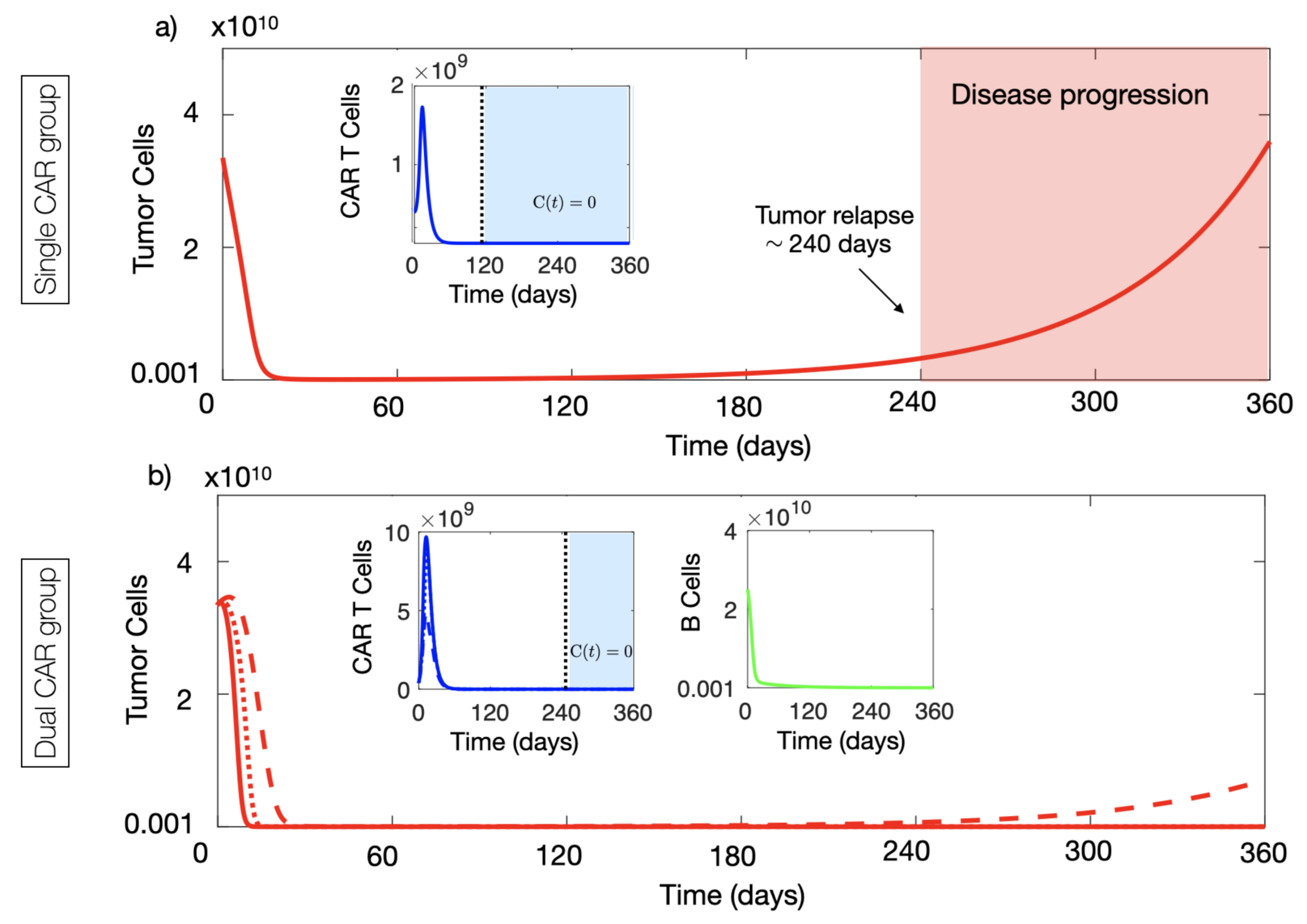
4.1. CAR-T Cells with Two Targets Provided Long-Time Tumour Control Advantages In Silico
4.2. Dual CAR-T Improves the Possibility of Therapy Success
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CAR | Chimeric antigen receptor |
| GBM | Glioblastoma |
Appendix A. Basic Properties of the Mathematical Model with Single CAR Group
Appendix A.1. Existence of Solutions
Appendix A.2. Equilibria of the Model and Local Stability Analysis

Appendix B. Basic Properties of the Mathematical Model with Dual CAR Group
Equilibria of the Model and Local Stability Analysis
References
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Yang, J.F.; Deng, B.P.; Zhao, X.J.; Zhang, X.; Lin, Y.H.; Wu, Y.N.P.; Deng, Z.L.; Zhang, Y.L.; Liu, S.H.; et al. High efficacy and safety of low-dose CD19− directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 2017, 31, 2587–2593. [Google Scholar] [CrossRef]
- Miliotou, A.N.; Papadopoulou, L.C. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr. Pharm. Biotechnol. 2018, 19, 5–18. [Google Scholar] [CrossRef]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; O’Rourke, D.M. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol. Ther. 2020, 205, 107419. [Google Scholar] [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D., Jr. Driving CARs to the clinic for solid tumors. Gene Ther. 2018, 25, 165–175. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, CH.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 21, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell. 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.H.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational Targeting Offsets Antigen Escape and Enhances Effector Functions of Adoptively Transferred T Cells in Glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; LimPrecision, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altrock, P.M.; Liu, L.L.; Michor, F. The mathematics of cancer: Integrating quantitative models. Nat. Rev. Cancer 2016, 15, 730–745. [Google Scholar] [CrossRef]
- Pérez-García, V.M.; Fitzpatrick, S.; Pérez-Romasanta, L.A.; Pesic, M.; Schucht, P.; Arana, E.; Sánchez-Gómez, P. Applied mathematics and nonlinear sciences in the war on cancer. Appl. Math. Nonlinear Sci. 2016, 1, 423–436. [Google Scholar] [CrossRef] [Green Version]
- Konstorum, A.; Vella, A.T.; Adler, A.J.; Laubenbacher, R.C. Addressing current challenges in cancer immunotherapy with mathematical and computational modelling. J. R. Soc. Interface 2017, 14, 20170150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, K.P. A review of mathematical models of cancer-immune interactions in the context of tumor dormancy. Adv. Exp. Med. Biol. 2013, 734, 201–234. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.; Enderling, H. From concept to clinic: Mathematically informed immunotherapy. Curr. Probl. Cancer 2016, 40, 68–83. [Google Scholar] [CrossRef]
- de Pillis, L.G.; Eladdadi, A.; Radunskaya, A.E. Modeling cancer-immune responses to therapy. J. Pharmacokinet. Pharmacodyn. 2014, 41, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Eftimie, R.; Bramson, J.L.; Earn, D.J.D. Interactions Between the Immune System and Cancer: A Brief Review of Non-spatial Mathematical Models. Bull. Math. Biol. 2011, 73, 2–32. [Google Scholar] [CrossRef]
- Norton, K.-A.; Gong, C.; Jamalian, S.; Popel, A.S. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes 2019, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Mahlbacher, G.E.; Reihmer, K.C.; Frieboes, H.B. Mathematical modeling of tumor-immune cell interactions. J. Theor. Biol. 2019, 469, 47–60. [Google Scholar] [CrossRef]
- Sahoo, P.; Yang, X.; Abler, D.; Maestrini, D.; Adhikarla, V.; Frankhouser, D.; Cho, H.; Machuca, V.; Wang, D.; Barish, M.; et al. Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data. J. R. Soc. Interface 2020, 17, 20190734. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.; Coquille, L.; Mayer, H.; Holzel, M.; Rogava, M.; Tuting, T.; Bovier, A. A stochastic model for immunotherapy of cancer. Sci. Rep. 2016, 6, 24169. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, G.J.; Locke, F.L.; Altrock, P.M. Evolutionary Dynamics of CAR T Cell Therapy. bioRxiv 2019, 717074. [Google Scholar] [CrossRef]
- Rodrigues, B.J.; Barros, L.R.C.; Almeida, R.C. Three-Compartment Model of CAR T-cell Immunotherapy. bioRxiv 2019, 779793. [Google Scholar] [CrossRef]
- Mostolizadeh, R.; Afsharnezhad, Z.; Marciniak-Czochra, A. Mathematical model of Chimeric Anti-gene Receptor (CAR) T cell therapy with presence of cytokine. Numer. Algebra Control Optim. 2018, 8, 63–80. [Google Scholar] [CrossRef] [Green Version]
- León-Triana, O.; Soukaina, S.; Calvo, G.F.; Belmonte-Beitia, J.; Chulián, S.; Martínez-Rubio, A.; Rosa, M.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. CAR T cell therapy in B-cell acute lymphoblastic leukaemia: Insights from mathematical models. Commun. Nonlinear Sci. Numer. Simul. 2021, 94, 105570. [Google Scholar]
- Pérez-García, V.M.; León-Triana, O.; Rosa, M.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. CAR T cells for T-cell leukemias: Insights from mathematical models. Commun. Nonlinear Sci. Numer. Simul. 2021, 96, 105684. [Google Scholar]
- Chulián, S.; Martínez-Rubio, A.; Rosa, M.; Pérez-García, V.M. Mathematical models of Leukaemia and its treatment: A review. SEMA J. Under review.
- Brown, M.P.; Ebert, L.M.; Gargett, T. Clinical chimeric antigen receptor-T cell therapy: A new and promising treatment modality for glioblastoma. Clin. Transl. Immunol. 2019, 8, e1050. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro-Oncology 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.R.; Wright, C.; et al. Bioactivity and safety of IL13Rα2- redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Stensjøen, A.L.; Solheim, O.; Kvistad, K.A.; Haberg, A.K.; Salvesen, O.; Berntsen, E.M. Growth dynamics of untreated glioblastomas in vivo. Neuro-Oncology 2015, 17, 1402–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-García, V.M.; Calvo, G.F.; Bosque, J.J.; León-Triana, O.; Jiménez, J.; Pérez-Beteta, J.; Belmonte-Beitia, J.; Valiente, M.; Zhu, L.; García-Gómez, P.; et al. Universal scaling laws rule explosive growth in human cancers. Nat. Phys. 2020, 16, 1232–1237. [Google Scholar] [CrossRef]
- Stein, A.M.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Levine, B.L.; Tomassian, L.; Shah, S.; et al. Optimization of IL13Ra2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, V.A.; Makalkin, I.A.; Taylor, M.A.; Perelson, A.S. Nonlinear dynamics of immunogenic tumors: Parameter estimation and global bifurcation analysis. Bull. Math. Biol. 1994, 56, 295–321. [Google Scholar] [CrossRef]
- Stein, A.M.; Grupp, S.A.; Levine, J.E.; Laetsch, T.W.; Pulsipher, M.A.; Boyer, M.W.; August, K.J.; Levine, B.L.; Tomassian, L.; Shah, S.; et al. Tisagenlecleucel Model-Based Cellular Kinetic Analysis of Chimeric Antigen Receptor-T Cells. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.L.; Fouser, L.A.; Jussif, J.; Fitz, L.; Deng, B.; Wood, C.R.; Collins, M.; Honjo, T.; Freeman, G.J.; Carreno, B.M. PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur. J. Immunol. 2002, 32, 634–643. [Google Scholar] [CrossRef]
- Radunskaya, A.; Kim, R.; Woods, T. Mathematical Modeling of Tumor Immune Interactions: A Closer Look at the Role of a PD-L1 Inhibitor in Cancer Immunotherapy. Spora J. Biomath. 2018, 4, 25–41. [Google Scholar] [CrossRef]
- Halkola, A.S.; Parvinen, K.; Kasanen, H.; Mustjoki, S.; Aittokallio, T. Modelling of killer T-cell and cancer cell subpopulation dynamics under immuno- and chemotherapies. J. Theor. Biol. 2020, 488, 110136. [Google Scholar] [CrossRef] [PubMed]
- Benchaib, M.A.; Bouchnita, A.; Volpert, V.; Makhoute, A. Mathematical Modeling Reveals That the Administration of EGF Can Promote the Elimination of Lymph Node Metastases by PD-1/PD-L1 Blockade. Front. Bioeng. Biotechnol. 2019, 7, 104. [Google Scholar] [CrossRef]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do they have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Fulcher, D.A.; Basten, A. B cell life span: A review. Immunol. Cell Biol. 1997, 75, 446–555. [Google Scholar] [CrossRef]
- Chen, X.; Jensen, P.E. The role of B lymphocytes as antigen-presenting cells. Arch. Immunol. Ther. Exp. 2008, 56, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.V.; Turtle, C.J. Toxicities of CD19 CAR-T cell immunotherapy. Am. J. Hematol. 2019, 94, S42–S49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome after Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.; Lin, Q.; Ma, G.; Yin, H.; Chen, S.; Lin, Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomed. Pharmacother. 2020, 121, 109625. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Value | Units | Source |
|---|---|---|---|---|
| Mitotic stimulation | ||||
| of CAR-T cells by | 0.2–0.9 | day | [33,50] | |
| tumour cells | ||||
| T cell concentration | Estimated | |||
| for half-maximal | 10 | cell | from [50] | |
| CAR-T cell proliferation | ||||
| Tumour inactivation rate | 0.01–0.99 | day | [53] | |
| CAR-T concentration | Estimated | |||
| for half-maximal | cell | from [53] | ||
| tumour inactivation | ||||
| Activated CAR-T | 7–30 | day | [51] | |
| cell lifetime | ||||
| Tumour growth rate | (0.001–0.2) | day | [46] | |
| Killing efficiency | day | |||
| of CAR-T cells | ∼ 10 | × cell | [33] | |
| against tumour | ||||
| Mitotic stimulation | ||||
| of CAR-T cells by | (0.2–0.9) | day | [33,50] | |
| CD19 | ||||
| B-cell concentration | Estimated | |||
| for half-maximal | 10 | cell | from [50] | |
| CAR-T cell proliferation | ||||
| B-lymphocyte | 30–60 | day | [58] | |
| lifetime | ||||
| Killing efficiency | day | Estimated | ||
| of CAR-T cells | ∼ 10 | × cell | from [57] | |
| against CD19 cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
León-Triana, O.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. Dual-Target CAR-Ts with On- and Off-Tumour Activity May Override Immune Suppression in Solid Cancers: A Mathematical Proof of Concept. Cancers 2021, 13, 703. https://doi.org/10.3390/cancers13040703
León-Triana O, Pérez-Martínez A, Ramírez-Orellana M, Pérez-García VM. Dual-Target CAR-Ts with On- and Off-Tumour Activity May Override Immune Suppression in Solid Cancers: A Mathematical Proof of Concept. Cancers. 2021; 13(4):703. https://doi.org/10.3390/cancers13040703
Chicago/Turabian StyleLeón-Triana, Odelaisy, Antonio Pérez-Martínez, Manuel Ramírez-Orellana, and Víctor M. Pérez-García. 2021. "Dual-Target CAR-Ts with On- and Off-Tumour Activity May Override Immune Suppression in Solid Cancers: A Mathematical Proof of Concept" Cancers 13, no. 4: 703. https://doi.org/10.3390/cancers13040703