
T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies



1
Centre de Recherche de l’Hôpital Maisonneuve-Rosemont, Montréal, QC H1T 2M4, Canada
2
Division of Hematology-Oncology, Hôpital Maisonneuve-Rosemont, Montréal, QC H1T 2M4, Canada
3
Department of Medicine, Université de Montréal, Montréal, QC H3T 1J4, Canada
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(4), 598; https://doi.org/10.3390/cancers13040598
Submission received: 30 December 2020
/
Revised: 29 January 2021
/
Accepted: 29 January 2021
/
Published: 3 February 2021
(This article belongs to the Section Cancer Immunology and Immunotherapy)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
T cells are immune cells that can be used to target infections or cancers. Adoptive T-cell immunotherapy leverages these properties and/or confers new features to T cells through ex vivo manipulations prior to their use in patients. However, as a “living drug,” the function of these cells can be hampered by several built-in physiological constraints and external factors that limit their efficacy. Manipulating T cells ex vivo can impart dysfunctional features to T cells through repeated stimulations and expansion, but it also offers many opportunities to improve the therapeutic potential of these cells, including emerging interventions to prevent or reverse T-cell dysfunction developing ex vivo or after transfer in patients. This review outlines the various forms of T-cell dysfunction, emphasizes how it affects various types of T-cell immunotherapy approaches, and describes current and anticipated strategies to limit T-cell dysfunction.
Abstract
Over the last decades, cellular immunotherapy has revealed its curative potential. However, inherent physiological characteristics of immune cells can limit the potency of this approach. Best defined in T cells, dysfunction associated with terminal differentiation, exhaustion, senescence, and activation-induced cell death, undermine adoptive cell therapies. In this review, we concentrate on how the multiple mechanisms that articulate the various forms of immune dysfunction impact cellular therapies primarily involving conventional T cells, but also other lymphoid subtypes. The repercussions of immune cell dysfunction across the full life cycle of cell therapy, from the source material, during manufacturing, and after adoptive transfer, are discussed, with an emphasis on strategies used during ex vivo manipulations to limit T-cell dysfunction. Applicable to cellular products prepared from native and unmodified immune cells, as well as genetically engineered therapeutics, the understanding and potential modulation of dysfunctional features are key to the development of improved cellular immunotherapies.
1. Introduction
Adoptive cell immunotherapy (ACT) is a promising approach to treat a variety of pathological states, including infections as well as both solid and hematologic cancers. Immune cells in ACT can be harvested from tumor resection/biopsy, from the patient’s own blood, or donated by a fully or partially human leukocyte antigen (HLA)-matched healthy donor. These cells are then injected into the patient after minimal or more extensive ex vivo manipulations. The oldest, and arguably still one of the most effective forms of ACT, is allogeneic hematopoietic cell transplantation, which most often requires only minimal cell handling and primarily leverages immunogenetic disparities between donor and recipient to treat hematopoietic cancers [1]. Such a prosaic form of ACT is associated with several shortcomings, including unpredictable therapeutic effect and immune complications in the form of graft-versus-host disease (GVHD). Increasingly, however, ex vivo procedures are used to both enhance “on-target” effects on cancer or infected cells and minimize immune complications. This review emphasizes these latter forms of immunotherapy that hinge on advanced ex vivo cellular manipulation procedures. Genetic cellular engineering has also been implemented to achieve optimal cell targeting and minimize off-target effects. In most cases, the manufacturing of therapeutic T-cell products requires T-cell stimulation and expansion, which may be conducive to the acquisition of dysfunctional features [2]. Similarly, continuous and/or chronic antigen encounters after patient infusion often further accentuate T-cell dysfunctionality. Thus, understanding the physiology behind the various T-cell dysfunctional states is fundamental to the design of optimal ACT protocols.
T-cell dysfunction is not inherently good nor bad, but must be considered as a central aspect of T-cell physiology. In the natural course of an immune response, a high number of T cells are rapidly generated to eliminate the foreign (most often microbial) antigens. After resolution of the threat, a contraction in the total number of T cells limits the risks associated with a sustained inflammatory response and restores homeostasis. Some lymphocytes are differentiated into long-term memory cells to protect the organism against future exposure to the same microorganism. During this process, T-cell differentiation is governed by the strength as well as the duration of the stimulation received by the T cell [3]. The T-cell receptor (TCR) complex, composed of TCRα/β chains and responsible for antigenic recognition, co-operate with a CD3 molecule responsible for transducing the activation signal through its immunoreceptor tyrosine-based activation motif (ITAM)-containing cytoplasmic tail [4]. Phosphorylation of these ITAMs by protein tyrosine kinases then allows other molecules to interact with the TCR complex [5]. Engagement of the TCR, aided by co-stimulatory molecules such as CD28 at the immunological synapse, then activates a wide range of intracellular pathways, including activator protein 1 (AP-1), nuclear factor of activated T-cells (NFAT), nuclear factor-kappa B (NF-κB), and mammalian target of rapamycin (mTOR) [6]. Although triggering of the TCR is a prerequisite for T-cell activation and differentiation, a sustained stimulation can lead to the loss of T-cell functions. In several settings, this is part of homeostatic processes that not only contribute to limiting systemic inflammation, but also protect the T cells against their own demise. Thus, dysfunctional T-cell states result from the sum of physiological countermeasures aimed at controlling T-cell responses and preventing hyperactivation. Several of these regulatory mechanisms are intrinsic to the T cells and others depend on extrinsic factors that can contribute to T-cell dysfunction or use T-cell inhibitory mechanisms to limit immune responses. T-cell dysfunction is defined by different transcriptional, phenotypic, and functional features and encompasses several cellular states such as terminal differentiation, exhaustion, senescence, and/or apoptosis [7,8]. Although the therapeutic opportunities potentially associated with the prevention and/or reversal of T-cell dysfunction are highly relevant to all forms of immunotherapies, including those that mobilize the patient’s own T-cell repertoire such as immune checkpoint blockade and bi-specific antibodies or engagers [9,10], this review focuses on the occurrence of T-cell dysfunction in ACT and the emerging strategies in the field to circumvent this problem.
2. T-Cell Dysfunction: Multifaceted Extension of T-Cell Physiology
The current use of T cells as therapy benefits from a large body of knowledge of T-cell dysfunction, obtained from both animal models and humans (extensively reviewed in [11,12,13,14]). This section summarizes the different mechanisms and cell states that can be regrouped under the general term of T-cell dysfunction. The extent to which these cellular states overlap or are exclusive still remains a matter of debate.
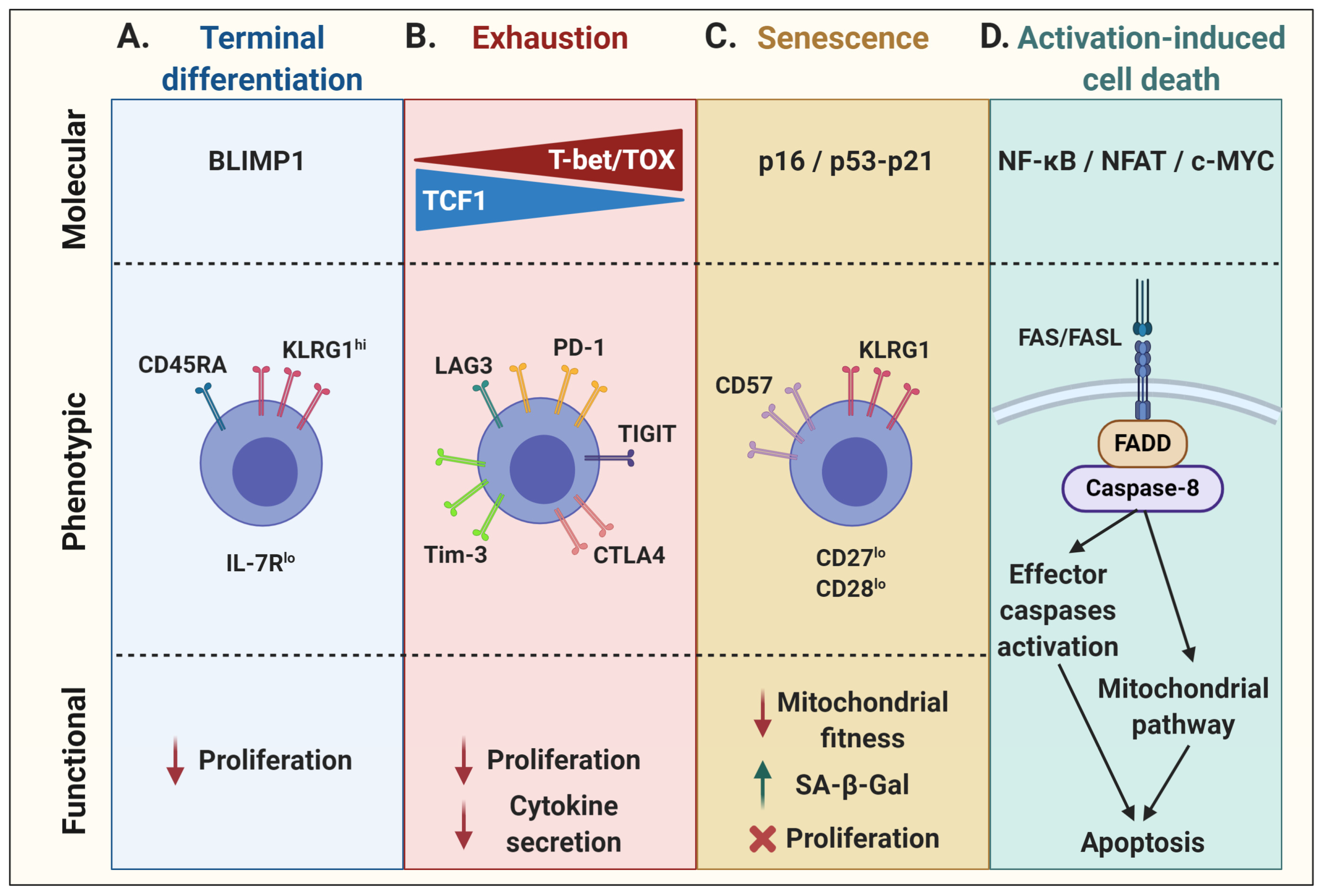
2.1. Terminal Effector Differentiation
In physiological conditions, although only a small number of cytotoxic T lymphocytes (CTLs) acquire memory properties following an immune response, a vast majority of effector cells enter a state of terminal effector differentiation. Mainly described in well-characterized mouse models of chronic infection, such as lymphocytic choriomeningitis virus (LCMV), short-lived terminally differentiated CTLs have been phenotypically associated with high expression of the NK cell marker killer cell lectin-like receptor G1 (KLRG1) and low expression of interleukin (IL)-7Rα occurring with the graded expression of the transcription factor T-bet [15,16]. On the one hand, KLRG1 expression impacts proliferative capacity through the decrease of AKT, cyclin D, and cyclin E activity, as well as through an increase in cyclin inhibitor p27 expression [17]. On the other hand, the lack of IL-7Rα prevents T-cell proliferation in response to homeostatic cytokines [18,19,20,21]. Moreover, these terminally differentiated T cells re-express the naïve T-cell marker CD45RA and lose their migration properties in addition to their proliferative capacity [16,22,23,24]. The transcriptional repressor B lymphocyte-induced maturation protein-1 (BLIMP1) may be the most determining factor orchestrating terminal effector differentiation of antigen-specific CD8+ T cells [25] (Figure 1A). In addition, it has been shown that hyperglycolysis can induce T-cell terminal differentiation whereas imposition of an oxidative metabolic program may promote T-cell quiescence and loss of effector function [26,27]. At the opposite end of the differentiation spectrum are the so-called early memory T cells (stem cell memory T cells—Tscm, and central memory T cells—Tcm), which have high proliferation potential as well as the capacity to self-renew and persist as long-term memory cells [3]. The skewing of differentiation towards early memory or terminal effector depends on several factors, including the strength and duration of T-cell stimulation as well as multiple intrinsic and extrinsic factors that imprint gene expression programs along with epigenetic marks that strengthen T-cell fates. As described below, modulation of metabolic pathways, cytokine signaling, and epigenetic programming can be used to favor early memory T-cell skewing in lieu of terminal effector T-cell differentiation.
2.2. Exhaustion
T-cell exhaustion, driven by chronic TCR signaling, is another evolutionarily conserved process to sustained antigen stimulation aimed at reducing the risks of immunopathology or autoreactivity [12]. Exhausted T cells express multiple inhibitory receptors such as PD1, TIM3, LAG3, CTLA4, and TIGIT, which also leads to a loss of proliferative capacity and effector functions. This state is associated with a gradual increase in the expression of T-bet and a concomitant decrease in TCF1 expression, ultimately rendering exhaustion irreversible [28]. Lately, the role of the calcineurin-dependent transcription factor NFAT and other NFAT-driven transcription factors, such as interferon regulatory factor 4 (IRF4), basic leucine zipper ATF-like transcription factor (BATF), nuclear receptor subfamily 4 group A (NR4A), and thymocyte selection-associated high mobility group box protein (TOX), have been associated with the expression of these checkpoint receptors as well as with the maintenance and survival of exhausted T cells [29,30,31,32,33]. Moreover, TOX expression can further shape the exhaustion transcriptional program and epigenetic landscape of T cells [11]. As a result, these cells have a reduced effector function, mainly shown by a hyporesponsiveness to stimulation and a decrease in cytokine secretion (Figure 1B).
2.3. Senescence
While more generally associated with aging, T-cell senescence is typically characterized by cell cycle arrest, activation of the DNA damage response, increased β-galactosidase activity, and dysfunctional mitochondria. These cells nonetheless remain viable and metabolically active [34,35,36]. T-cell senescence is associated with a decrease in the expression of co-stimulatory molecules, such as CD28 and CD27, and an increase in KLRG1 and/or CD57 expression [37,38]. As the T cells divide due to repeated stimulation, they may enter a stage of replicative senescence and lose their proliferation capacity as a result of telomere erosion and a loss of telomerase activity [39,40,41]. Alternatively, a state of “premature” senescence may develop in T cells that sustain DNA damage. In both instances, the expression of cell cycle regulators is upregulated. On one side, the tumor suppressor p53 can activate the cyclin-dependent kinase inhibitor p21, which is responsible for initial cell cycle arrest by inhibiting cyclin-dependent kinase 2 (CDK2), thus preventing S-phase entry. On the other side, upregulation of the cyclin-dependent kinase inhibitor p16 can inhibit the binding of CDK4/6 to cyclin D, restraining this complex to phosphorylate the transcriptional regulator retinoblastoma (RB) protein and preventing cell cycle transition from G1 to the S phase, leading to senescence [42,43,44,45,46,47]. Whereas p16 is mainly associated with cell stress and/or DNA damage, and thus implicated in premature senescence, telomere attrition generally induces the p53–p21 axis (Figure 1C) [42,43,44,45,46]. In addition, telomere-independent, DNA damage-associated cellular senescence has also been linked to p38 mitogen-activated protein kinase (p38MAPK) signaling in activated T cells [48,49].
2.4. Activation-Induced Cell Death
The maintenance of peripheral immune tolerance by activation-induced cell death (AICD) is another key physiological process in T-cell biology [50]. Indeed, TCR stimulation can result in the initiation of apoptosis. Although ligation of TNF-α/TNF receptor 1 and TRAIL/DR4/DR5 have been shown contribute to AICD [51,52], FAS/FASL is the prototype receptor/ligand pair of the extrinsic pathway of apoptosis [53,54]. FASL expression in activated T cells is regulated by several transcription factors [50]. Ensuing TCR stimulation, NF-κB signaling induces the transcription of various target genes such as FASL [55]. Along the same lines, NFAT consensus sequences can be found on the FASL promoter to regulate its expression, which also requires the co-operation of AP-1 and secretory protein-1 (SP-1) [56]. In addition, FASL expression by T cells can be regulated by c-MYC following its dimerization with its partner molecule MAX to form an active complex for the transcriptional regulation of the FASL gene [57]. TGFβ has been shown to control FASL expression by downregulating c-MYC expression, which inhibits T-cell AICD [58]. Upon FAS engagement, a death-inducing signaling complex forms with the adaptor protein FAS-associated death domain (FADD) and procaspase-8, leading to the activation of caspase-8 and subsequent activation of effector caspases, resulting in apoptosis [59]. Activated caspase-8 can further activate a mitochondria-mediated pathway by specific cleavage of the BH3-only B-cell lymphoma-2 (BCL-2) family member BID [60,61]. This converges towards the intrinsic apoptosis pathway, involving other pro-apoptotic Bcl-2 family members, such as BIM, in mitochondria [62,63]. Conversely, T-cell survival mediated by the anti-apoptotic BCL-2 protein was shown to be largely dependent on the IL-7 homeostatic effect [64,65]. As for the other dysfunctional cell states, the nature as well as signal strength of the T-cell stimulation greatly influences cell survival [66,67] (Figure 1D).
3. Biology Meets Therapy: T-Cell Dysfunction in Adoptive Cell Therapy
This section reviews the current ACT approaches and how the notions of T-cell dysfunction integrate in both the manufacturing phase and the post-infusion period. Approaches aimed at mitigating or reversing T-cell dysfunction are also discussed.
3.1. Conventional T Cells
Adoptive transfer of ex vivo expanded T cells from the natural repertoire is a promising approach to treat a variety of cancers. However, this requires stimulatory signals to promote in vitro proliferation of T cells that impart a T-cell differentiation program that is susceptible to inducing T-cell dysfunction. Hence, understanding how T-cell dysfunction develops or is programmed by the ex vivo expansion process is imperative. Strategies to promote optimal differentiation and functionality are susceptible to promoting better function and persistence of the transferred cells.
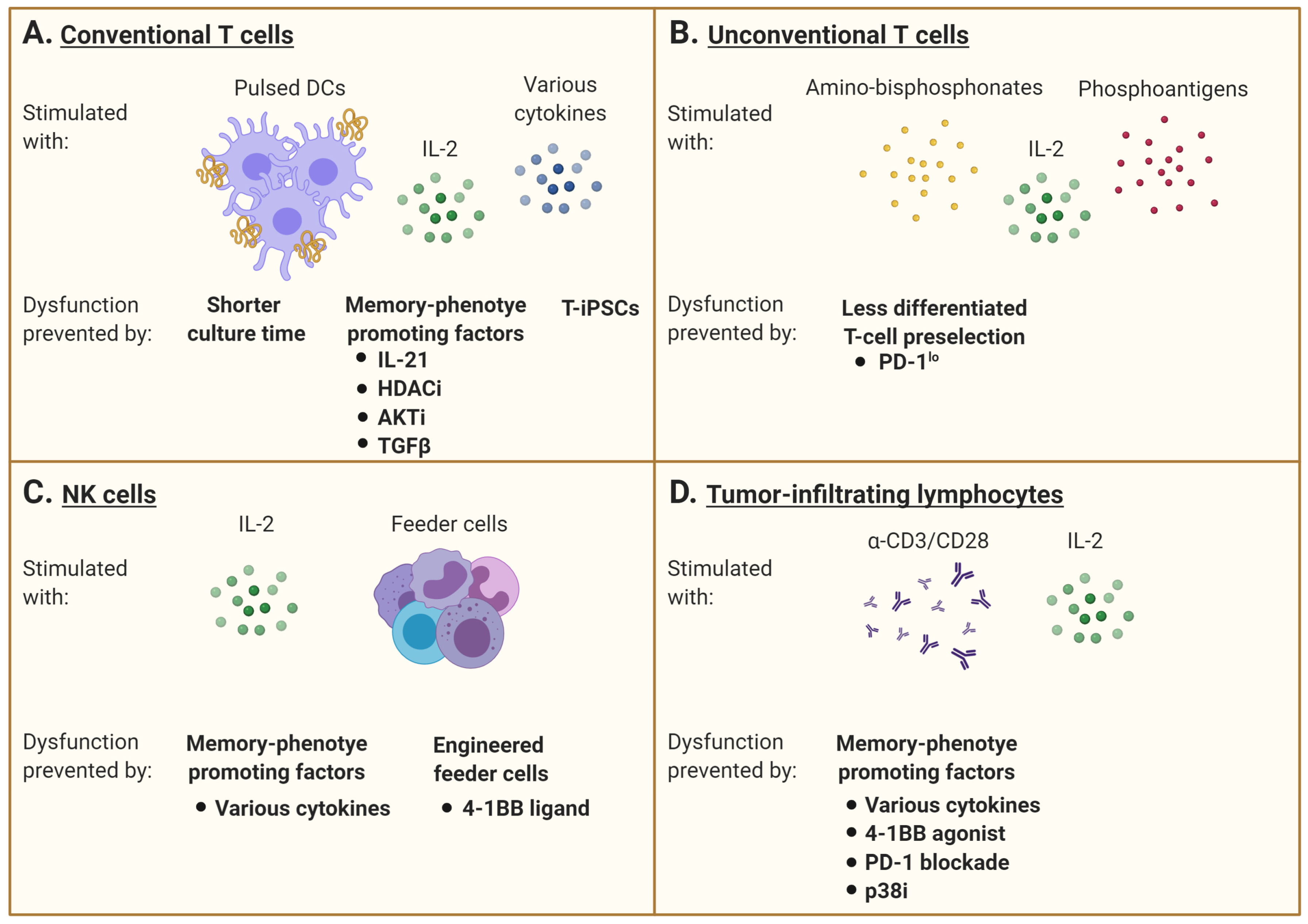
Allogenic hematopoietic cell transplantation is not only the first established form of ACT, it has also served as an ideal setting to advance the field. One of the greatest successes of ACT in the wake of allogeneic hematopoietic cell transplantation is the treatment of immunosuppression-associated opportunistic virus reactivations. Antiviral T cells expanded ex vivo have proven to be highly effective in controlling such viral reactivations in both hematopoietic and organ transplantation [68,69,70,71,72]. Over the years, manufacture duration for these virus-specific T-cell products has gone from three months to around 10 days [73]. It is also possible to directly enrich virus-specific T cells from a healthy donor apheresis product and administer the T cells without further expansion [74]. Most of the published experience with virus-specific T cells reports on the generation of cellular products manufactured from a robust memory repertoire in healthy donors. Such memory cells readily expand in the culture and show limited evidence of T-cell dysfunction at the time of infusion. In hematopoietic cell transplantation, long-term persistence of the transferred cells, thereby establishing long-lasting memory in the recipients, has been shown when the virus-specific T cells are prepared from the original stem cell donor [69]. However, the stimulation and expansion of T cells from naïve repertoire has proven to be more difficult. Although clearly feasible for virus-specific T cells [75,76], the generation and expansion of T cells targeting tumor-associated antigens (TAA) or transplantation antigens from naïve repertoires require more elaborate culture processes [1,2,77,78,79].
Alloreactive donor T cells can recognize major histocompatibility complex (MHC)-bound polymorphic peptides derived from the host proteome, known as minor histocompatibility antigens (MiHAs) [80,81,82,83]. Most of the molecularly characterized MiHAs are encoded by autosomal genes that differ between patient and donor secondary to germline-encoded non-synonymous single nucleotide polymorphisms (ns-SNP). Although potent immunogenic antigens, up to five rounds of ex vivo weekly stimulation with antigen-loaded dendritic cells and the presence of interleukin (IL)-2 are often needed to generate high numbers of specific CD8+ T cells [2]. However, despite the immunogenic nature of these antigens, protocols often generate dysfunctional cells that highly impede cell functionality and persistence after adoptive transfer [78,79]. Repeated ex vivo antigen exposure in the context of stimulatory cytokines eventually blunts T-cell growth and leads to terminal differentiation and exhaustion marker expression, especially in antigen-specific T cells in the culture [2].
It is now generally accepted that optimal therapeutic effects are achieved when the ex vivo expanded T cells maintain features associated with early memory differentiation (Tscm or Tcm) [3,84]. The clinical outcome might thus depend more on the antigen-specific T-cell early differentiation phenotype, leading to a better ability to proliferate and persist in vivo, rather than on the bulk number of infused cells [85]. As such, careful design of ex vivo culture conditions may promote the acquisition of a favorable differentiation status. Exogenous cytokines, small molecules altering cell signaling, metabolic modulation, and epigenetic modification during the ex vivo priming and expansion phase may confer early memory features [86]. We and others have demonstrated that exogenous exposure to IL-21 can limit terminal differentiation of antigen-specific T cells, which can increase their in vivo persistence and promote phenotypic and functional characteristics associated with long-lived memory T cells [77,87]. The histone deacetylase inhibitor (HDACi) combined with IL-21 can also reprogram differentiated CD8+ T cells into central memory-like T cells. This is achieved through an increase in histone H3 acetylation and chromatin accessibility at the CD28 promoter region. It is then followed by an IL-21-mediated phosphorylation of the signal transducer and activator of transcription 3 (STAT3) binding to the CD28 region, and a resulting memory-associated transcriptional signature [88]. As the epigenetic landscape of the terminal effector and exhausted T cells is being defined, it is likely that several interventions involving epigenetic modulation are introduced in T-cell manufacturing protocols [89,90]. As such, the inhibition of DNA methylation programs responsible for exhaustion-associated signaling can be a strategy to improve T-cell function [91]. A dominant feature of memory differentiation promoting interventions is the mitigation of activation signals. A brief exposure to the immunosuppressive cytokine transforming factor beta (TGFβ) promotes the Tcm differentiation of ex vivo expanded human T cells [86]. Along the same lines, AKT inhibition has been shown to increase early T-cell memory features [92]. The association of AKT inhibition and early memory T-cell differentiation also highlights how the modulation of pathways associated with cell growth and metabolism are attractive targets for programming T-cell fates. Likewise, limiting glycolysis at the expense of oxidative phosphorylation during ex vivo T-cell expansion has been suggested to improve the fitness of adoptively transferred T cells (reviewed in [26]), and limiting reactive oxygen species metabolism with N-acetylcysteine during T-cell manufacturing can favor early memory T-cell formation [93].
Nonetheless, relying solely on phenotyping may be misleading. The proportion of effector memory or central memory phenotype cells does not necessarily correlate with ex vivo loss of antigen-specific cells or a decline in their functionality [2]. Although repeated antigenic stimulation of MiHA-specific T cells may lead to terminal differentiation, prolonging the expansion phase in the absence of antigenic stimulation can decrease T-cell proliferation despite limited expression of inhibitory receptors and the preservation of polyfunctional cytokine secretion by the remaining antigen-reactive cells. Thus, the analysis of both phenotypic and functional properties of T cells prior to ACT may best inform about the potency of the T-cell product [85]. In addition, it has been shown that a fraction of terminally differentiated melanoma-specific or leukemia-specific CTL clones after ex vivo expansion appears to revert back to a central memory type in vivo after ACT, potentially conferring clinical benefits [77,94].
As an alternative to limiting the development of dysfunction, some groups have concentrated on cell reprogramming. A terminally differentiated or exhausted T cell may be induced into a pluripotent stem cell (iPSC), which enables re-differentiation into a naïve or central-memory phenotype T cell with the re-expression of CCR7, CD27, and CD28 and no exhaustion markers [95,96,97,98]. An interesting characteristic of iPCSs generated from lymphocytes is their ability to keep the rearranged TCR loci of the parental cells, which remain unchanged during in vitro differentiation [95,97]. As such, antigen-specific CTL clones can be preselected and reprogrammed into iPSC (T-iPSCs) with the Yamanaka transcription factors (Oct3/4, SOX2, KLF4, and c-MYC) [95,96,97,99,100]. These new CTLs then have longer telomeres than the original cells, with higher proliferative and functional capacities [95]. Furthermore, exhausted T cells turned into T-iPSCs are functionally able to respond to antigen-specific stimulation [101]. However, a caveat with the use of iPSC in cell therapy is the risk of only partial re-differentiation and teratoma formation post-transfer (Figure 2A).
3.2. Unconventional T Cells
γδ TCR-expressing T lymphocytes is another T-cell subtype with effector and regulatory functions and the ability to infiltrate tumors. Adoptive cell therapy with human γδ T cells expressing a Vγ2Vδ2 TCR has shown promise because of their capacity to recognize and kill most types of tumors in an MHC-unrestricted manner [102]. Vγ9Vδ2 T cells are relatively abundant in human blood and can be easily ex vivo expanded in response to amino-bisphosphonates (N-BPs) or phosphoantigens (PAgs). However, culture conditions, timing, and dosage of N-BPs or PAgs, as well as added co-stimulators such as IL-2, may result in different phenotypes and effector cell characteristics [103,104]. Results of in vitro expansion are often highly donor dependent and may also predict the respective in vivo expansion efficacy, which can be additionally restricted in cancer patients. Currently, optimal doses of N-BPs or PAgs as well as IL-2 have not yet been determined for ex vivo expansion. The efficacy of stimulation may depend on drug concentration as well as duration of exposure, which have to be individualized [105]. The role of additional systemic application of N-BPs in the context of adoptive cell transfer strategies also remains elusive. On one side, it has been reported to promote the engraftment of ex vivo-stimulated and adoptively transferred human cells in mice, but on the other side, there are indications that repetitive application of these drugs in vivo induces Vγ9Vδ2 T-cell exhaustion [103]. As an alternative, some groups tried to adoptively transfer PD-1lo Vδ2+ T cells to bypass the tumor immunosupressive environment in vivo (Figure 2B) [106].
Other T cells with “innate-like” characteristics can recognize vitamin metabolites, small phosphoantigens, and lipid antigens presented within various highly conserved and non-polymorphic MHC class I-like molecules [107,108,109,110]. One of the best characterized subsets is invariant natural killer T (iNKT) cells, which recognize lipid antigens bound within the antigen-presenting molecule CD1d [111]. These cells utilize the near-germline TCRα rearrangement Vα24-Jα18 combined with a limited TCRβ repertoire and are functionally defined by their ability to respond to galactosylceramide (α-GalCer) when presented by CD1d molecules [107,108,109,110,112,113]. They are potent cytokine secretors that bridge innate and adaptive immunity [109,111]. These cells have thus been investigated as cell transfer therapy products [114,115,116,117,118,119]. However, whether these cells develop dysfunctional features prior, during, and after therapy is still unclear. Nevertheless, since iNKT cells have been shown to limit GVHD [120], there is a growing interest to use them as a platform for cell engineering [121].
The development of ACT using other lymphoid cells is also rapidly expanding. Innate lymphoid cells (ILCs) derive from common lymphoid progenitors in the bone marrow that lack other lineage markers and genetically rearranged antigen receptors. They are defined according to their cytokine production pattern as well as unique transcription factors [122,123,124]. Group 1 ILCs (ILC1s) secrete IFNγ and express the transcription factor T-bet, whereas ILC2s produce IL-5 and IL-13 and require expression of Gata3 [125,126,127,128]. ILC3s generate IL-22 and IL-17 and are defined by the expression of RORγt, as with lymphoid tissue-inducer cells, which also express IL-7Rα [129,130,131]. Since ILCs can respond to many danger signals such as innate immune cells and secrete cytokines such as T cells despite the lack of TCR, different therapeutic approaches aim at targeting these cells in vivo to improve the efficacy of tumor immunotherapies [132]. Although the mechanisms underlying dysfunction in these cell subtypes may not be as well understood as for conventional T cells, several strategies emerge to enhance both the in vitro expansion of these cells and their therapeutic potential.
3.3. NK Cells
Natural killer (NK) cells can produce a vast array of cytokines/chemokines and are key players in immune responses against tumors and infected cells. NK cells can also directly regulate T-cell responses as well as modulate antigen-presenting cell activation. In solid tumors, NK cell secretion of CC-chemokine ligand 5 (CCL5), XC-chemokine ligand 1 (XCL1) and XCL2, and IFNγ can promote the recruitment of dendritic cells and further mediate their activation, which have been shown to improve patient outcome [133,134]. However, as with T cells, NK cells can become exhausted, and blockading TIGIT has demonstrated potential in preventing NK-cell dysfunction. Hence, a direct checkpoint blockade in NK cells may result in a more potent tumor-specific T-cell response in an NK cell-dependent manner [135,136].
Activated NK cells also upregulate many receptors, which can be shaped according to culture conditions and media supplementation with cytokines such as IL-2, IL-12, IL-15, IL-18, or IL-21 and Type I IFNs [137,138,139,140,141,142,143,144]. Hence, ex vivo modulation of NK-cell receptor expression has also been extensively investigated to overcome dysfunction. In general, PBMCs are first depleted for CD3+ cells and enriched for CD56+ cells, then cultured in medium containing IL-2 for up to two weeks [140,145]. This ex vivo stimulation induces NK-cell cytokine secretion, STAT3/AKT signaling, and upregulation of the NKG2D receptor [146]. IL-2 can also enhance NK-cell response to IL-12 by increasing the expression of its receptor [147]. IL-15 supplementation was later used to inhibit activation-induced cell death, and activate the mTOR pathway and stress-activated genes, which confer better anti-tumor capacity [148,149,150]. However, continuous IL-15 signaling has been linked to functional NK-cell exhaustion by decreased fatty acid oxidation [151]. Moreover, it was shown that IL-12-mediated IFNγ production of NK cells requires priming with IL-18, a cytokine also known to enhance IL-15-induced NK-cell proliferation [152,153]. Finally, IL-21 has been used to further increase NK-cell proliferation and effector functions, even though it can trigger apoptosis in vitro [154,155,156,157]. Other studies demonstrated that the duration of NK cell exposure to IL-21 is in fact critical [158,159]. Since NK cells can also express the inhibitory receptor PD-1, it has been found that a blockade of the PD-1/PD-L1 axis can improve NK cell-mediated immunity to tumors, and this response is indispensable for the full therapeutic effect of immunotherapy [160].
Another issue to consider is the NK-cell expansion and functional status from heavily pretreated and thus immunocompromised patients, which are much poorer than for allogeneic NK cells [161]. Other than cytokine supplementation, investigation on feeder cells required for in vitro culture is underway. In a phase I clinical trial (clinicaltrials.gov #NCT02481934), autologous NK cells were activated by an engineered K562 cell-expressed membrane-bound form of IL-15 and 4-1BB ligand [162]. NK cells can also be derived from umbilical cord blood as an allogenic cell source (clinicaltrials.gov #NCT01729091) (Figure 2C).
3.4. Tumor-Infiltrating Lymphocytes
Tumor-infiltrating lymphocytes (TIL) are composed of antigen-experienced and “passenger” T cells found at the tumor site. The tumor reactive T cells are subjected to repeated antigen encounters. When harvested for immunotherapy purposes, these cells already exhibit signs of dysfunction that may in fact identify the cancer-reactive T cells [163]. Still, they can be expanded prior to re-infusion into the patient. Although standard protocols use anti-CD3 stimulation with IL-2, generating T cells with a more advanced differentiation state, some groups focused on a cytokine combination cocktail during the expansion phase to increase cell functionality or on the selection of less differentiated TILs among the tumor [23,164,165,166].
TILs found in solid tumors indeed represent a heterogeneous population. It was recently discovered that TILs can be divided into two functionally distinct subsets [167]. The first is the most abundant and is constituted of a clonally related terminally differentiated population that expresses high levels of inhibitory receptors. The other is a TCF1+ stem-like CD8+ T-cell population, which is suggested to be a major factor in the success or failure to eradicate a tumor depending on their ability to be sufficiently stimulated by an antigen-presenting-cell niche and to continuously produce terminally differentiated CD8+ T cells within the tumor [167].
It has also been found that the presence of the integrin αEβ7 (CD103), characteristic of tissue-resident memory T cells (Trm), is positively associated with cytokine production, whereas expression of the transcription factor EOMES is negatively associated with TIL function, suggesting a competition between an antitumor CD103+ Trm-like and an exhaustion program [168]. CD69+CD103+ Trm cells usually reside in non-lymphoid tissues and function as a first line of defense against secondary infections. In addition to their unique anatomic location, Trm have distinct transcriptional profiles showing the upregulation of inhibitory receptors such as PD-1. However, human Trm have an enhanced capacity for production of certain cytokines and regulatory molecules and a decreased turnover compared to circulating effector memory T cells, suggesting long-term maintenance in situ [169]. Furthermore, it has been demonstrated that the transcription factor BHLHE40 is specifically required for both Trm and TIL development as well as their polyfunctionality by sustaining mitochondrial fitness and a functional epigenetic state. Local PD-1 signaling in the tumor microenvironment inhibits TIL BHLHE40 expression, and BHLHE40 is critical for TIL reinvigoration following anti-PD-L1 blockade [170].
Another approach to improve TIL fitness is to target the member of the tumor necrosis factor receptor superfamily T-cell co-stimulatory receptor 4-1BB (CD137). As 4-1BB is frequently present on non-exhausted CD8+ TILs, a 4-1BB agonist and a PD-1 blockade demonstrated a synergistic survival benefit in a CD8+ T-cell dependent manner. As such, combined treatment decreased TIL exhaustion and improved TIL functionality in a glioblastoma model [171]. Similarly, a PD-1 blockade and 4-1BB stimulation were demonstrated to be an effective strategy to improve pancreatic tumor-reactive TIL yield [172].
Following tumor infiltration, T cells interact with other immune, cancer, and stromal cells within the tumor microenvironment (TME). Cells comprised in this environment may develop premature senescence caused by external factors such as TME metabolic changes or drug and radiation therapy [173]. Senescent cells stay metabolically active but cease to proliferate [174,175]. Another important characteristic of senescent cells is that the expression of many genes changes during senescence and gives rise to what is called the senescence-associated secretory phenotype (SASP) [176]. Senescent cells secrete numerous biologically active factors, including cytokines, chemokines, growth factors, and proteases [177,178]. Because these secreted factors act in autocrine and paracrine manners and have pleiotropic effects on surrounding cells, they may virtually affect any cell type within the tumor microenvironment, including infiltrating T cells. Indeed, tumor-induced senescence in T cells can be reproduced in vitro by briefly incubating cells in conditions of a low tumor-to-T-cell ratio. Furthermore, senescent T lymphocytes become able to suppress the proliferation of normal T cells and promote tumor immune evasion [179]. In anti-CD3 + IL-2 stimulated T cells, the inhibition of p38MAPK signaling proved to be helpful in reversing the senescence phenotype of CD8+ T cells by increasing their proliferation and functionality [48,49] (Figure 2D).
3.5. T-Cell Receptor (TCR) Transgenic Cells
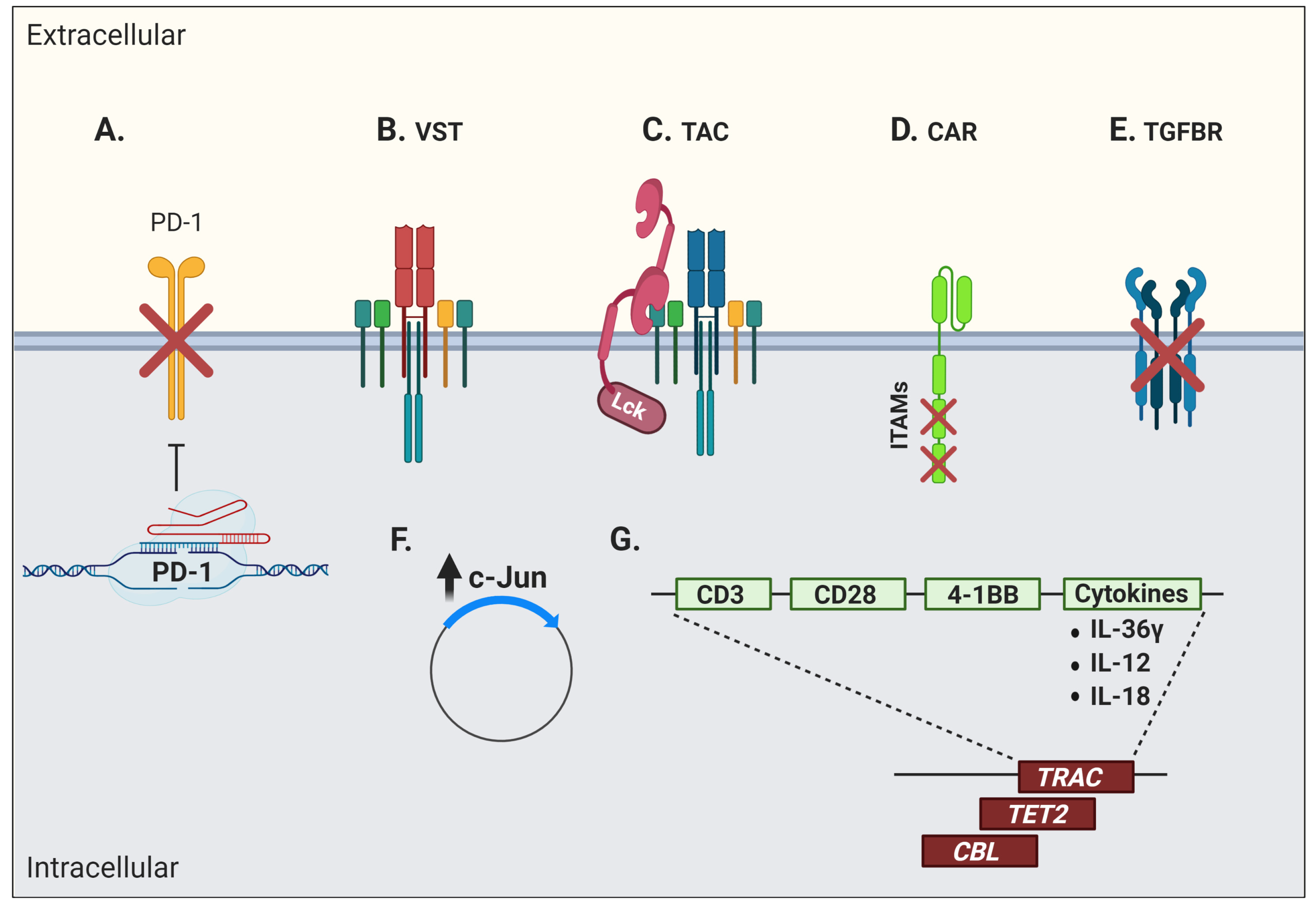
Genetic engineering offers several possibilities, such as conferring new antigenic specificities to T cells and circumventing certain limitations linked to T-cell dysfunction. These approaches provide many advantages, as they minimise culture duration and offer the possibility of directly modulating key signaling pathways within engineered cells to reduce dysfunction. Among these strategies is the introduction of an artificial TCR into antigen-specific conventional T cells. Although it can confer more potent antitumor activity [180], TCR gene transfer is fraught with the risk of inappropriate pairing between exogenous and endogenous TCR chains. Several strategies have thus been used to avoid mispairing that can result in suboptimal activity and novel off-target antigen reactivity, potentially leading to harmful immune side effects [181]. Moreover, the transduced TCR must successfully compete with the endogenous TCR chains to form a ternary complex with the CD3 signaling complex [182]. Hence, the native TCRβ gene, or simultaneous TCRα/β genes, have been knocked out using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology prior to transduction with a cancer-specific receptor of choice, resulting in a stronger and more polyfunctional response in engineered T cells when tested against target cancer cell lines [183,184]. Remarkably, the combination of CRISPR/Cas9 and TCR transgenic therapy has recently been used to knock out both the endogenous TCR chains and the negative co-signaling receptor PD-1 to redirected T cells bearing a transgenic TCR targeting a TAA from NY-ESO-1 [185] (Figure 3A).
The introduction of a transgenic TCR into virus-specific T cells (VSTs) to redirect their specificity towards cancer antigens has also been investigated [186,187,188,189,190] (Figure 3B). However, expression of a transgenic TCR often results in the downregulation of the endogenous TCR, which unfortunately leads to a reduced anti-viral reactivity [186,187,188,189,190,191]. This is in part explained by the competition for TCR signaling components by the endogenous and exogenous TCRs, and as possible consequences, one may expect lessened control of viral reactivations post-transplantation and poor capacity of TCR-transgenic VSTs to re-expand in vivo upon viral reactivation or vaccination [190,191]. The addition of CD8αβ to the transgenic TCR vector has been used to rescue endogenous MHC class I-restricted anti-viral TCR function [192,193]. These TCR-transgenic VSTs have a predominant central-memory phenotype and their anti-viral reactivity is preserved together with their anti-tumor function [194]. The insertion of the CD8αβ co-receptor also improved antigen recognition by the TCR/MHC complex, recruitment of the tyrosine kinase Lck to the immune synapse, and proper activation of signaling components for T-cell activation, in addition to allowing CD4+ T cells to recognize MHC class I-restricted antigens [195,196].
Another strategy to redirect T cells in a TCR-dependent, MHC-independent manner has been the use of a T-cell antigen coupler (TAC) composed of an antigen-binding domain, a TCR-recruitment domain, and a co-receptor domain (Figure 3C) [197]. This design recapitulates the architecture of a TCR complex and engages natural cellular pathways. In addition, these modified cells do not show signs of tonic signaling and display a less differentiated phenotype, which results in a potent T-cell product. These TAC-T cells also showed efficient tumor tissue infiltration at early time points post-ACT and great anti-tumor efficacy in a pre-clinical in vivo model of a solid tumor [197]. Along the same lines, another group fused TCR subunits to an antibody-based binding domain to reprogram T-cell specificity in an HLA-independent manner while still taking advantage of the endogenous TCR signaling. These cells showed tumor cell killing in vivo, although they were less efficient in cytokine production, suggesting some degree of dysfunctionality or impaired activation [198].
3.6. Chimeric Antigen Receptor (CAR) T Cells
Engineered T cells for the expression of an artificial receptor emerged in 1989 [199]. This first-generation chimeric antigen receptor (CAR) T cell, composed of a CD3ζ chain containing three ITAMs for TCR-like signal transduction fused with single chain fragment of variable region (scFv) antibody, could support T-cell activation and cytotoxicity, but with very limited persistence and in vivo antitumor efficacy [5,200,201]. Second-generation CARs therefore incorporated the two-signal model of T-cell activation by modifying CAR vectors to include a CD28 or 4-1BB co-stimulatory domain providing signals for T-cell effector function, proliferation, and more importantly, persistence [202,203]. Although complicated by massive activation and cytokine release syndrome, second-generation CD19-targeting CAR T cells rapidly entered routine clinical care. Third-generation CAR T cells further incorporate more than one co-stimulatory domain and other modifications or include the inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities [204]. However, lack of CAR T-cell long-term persistence, poor expansion after ACT, and tumor immune escape remain cardinal limitations of this form of therapy [205]. Hence, several issues pertaining to intrinsic biologic T-cell defaults impact the outcome of CAR-based treatments. Tonic CAR CD3ζ phosphorylation, triggered by antigen-independent clustering of scFv, has been shown to induce early CAR T-cell exhaustion. Moreover, integration of CD28 co-stimulation into the CAR vector seems to increase, whereas 4-1BB co-stimulation limits exhaustion induced by persistent CAR signaling [206]. Indeed, stimulation of CD28/CD3ζ CARs activates faster with larger-magnitude changes in protein phosphorylation, which correlates with an effector T-cell phenotype. In contrast, 4-1BB/CD3ζ CAR T cells preferentially express T-cell memory-associated genes and exhibit sustained antitumor activity against established tumors in vivo [207]. Another way to address the issue of tonic signaling is to calibrate the number of ITAMs on the CD3 moiety. Although the presence of one, two, or three functional ITAMs does not impede in vitro function, a single ITAM-containing CAR can outperform the other forms in vivo (Figure 3D). Remarkably, this modified vector also favors persistence of highly functional CAR T cells, inducing long-lived memory cells with effective anti-tumor properties [208].
Beyond concerns pertaining to T-cell engineering, the quality of the “input” material at the beginning of the manufacturing process impacts clinical outcomes. Autologous CAR T-cell efficacy greatly depends on the functional capacity of patients’ endogenous T cells. Indeed, studies have shown that T-cell fitness diminishes throughout the progression of diseases such as chronic lymphocytic leukemia (CLL), implying impaired proliferative capacity, a dysfunctional phenotype, and a decreased T-cell cytotoxicity, which impact the generation of CAR T cells [209,210,211,212,213,214]. Furthermore, molecular and functional T-cell defects are also acquired by a co-culture of previously healthy T cells with CLL cells [213,215,216]. Among these defects, impairment of mitochondrial biogenesis and fitness, accompanied by reduced glucose transporter 1 (GLUT1) reserves, has been identified, which negatively correlates with the persistence of the transferred CAR T cells and clinical outcome [217]. It was further shown that the frequency of memory T cells, defined by a CD8+CD45RO-CD27+ population, in the pre-manufacturing leukapheresis product was significantly associated with clinical response [218,219]. Similarly, cell features and the magnitude of ex vivo expansion when harvested after a response to induction therapy to manufacture B-cell maturation antigen (BCMA)-specific CAR T cells would be expected to be more clinically effective compared to a leukapheresis product from relapsed/refractory multiple myeloma [220]. Finally, despite the generation of a lesser number of cells, the beneficial effects of reduced culture duration manifests in improved in vitro proliferation and effector function, which directly correlates with improved engraftment and anti-tumor function in vivo, even at a six-fold lower dose [221].
The blockade of immunosuppressive pathways has also been investigated to confer superior functionality in CAR T cells. As such, it was demonstrated that exposure to TGFβ impairs proliferation as well as cytokine production of receptor tyrosine kinase-like orphan receptor 1 (ROR1)-specific CAR T cells co-cultured with ROR1-expressing triple-negative breast cancer cells. Thus, the blocking of TGFβ receptor signaling with a specific kinase inhibitor could promote better anti-tumor function in vitro [222]. TGF-β is abundant in several cancer microenvironments (secreted by the tumor cells themselves, stromal cells, or infiltrating immunosuppressive cells such as myeloid derived suppressor cells—MDSC) and is a prime target for enhancing ACT. However, this is to be undertaken with caution, as TGFβ can also be beneficial for T-cell memory differentiation in certain contexts [223]. After adoptive transfer, the overexpression of a dominant negative TGFβ receptor or hybrid receptors converting an inhibitory signal (such as PD-1) into a stimulatory signal are additional strategies to enhance ACT efficacy (Figure 3E) [224,225]. Along the same lines, genetic ablation of negative co-signaling molecules such as PD-1 in CAR T cells, or CAR T cells secreting anti-PD-1 antibodies, is currently being investigated in clinical trials (clinicaltrials.gov #NCT04213469, #NCT04489862) (Figure 3A).
Studies on CAR metabolic pathway activation revealed that AP-1, a transcription factor composed of dimers of c-Jun and c-Fos, was highly solicited. Its activity is regulated by extracellular signals that can repress or activate its transcription. The formation of the AP-1 complex downstream of the TCR signaling induces IL-2 transcription, among other factors [226,227]. CAR T-cell exhaustion has also been associated with a profound defect in the production of IL-2, along with increased chromatin accessibility of AP-1 transcription factor motifs and overexpression of the basic leucine zipper (bZIP) and IRF transcription factors [33,228,229]. Thus, overexpression of c-Jun rendered CAR T cells resistant to exhaustion, enhanced their expansion potential, increased their functional capacity, and diminished their terminal differentiation [230] (Figure 3F).
Fourth-generation CARs, also called “T cells redirected for antigen-unrestricted cytokine-initiated killing” or TRUCKs, are armored to improve cell fitness by inserting genes coding for other molecules such as cytokines, into the CAR vector [231] (Figure 3G). Interleukin-36γ (IL-36γ), for instance, showed significantly improved CAR T-cell expansion and persistence, and resulted in superior tumor eradication compared to conventional CAR T cells. The enhanced cellular function by IL-36γ was mediated in an autocrine manner. Furthermore, the activation of endogenous antigen-presenting and T cells by IL-36γ promoted a secondary anti-tumor response, which delayed the progression of an antigen-negative tumor challenge [232]. IL-18-armored CAR T cells have also demonstrated enhanced proliferation and persistence in pre-clinical models, in addition to inducing a broadened anti-tumor response through endogenous immune effectors [233,234,235]. Similarly, a CAR T cell secreting the pro-inflammatory cytokine IL-12 demonstrated an improved cytotoxicity and the ability to overcome an immune inhibitory microenvironment in several models, but a clinical trial using IL-12-secreting TILs revealed the high risk of toxicity of this approach if IL-12 secretion is not limited in time and space [236,237]. An attractive strategy to limit cytokine secretion into activated T cells is to use a NFAT-inducible system [238].
As seen with conventional T cells, the use of endogenous TCR signaling has been investigated to improve the expansion and function of CAR T cells. As such, virus-specific T cells were modified with a CD19-specific CAR vector and infused into patients without prior cytoreductive chemotherapy. This approach is attractive for two reasons: It leverages the qualities of long-lived memory cells and can use viral reactivation as an adjuvant. In patients with viral reactivation, a striking proliferation of CAR T cells was observed with an associated depletion of CD19-expressing B cells, suggesting that dual TCR and CAR stimulation can potentiate engineered cell expansion [239].
Since CAR constructs are usually generated with the use of viral vectors, integration may result in adverse effects, such as oncogenic transformation or uncontrolled growth, transgene expression, or transcriptional silencing [240]. In a case report of CD19-specific CAR T-cell infusion for CLL treatment, an impressive clinical response was associated with the persistence of one major T-cell clone. It was further shown that the random integration of the CAR vector had disrupted the methylcytosine dioxygenase TET2 gene, which led to an epigenetic profile consistent with altered T-cell differentiation and a central memory phenotype [241]. In a case of anti-CD22 CAR T-cell therapy to treat B-cell acute lymphoblastic leukemia (ALL), a dominant T-cell clone containing a copy of the vector integrated into the second intron of the E3 ubiquitin-protein ligase CBL gene was discovered. A loss of CBL has been associated with a reduction in the threshold for T-cell activation and dependence on co-stimulation [242]. As such, integration of the CAR vector in this region resulted in a dominant-negative effect with the normal CBL (and/or its homolog CBL-B) function, thus contributing to the hyperexpansion in response to a small amount of antigen [243]. Hence, attempts to direct the CAR vector into specific DNA regions are obviously of interest. Indeed, the targeted integration of a CD19-specific CAR vector into the T-cell receptor α constant (TRAC) locus resulted in a more physiologic CAR expression, therefore limiting tonic CAR signaling and delaying effector T-cell differentiation and exhaustion [244] (Figure 3G).
Given that iPSCs can be easily amenable to genetic transformations in vitro, T-iPSCs can be genetically modified to augment their applicability, potency, and persistence and offer a great advantage compared to primary cells [245]. Thus, antigen specificity could be assigned to T-iPSCs by means of a chimeric receptor [246]. However, optimization is still needed, as these first iPSC-derived CAR-expressing T cells were still prone to more a terminally differentiated phenotype [246].
3.7. Other Engineered Cell Types
Other cell types have also gained interest for CAR engineering. For instance, CAR-modified NK cells show a better safety profile than CAR T cells [247]. Furthermore, their shorter half-life, the smaller array of secreted cytokines limiting the possibility of cytokine release syndrome, their CAR-independent natural killing activity, and their greater potential to be made as an “off-the-shelf” cellular product are among the most attractive features of CAR-NK cells [248,249,250,251]. Nonetheless, late differentiation and exhaustion remain an issue in CAR-NK cell ex vivo expansion protocols. As for CAR T cells, memory-like CAR-NK cells could be promoted when membrane-bound IL-21 was added with feeder cells [252]. Attempts have also been made to increase the lifespan of CAR-NK cells in vivo through an IL-15-armored CAR. A phase I/II study (clinicaltrials.gov #NCT03056339) investigating NK cells engineered with a vector containing an anti-CD19 CAR, IL-15, and an inducible suicide gene revealed a high response rate with an excellent toxicity profile [253]. Lastly, the use of cord blood NK cells as the primary cell source could generate greater numbers of CAR-NK cells, thus potentially reducing overall culture time per patient.
Recently, CAR-macrophages have also entered the field. As professional antigen-presenting cells, these modified cells can be reprogramed to direct their phagocytic activity against cancer cells. CAR-macrophages also show beneficial bystander effects by being able to activate dendritic cells and recruit CD8+ T cells to the tumor site [254].
4. Summary and Conclusions
Adoptive cell therapy is a promising approach that can be adapted to target virtually any cancer type or infection in a very dynamic and durable manner. Indeed, T cells as living drugs can provide long-term protection against relapse and can cure diseases that are otherwise incurable. In order to fulfill the promise of ACT, the limitations imposed by the various forms of T-cell dysfunction need to be overcome. These interventions depend on our evolving knowledge of the physiological processes governing immune cell differentiation. Important aspects, such as the plasticity and overlap between dysfunctional states, as well as how these cellular processes progress, remain elusive. The effects of manipulations aimed at these mechanisms may also induce compensatory pathways, making these interventions unpredictable. Hence, research in the physiological mechanisms leading to cell dysfunction must be integrated with translational research in T-cell manufacturing as well as carefully designed clinical trials to reveal the most impactful biological processes to target in order to improve T-cell therapies.
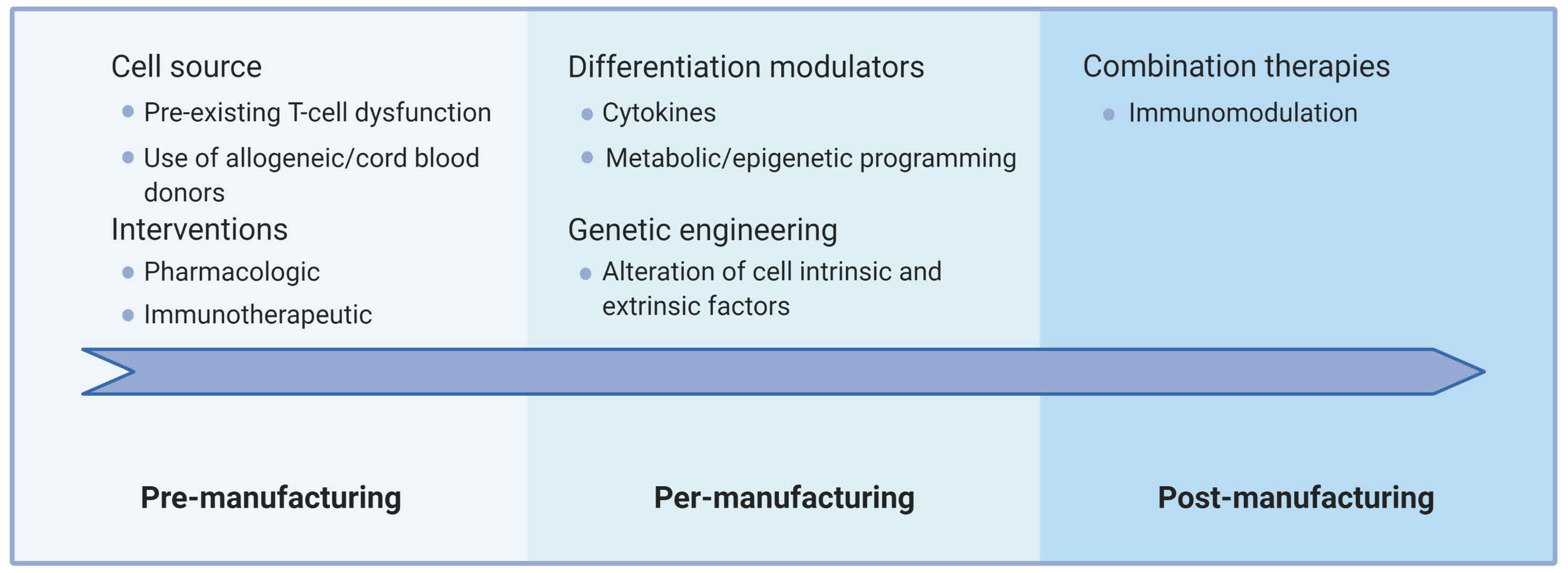
Generating such improved immunotherapies requires innovations along two main axes. First, interventions must be directed at the immune cells themselves (intrinsic factors) and/or the multiple cell-extrinsic factors that impact T-cell physiology. Second, improvements may include interventions aimed at the various steps of the ACT procedure, namely, pre-manufacturing (source material), per-manufacturing (cell expansion/differentiation/modification), and post-manufacturing (after adoptive transfer). Figure 4 conceptually summarizes how this can be articulated currently in ACT settings.
The source material is crucial since dysfunctional cells are already less likely to adequately support strong ex vivo proliferation signals. As such, harvesting less differentiated cells, or using healthy donor cells, facilitates the manufacturing of fit T cells. Whether patient-directed interventions could improve the status of the harvested T cells beyond collecting patients early in their treatment history is an active area of research. For example, Ibrutinib therapy prior to T-cell collection can improve CAR T-cell production in CLL patients [255], highlighting that ACT includes many steps before T-cell injection. Alternative approaches include using healthy allogeneic donors with the caveats imposed by histo-incompatibility (reviewed in [256,257]) and rejuvenation through iPSC generation. The manufacturing phase offers several options that involve the modulation of culture conditions and/or modification of the cells themselves. Several of these have already entered the clinical stage, such as T-cell memory promoting cytokines and genetic ablation of immune checkpoints. It is to be expected that further advances, such as epigenetic programming, will rapidly enter manufacturing protocols. Beyond impacting intrinsic T-cell processes, genetic engineering offers many more opportunities. Adoptively transferred T cells can not only be engineered to resist the immunosuppressive cancer microenvironment (with dominant-negative cytokine receptors, for instance), but can also be modified to enhance trafficking to tumors and alter this microenvironment (through cytokine secretion or secretion of a decoy receptor, among others) [258]. Such “armored” T cells have the potential to reprogram the microenvironment and have an effect reaching beyond the optimization of T-cell function. Similar effects may be achieved through the rationale design of combination therapies that include standard cancer treatments such as radiotherapy, immunological cell death-causing chemotherapy, oncolytic viruses, and other immunotherapeutic approaches [259,260,261,262]. The combination of ACT with a PD-1 blocking antibody is already undergoing clinical evaluation in large trials. However, interference with the homeostatic roles of T-cell dysfunction mechanisms may also be fraught with the danger of triggering unwanted reactivity or even promote further exhaustion or terminal differentiation [263]. Thus, combination therapies or inducible systems to interfere with dysfunctional features more precisely in space and time might be required to fully harness the potential of ACT. It is to be expected that combination therapies involving ACT and advanced engineering cellular protocols will be a major focus of basic and clinical investigation in the next decades.
Author Contributions
Both V.J. and J.-S.D. performed the literature review and wrote, reviewed, and approved the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Richard and Edith Strauss Foundation and the Leukemia/Lymphoma Society of Canada (#622735) operating grants held by J.-S.D.
Acknowledgments
We are grateful to the Delisle laboratory members for helpful discussion and we apologize to authors we could not cite. J.-S.D. and V.J. hold an FRQS senior clinician researcher career award and a FRQS postdoctoral fellowship, respectively. J.-S.D. is a Cole Foundation Early Career Transition Award laureate as well as a member of the ThéCell network and of the Canadian Donation and Transplant Research Program.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Janelle, V.; Rulleau, C.; Del Testa, S.; Carli, C.; Delisle, J.-S. T-Cell Immunotherapies Targeting Histocompatibility and Tumor Antigens in Hematological Malignancies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Janelle, V.; Carli, C.; Taillefer, J.; Orio, J.; Delisle, J.-S. Defining novel parameters for the optimal priming and expansion of minor histocompatibility antigen-specific T cells in culture. J. Transl. Med. 2015, 13, 123. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef]
- Samelson, L.E.; Patel, M.D.; Weissman, A.M.; Harford, J.B.; Klausner, R.D. Antigen activation of murine T cells induces tyrosine phosphorylation of a polypeptide associated with the T cell antigen receptor. Cell 1986, 46, 1083–1090. [Google Scholar] [CrossRef]
- Bu, J.Y.; Shaw, A.S.; Chan, A.C. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc. Natl. Acad. Sci. USA 1995, 92, 5106–5110. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hogg, G.D.; DeNardo, D.G. Rethinking immune checkpoint blockade: ‘Beyond the T cell’. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef]
- Dahlén, E.; Veitonmäki, N.; Norlén, P. Bispecific antibodies in cancer immunotherapy. Ther. Adv. Vaccines Immunother. 2018, 6, 3–17. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2015, 37, 457–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Henson, S.M.; Lanna, A. Senescence of T Lymphocytes: Implications for Enhancing Human Immunity. Trends Immunol. 2016, 37, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Petit, P.F.; Van den Eynde, B.J. Apoptosis of tumor-infiltrating T lymphocytes: A new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019, 68, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.W.; Rutishauser, R.L.; Joshi, N.S.; Haberman, A.M.; Kaech, S.M. Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J. Immunol. 2010, 185, 5315–5325. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef]
- Henson, S.M.; Franzese, O.; Macaulay, R.; Libri, V.; Azevedo, R.I.; Kiani-Alikhan, S.; Plunkett, F.J.; Masters, J.E.; Jackson, S.; Griffiths, S.J.; et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 2009, 113, 6619–6628. [Google Scholar] [CrossRef]
- Martin, M.D.; Badovinac, V.P. Defining Memory CD8 T Cell. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Sarkar, S.; Kalia, V.; Haining, W.N.; Konieczny, B.T.; Subramaniam, S.; Ahmed, R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med. 2008, 205, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.C.; Matthews, S.; Yap, G.S. IL-12 Signaling Drives CD8+ T Cell IFN-γ Production and Differentiation of KLRG1+ Effector Subpopulations during Toxoplasma gondii Infection. J. Immunol. 2008, 180, 5935–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, D.T.; Manders, P.; Wagner, S.D. Arrested Differentiation, the Self-Renewing Memory Lymphocyte, and Vaccination. Science 2001, 293, 248. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzavecchia, A.; Sallusto, F. Progressive differentiation and selection of the fittest in the immune response. Nat. Rev. Immunol. 2002, 2, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Rutishauser, R.L.; Martins, G.A.; Kalachikov, S.; Chandele, A.; Parish, I.A.; Meffre, E.; Jacob, J.; Calame, K.; Kaech, S.M. Transcriptional Repressor Blimp-1 Promotes CD8+ T Cell Terminal Differentiation and Represses the Acquisition of Central Memory T Cell Properties. Immunity 2009, 31, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Kishton, R.J.; Restifo, N.P. Metabolic reprograming of anti-tumor immunity. Curr. Opin. Immunol. 2017, 46, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825–841.e828. [Google Scholar] [CrossRef]
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 2015, 42, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141.e1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennifer, P.C.; Rita, B.E. T Cell Replicative Senescence in Human Aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1--more than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Larbi, A. Markers of T Cell Senescence in Humans. Int. J. Mol. Sci. 2017, 18, 1742. [Google Scholar] [CrossRef] [Green Version]
- Effros, R.B.; Pawelec, G. Replicative senescence of T cells: Does the Hayflick Limit lead to immune exhaustion? Immunol. Today 1997, 18, 450–454. [Google Scholar] [CrossRef]
- Effros, R.B. Replicative senescence in the immune system: Impact of the Hayflick limit on T-cell function in the elderly. Am. J. Hum. Genet. 1998, 62, 1003–1007. [Google Scholar] [CrossRef] [Green Version]
- Hodes, R.J.; Hathcock, K.S.; Weng, N.P. Telomeres in T and B cells. Nat. Rev. Immunol. 2002, 2, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Weinberg, R.A. When cells get stressed: An integrative view of cellular senescence. J. Clin. Investig. 2004, 113, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Shawi, M.; Autexier, C. Telomerase, senescence and ageing. Mech. Ageing Dev. 2008, 129, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Chen, X. Tumor suppression by p53: Making cells senescent. Histol. Histopathol. 2010, 25, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8⁺ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef]
- Gurusamy, D.; Henning, A.N.; Yamamoto, T.N.; Yu, Z.; Zacharakis, N.; Krishna, S.; Kishton, R.J.; Vodnala, S.K.; Eidizadeh, A.; Jia, L.; et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 2020, 37, 818–833.e819. [Google Scholar] [CrossRef]
- Arakaki, R.; Yamada, A.; Kudo, Y.; Hayashi, Y.; Ishimaru, N. Mechanism of activation-induced cell death of T cells and regulation of FasL expression. Crit. Rev. Immunol. 2014, 34, 301–314. [Google Scholar] [CrossRef]
- Zheng, L.; Fisher, G.; Miller, R.E.; Peschon, J.; Lynch, D.H.; Lenardo, M.J. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995, 377, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.I.; Devadas, S.; Zhang, X.; Zhang, L.; Keegan, A.; Greeneltch, K.; Solomon, J.; Wei, L.; Das, J.; Sun, E.; et al. The role of activation-induced cell death in the differentiation of T-helper-cell subsets. Immunol. Res. 2003, 28, 285–293. [Google Scholar] [CrossRef]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.-I.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Suda, T.; Takahashi, T.; Golstein, P.; Nagata, S. Molecular cloning and expression of the fas ligand, a novel member of the tumor necrosis factor family. Cell 1993, 75, 1169–1178. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, J.; Mittelstadt, P.R.; Mages, H.W.; Gerth, A.J.; Kroczek, R.A.; Ashwell, J.D.; Glimcher, L.H. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity 2000, 12, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Brunner, T.; Kasibhatla, S.; Pinkoski, M.J.; Frutschi, C.; Yoo, N.J.; Echeverri, F.; Mahboubi, A.; Green, D.R. Expression of Fas ligand in activated T cells is regulated by c-Myc. J. Biol. Chem. 2000, 275, 9767–9772. [Google Scholar] [CrossRef] [Green Version]
- Genestier, L.; Kasibhatla, S.; Brunner, T.; Green, D.R. Transforming growth factor beta1 inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J. Exp. Med. 1999, 189, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Thome, M.; Tschopp, J. TCR-induced NF-kappaB activation: A crucial role for Carma1, Bcl10 and MALT1. Trends Immunol. 2003, 24, 419–424. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, M.; Schwarz, M.; Zaltsman, Y.; Eini, T.; Niv, H.; Pietrokovski, S.; Gross, A. Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol. Cell. Biol. 2005, 25, 4579–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akashi, K.; Kondo, M.; von Freeden-Jeffry, U.; Murray, R.; Weissman, I.L. Bcl-2 Rescues T Lymphopoiesis in Interleukin-7 Receptor–Deficient Mice. Cell 1997, 89, 1033–1041. [Google Scholar] [CrossRef] [Green Version]
- Maraskovsky, E.; O’Reilly, L.A.; Teepe, M.; Corcoran, L.M.; Peschon, J.J.; Strasser, A. Bcl-2 Can Rescue T Lymphocyte Development in Interleukin-7 Receptor–Deficient Mice but Not in Mutant rag-1−/− Mice. Cell 1997, 89, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Bouillet, P.; O’Reilly, L.A. CD95, BIM and T cell homeostasis. Nat. Rev. Immunol. 2009, 9, 514–519. [Google Scholar] [CrossRef]
- Hughes, P.D.; Belz, G.T.; Fortner, K.A.; Budd, R.C.; Strasser, A.; Bouillet, P. Apoptosis Regulators Fas and Bim Cooperate in Shutdown of Chronic Immune Responses and Prevention of Autoimmunity. Immunity 2008, 28, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Craddock, J.; Heslop, H.E. Adoptive cellular therapy with T cells specific for EBV-derived tumor antigens. Update Cancer Ther. 2008, 3, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Houghtelin, A.; Bollard, C.M. Virus-Specific T Cells for the Immunocompromised Patient. Front. Immunol. 2017, 8, 1272. [Google Scholar] [CrossRef] [Green Version]
- Heslop, H.E.; Slobod, K.S.; Pule, M.A.; Hale, G.A.; Rousseau, A.; Smith, C.A.; Bollard, C.M.; Liu, H.; Wu, M.F.; Rochester, R.J.; et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010, 115, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, U.; Keirnan, J.M.; Katari, U.L.; Yanagisawa, R.; Christin, A.S.; Huye, L.E.; Perna, S.K.; Ennamuri, S.; Gottschalk, S.; Brenner, M.K.; et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol. Ther. 2012, 20, 1622–1632. [Google Scholar] [CrossRef] [Green Version]
- Icheva, V.; Kayser, S.; Wolff, D.; Tuve, S.; Kyzirakos, C.; Bethge, W.; Greil, J.; Albert, M.H.; Schwinger, W.; Nathrath, M.; et al. Adoptive transfer of epstein-barr virus (EBV) nuclear antigen 1-specific t cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J. Clin. Oncol. 2013, 31, 39–48. [Google Scholar] [CrossRef]
- Hanley, P.J.; Melenhorst, J.J.; Nikiforow, S.; Scheinberg, P.; Blaney, J.W.; Demmler-Harrison, G.; Cruz, C.R.; Lam, S.; Krance, R.A.; Leung, K.S.; et al. CMV-specific T cells generated from naïve T cells recognize atypical epitopes and may be protective in vivo. Sci. Transl. Med. 2015, 7, 285ra263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, G.; Carli, C.; Lamarche, C.; Rulleau, C.; Bonnaure, G.; Néron, S.; Delisle, J.-S. Leukoreduction system chambers are a reliable cellular source for the manufacturing of T-cell therapeutics. Transfusion 2019, 59, 1300–1311. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 2013, 5, 174ra127. [Google Scholar] [CrossRef] [Green Version]
- Warren, E.H.; Fujii, N.; Akatsuka, Y.; Chaney, C.N.; Mito, J.K.; Loeb, K.R.; Gooley, T.A.; Brown, M.L.; Koo, K.K.W.; Rosinski, K.V.; et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood 2010, 115, 3869–3878. [Google Scholar] [CrossRef] [Green Version]
- Meij, P.; Jedema, I.; van der Hoorn, M.A.; Bongaerts, R.; Cox, L.; Wafelman, A.R.; Marijt, E.W.A.; Willemze, R.; Falkenburg, J.H.F. Generation and administration of HA-1-specific T-cell lines for the treatment of patients with relapsed leukemia after allogeneic stem cell transplantation: A pilot study. Haematologica 2012, 97, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Granados, D.P.; Rodenbrock, A.; Laverdure, J.P.; Côté, C.; Caron-Lizotte, O.; Carli, C.; Pearson, H.; Janelle, V.; Durette, C.; Bonneil, E.; et al. Proteogenomic-based discovery of minor histocompatibility antigens with suitable features for immunotherapy of hematologic cancers. Leukemia 2016, 30, 1344. [Google Scholar] [CrossRef]
- Roopenian, D.; Choi, E.Y.; Brown, A. The immunogenomics of minor histocompatibility antigens. Immunol. Rev. 2002, 190, 86–94. [Google Scholar] [CrossRef]
- Mullally, A.; Ritz, J. Beyond HLA: The significance of genomic variation for allogeneic hematopoietic stem cell transplantation. Blood 2007, 109, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilberberg, J.; Feinman, R.; Korngold, R. Strategies for the identification of T cell-recognized tumor antigens in hematological malignancies for improved graft-versus-tumor responses after allogeneic blood and marrow transplantation. Biol. Blood Marrow Transplant. 2015, 21, 1000–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Riddell, S.R.; Schumacher, T.N. Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med. 2015, 7, 280–287. [Google Scholar] [CrossRef]
- Dahmani, A.; Janelle, V.; Carli, C.; Richaud, M.; Lamarche, C.; Khalili, M.; Goupil, M.; Bezverbnaya, K.; Bramson, J.L.; Delisle, J.S. TGFβ Programs Central Memory Differentiation in Ex Vivo-Stimulated Human T Cells. Cancer Immunol Res. 2019, 7, 1426–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orio, J.; Carli, C.; Janelle, V.; Giroux, M.; Taillefer, J.; Goupil, M.; Richaud, M.; Roy, D.-C.; Delisle, J.-S. Early exposure to interleukin-21 limits rapidly generated anti–Epstein-Barr virus T-cell line differentiation. Cytotherapy 2015, 17, 496–508. [Google Scholar] [CrossRef]
- Wang, J.; Hasan, F.; Frey, A.C.; Li, H.S.; Park, J.; Pan, K.; Haymaker, C.; Bernatchez, C.; Lee, D.A.; Watowich, S.S.; et al. Histone Deacetylase Inhibitors and IL21 Cooperate to Reprogram Human Effector CD8+; T Cells to Memory T Cells. Cancer Immunol Res. 2020, 8, 794. [Google Scholar] [CrossRef] [Green Version]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front. Immunol. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Waart, A.B.; van de Weem, N.M.P.; Maas, F.; Kramer, C.S.M.; Kester, M.G.D.; Falkenburg, J.H.F.; Schaap, N.; Jansen, J.H.; van der Voort, R.; Gattinoni, L.; et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 2014, 124, 3490–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapuis, A.G.; Thompson, J.A.; Margolin, K.A.; Rodmyre, R.; Lai, I.P.; Dowdy, K.; Farrar, E.A.; Bhatia, S.; Sabath, D.E.; Cao, J.; et al. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zúñiga-Pflücker, J.C.; Keller, G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef] [Green Version]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Nishimura, T.; Yamazaki, S.; Yamaguchi, T.; Kawana-Tachikawa, A.; Hayama, T.; Nakauchi, Y.; Ando, J.; Ota, Y.; Takahashi, S.; et al. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Rep. 2015, 5, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, F.; Velghe, I.; Vanwalleghem, L.; De Smedt, M.; Van Coppernolle, S.; Taghon, T.; Moore, H.D.; Leclercq, G.; Langerak, A.W.; Kerre, T.; et al. Generation of T Cells from Human Embryonic Stem Cell-Derived Hematopoietic Zones. J. Immunol. 2009, 182, 6879. [Google Scholar] [CrossRef]
- Chan, J.; Kim, P.Y.; Kranz, E.; Nagaoka, Y.; Lee, Y.; Wen, J.; Elsaesser, H.J.; Qin, M.; Brooks, D.G.; Ringpis, G.E.; et al. Purging Exhausted Virus-Specific CD8 T Cell Phenotypes by Somatic Cell Reprogramming. AIDS Res. Hum. Retrovir. 2017, 33, S59–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Murata-Hirai, K.; Iwasaki, M.; Matsumoto, K.; Hayashi, K.; Kumagai, A.; Nada, M.H.; Wang, H.; Kobayashi, H.; Kamitakahara, H.; et al. Expansion of human γδ T cells for adoptive immunotherapy using a bisphosphonate prodrug. Cancer Sci. 2018, 109, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeres, T.; Smetak, M.; Pretscher, D.; Wilhelm, M. Improving the Efficiency of Vγ9Vδ2 T-Cell Immunotherapy in Cancer. Front. Immunol. 2018, 9, 800. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Lal, G. Regulatory and effector functions of gamma–delta (γδ) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int. J. Cancer 2016, 139, 976–985. [Google Scholar] [CrossRef]
- Nada, M.H.; Wang, H.; Workalemahu, G.; Tanaka, Y.; Morita, C.T. Enhancing adoptive cancer immunotherapy with Vγ2Vδ2 T cells through pulse zoledronate stimulation. J. Immunother. Cancer 2017, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Romero-Masters, J.C.; Hudson, A.W.; Gendron-Fitzpatrick, A.; Kenney, S.C.; Gumperz, J.E. Adoptively transferred Vγ9Vδ2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, M. When less is more: T lymphocyte populations with restricted antigen receptor diversity. J. Immunol. 2014, 193, 975–976. [Google Scholar] [CrossRef] [Green Version]
- Van Rhijn, I.; Moody, D.B. Donor Unrestricted T Cells: A Shared Human T Cell Response. J. Immunol. 2015, 195, 1927–1932. [Google Scholar] [CrossRef] [Green Version]
- Salio, M.; Silk, J.D.; Jones, E.Y.; Cerundolo, V. Biology of CD1- and MR1-restricted T cells. Annu. Rev. Immunol. 2014, 32, 323–366. [Google Scholar] [CrossRef]
- Constantinides, M.G.; Bendelac, A. Transcriptional regulation of the NKT cell lineage. Curr. Opin. Immunol. 2013, 25, 161–167. [Google Scholar] [CrossRef]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Brossay, L.; Chioda, M.; Burdin, N.; Koezuka, Y.; Casorati, G.; Dellabona, P.; Kronenberg, M. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J. Exp. Med. 1998, 188, 1521–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exley, M.A.; Lynch, L.; Varghese, B.; Nowak, M.; Alatrakchi, N.; Balk, S.P. Developing understanding of the roles of CD1d-restricted T cell subsets in cancer: Reversing tumor-induced defects. Clin. Immunol. 2011, 140, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohashi, S.; Ishikawa, A.; Ishikawa, E.; Otsuji, M.; Iizasa, T.; Hanaoka, H.; Shimizu, N.; Horiguchi, S.; Okamoto, Y.; Fujii, S.; et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer. Res. 2006, 12, 6079–6086. [Google Scholar] [CrossRef] [Green Version]
- Kunii, N.; Horiguchi, S.; Motohashi, S.; Yamamoto, H.; Ueno, N.; Yamamoto, S.; Sakurai, D.; Taniguchi, M.; Nakayama, T.; Okamoto, Y. Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. 2009, 100, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Horiguchi, S.; Kurosaki, M.; Kunii, N.; Nagato, K.; Hanaoka, H.; Shimizu, N.; Ueno, N.; Yamamoto, S.; Taniguchi, M.; et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 2011, 138, 255–265. [Google Scholar] [CrossRef]
- Exley, M.A.; Friedlander, P.; Alatrakchi, N.; Vriend, L.; Yue, S.; Sasada, T.; Zeng, W.; Mizukami, Y.; Clark, J.; Nemer, D.; et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin. Cancer Res. 2017, 23, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Exley, M.A.; Hou, R.; Shaulov, A.; Tonti, E.; Dellabona, P.; Casorati, G.; Akbari, O.; Akman, H.O.; Greenfield, E.A.; Gumperz, J.E.; et al. Selective activation, expansion, and monitoring of human iNKT cells with a monoclonal antibody specific for the TCR alpha-chain CDR3 loop. Eur. J. Immunol. 2008, 38, 1756–1766. [Google Scholar] [CrossRef]
- Exley, M.A.; Wilson, S.B.; Balk, S.P. Isolation and Functional Use of Human NKT Cells. Curr. Protoc. Immunol. 2017, 119, 14.11.11–14.11.20. [Google Scholar] [CrossRef]
- Pillai, A.B.; George, T.I.; Dutt, S.; Teo, P.; Strober, S. Host NKT Cells Can Prevent Graft-versus-Host Disease and Permit Graft Antitumor Activity after Bone Marrow Transplantation. J. Immunol. 2007, 178, 6242. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B.J.; Choi, J.E.; Exley, M.A. Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front. Immunol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.; Vosshenrich, C.A.; Di Santo, J.P. Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 2015, 15, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [Green Version]
- Ibiza, S.; García-Cassani, B.; Ribeiro, H.; Carvalho, T.; Almeida, L.; Marques, R.; Misic, A.M.; Bartow-McKenney, C.; Larson, D.M.; Pavan, W.J.; et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 2016, 535, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Withers, D.R. Lymphoid tissue inducer cells. Curr. Biol. 2011, 21, R381–R382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis E Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallmer, K.; Oxenius, A. Recognition and Regulation of T Cells by NK Cells. Front. Immunol. 2016, 7, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Lim, O.; Lee, Y.; Chung, H.; Her, J.H.; Kang, S.M.; Jung, M.-y.; Min, B.; Shin, H.; Kim, T.M.; Heo, D.S.; et al. GMP-compliant, large-scale expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells in vitro and in vivo. PLoS ONE 2013, 8, e53611. [Google Scholar] [CrossRef]
- Torelli, G.F.; Rozera, C.; Santodonato, L.; Peragine, N.; D’Agostino, G.; Montefiore, E.; Napolitano, M.R.; Monque, D.M.; Carlei, D.; Mariglia, P.; et al. A good manufacturing practice method to ex vivo expand natural killer cells for clinical use. Blood Transfus. 2015, 13, 464–471. [Google Scholar] [CrossRef]
- Alici, E.; Sutlu, T.; Björkstrand, B.; Gilljam, M.; Stellan, B.; Nahi, H.; Quezada, H.C.; Gahrton, G.; Ljunggren, H.G.; Dilber, M.S. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood 2008, 111, 3155–3162. [Google Scholar] [CrossRef] [Green Version]
- Koehl, U.; Brehm, C.; Huenecke, S.; Zimmermann, S.Y.; Kloess, S.; Bremm, M.; Ullrich, E.; Soerensen, J.; Quaiser, A.; Erben, S.; et al. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front. Oncol. 2013, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Ortaldo, J.R.; Winkler-Pickett, R.; Wigginton, J.; Horner, M.; Bere, E.W.; Mason, A.T.; Bhat, N.; Cherry, J.; Sanford, M.; Hodge, D.L.; et al. Regulation of ITAM-positive receptors: Role of IL-12 and IL-18. Blood 2006, 107, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, S.J.; Marusina, A.I.; Pathmanathan, I.; Borrego, F.; Coligan, J.E. IL-21 down-regulates NKG2D/DAP10 expression on human NK and CD8+ T cells. J. Immunol. 2006, 176, 1490–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becknell, B.; Caligiuri, M.A. Interleukin-2, interleukin-15, and their roles in human natural killer cells. Adv. Immunol. 2005, 86, 209–239. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.P.; Trinchieri, G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 155–168. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Huenecke, S.; Zimmermann, S.Y.; Kloess, S.; Esser, R.; Brinkmann, A.; Tramsen, L.; Koenig, M.; Erben, S.; Seidl, C.; Tonn, T.; et al. IL-2-driven regulation of NK cell receptors with regard to the distribution of CD16+ and CD16- subpopulations and in vivo influence after haploidentical NK cell infusion. J. Immunother. 2010, 33, 200–210. [Google Scholar] [CrossRef]
- Wang, K.S.; Frank, D.A.; Ritz, J. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood 2000, 95, 3183–3190. [Google Scholar] [CrossRef]
- Mao, Y.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Masvidal, L.; Liang, S.; Murray, S.; Larsson, O.; et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 2016, 128, 1475–1489. [Google Scholar] [CrossRef] [Green Version]
- Fehniger, T.A.; Cooper, M.A.; Caligiuri, M.A. Interleukin-2 and interleukin-15: Immunotherapy for cancer. Cytokine Growth Factor Rev. 2002, 13, 169–183. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, A.; Chu, S.; McElmurry, R.; Cooley, S.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous IL-15 Signaling Leads to Functional Exhaustion of Human Natural Killer Cells through Metabolic Changes That Alters Their In Vivo Anti-Tumor Activity. Blood 2016, 128, 551. [Google Scholar] [CrossRef]
- French, A.R.; Holroyd, E.B.; Yang, L.; Kim, S.; Yokoyama, W.M. IL-18 acts synergistically with IL-15 in stimulating natural killer cell proliferation. Cytokine 2006, 35, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Chaix, J.; Tessmer, M.S.; Hoebe, K.; Fuséri, N.; Ryffel, B.; Dalod, M.; Alexopoulou, L.; Beutler, B.; Brossay, L.; Vivier, E.; et al. Cutting edge: Priming of NK cells by IL-18. J. Immunol. 2008, 181, 1627–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMichael, E.L.; Jaime-Ramirez, A.C.; Guenterberg, K.D.; Luedke, E.; Atwal, L.S.; Campbell, A.R.; Hu, Z.; Tatum, A.S.; Kondadasula, S.V.; Mo, X.; et al. IL-21 Enhances Natural Killer Cell Response to Cetuximab-Coated Pancreatic Tumor Cells. Clin. Cancer. Res. 2017, 23, 489–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannello, A.; Boulassel, M.-R.; Samarani, S.; Tremblay, C.; Toma, E.; Routy, J.-P.; Ahmad, A. IL-21 enhances NK cell functions and survival in healthy and HIV-infected patients with minimal stimulation of viral replication. J. Leukoc. Biol. 2010, 87, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Kasaian, M.T.; Whitters, M.J.; Carter, L.L.; Lowe, L.D.; Jussif, J.M.; Deng, B.; Johnson, K.A.; Witek, J.S.; Senices, M.; Konz, R.F.; et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: A mediator of the transition from innate to adaptive immunity. Immunity 2002, 16, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ye, L.J.; Ren, H.L.; Huyan, T.; Li, J.; Shi, J.L.; Huang, Q.S. Multiple effects of IL-21 on human NK cells in ex vivo expansion. Immunobiology 2015, 220, 876–888. [Google Scholar] [CrossRef]
- Granzin, M.; Stojanovic, A.; Miller, M.; Childs, R.; Huppert, V.; Cerwenka, A. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. Oncoimmunology 2016, 5, e1219007. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.P.; Jang, Y.Y.; Kim, S.; Koh, S.S.; Lee, J.J.; Kim, J.S.; Thi Phan, M.T.; Shin, D.J.; Shin, M.G.; Lee, S.H.; et al. Effect of exposure to interleukin-21 at various time points on human natural killer cell culture. Cytotherapy 2014, 16, 1419–1430. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leivas, A.; Perez-Martinez, A.; Blanchard, M.J.; Martín-Clavero, E.; Fernández, L.; Lahuerta, J.J.; Martinez-Lopez, J. Novel treatment strategy with autologous activated and expanded natural killer cells plus anti-myeloma drugs for multiple myeloma. Oncoimmunology 2016, 5, e1250051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Ji, Y.; Gattinoni, L.; Zhang, L.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol. Immunother. 2013, 62, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, S.; Hernandez, J.; Vence, L.; Hwu, P.; Radvanyi, L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J. Immunol. 2010, 184, 452–465. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Brien, S.M.; Klampatsa, A.; Thompson, J.C.; Martinez, M.C.; Hwang, W.-T.; Rao, A.S.; Standalick, J.E.; Kim, S.; Cantu, E.; Litzky, L.A.; et al. Function of Human Tumor-Infiltrating Lymphocytes in Early-Stage Non–Small Cell Lung Cancer. Cancer Immunol. Res. 2019, 7, 896. [Google Scholar] [CrossRef]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhu, B.; Son, Y.M.; Wang, Z.; Jiang, L.; Xiang, M.; Ye, Z.; Beckermann, K.E.; Wu, Y.; Jenkins, J.W.; et al. The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 2019, 51, 491–507.e497. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Dechant, C.; Cui, X.; Chongsathidkiet, P.; Wilkinson, D.; Waibl-Polania, J.; Sanchez-Perez, L.; Fecci, P.E. 4-1BB Agonism Averts TIL Exhaustion and Licenses PD-1 Blockade in Glioblastoma and Other Intracranial Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1349–1358. [Google Scholar] [CrossRef]
- Hall, M.; Liu, H.; Malafa, M.; Centeno, B.; Hodul, P.J.; Pimiento, J.; Pilon-Thomas, S.; Sarnaik, A.A. Expansion of tumor-infiltrating lymphocytes (TIL) from human pancreatic tumors. J. Immunother. Cancer 2016, 4, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Stachura, P.; Xu, H.C.; Bhatia, S.; Borkhardt, A.; Lang, P.A.; Pandyra, A.A. Senescent Tumor CD8(+) T Cells: Mechanisms of Induction and Challenges to Immunotherapy. Cancers 2020, 12, 2828. [Google Scholar] [CrossRef]
- Roninson, I.B.; Broude, E.V.; Chang, B.D. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resistance Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2001, 4, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Andersen, J.; Kapahi, P.; Melov, S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin. Cancer Biol. 2011, 21, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-Induced Senescent T Cells with Suppressor Function: A Potential Form of Tumor Immune Evasion. Cancer Res. 2008, 68, 870. [Google Scholar] [CrossRef] [Green Version]
- Dossett, M.L.; Teague, R.M.; Schmitt, T.M.; Tan, X.; Cooper, L.J.; Pinzon, C.; Greenberg, P.D. Adoptive immunotherapy of disseminated leukemia with TCR-transduced, CD8+ T cells expressing a known endogenous TCR. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 742–749. [Google Scholar] [CrossRef]
- Zhang, L.; Morgan, R.A. Genetic engineering with T cell receptors. Adv. Drug Deliv. Rev. 2012, 64, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, M.; King, J.W.; Xue, S.A.; Voisine, C.; Holler, A.; Wright, G.P.; Waxman, J.; Morris, E.; Stauss, H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, L.T.; Reijmers, R.M.; Wouters, A.K.; Kweekel, C.; Remst, D.F.G.; Pothast, C.R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Simultaneous Deletion of Endogenous TCRαβ for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol. Ther. 2020, 28, 64–74. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Heemskerk, M.H.M.; Hoogeboom, M.; Hagedoorn, R.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F. Reprogramming of Virus-specific T Cells into Leukemia-reactive T Cells Using T Cell Receptor Gene Transfer. J. Exp. Med. 2004, 199, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Marieke, G.; van Egmond, H.M.E.; Helen, B.-P.; van der Hoorn, M.A.W.G.; Renate, S.H.; Michel, G.D.K.; Nikolai, S.; Roel, W.; Falkenburg, J.H.F.; Mirjam, H.M.H. Genetic engineering of virus-specific T cells with T-cell receptors recognizing minor histocompatibility antigens for clinical application. Haematologica 2008, 93, 1535–1543. [Google Scholar] [CrossRef]
- Marleen, M.V.L.; de Renate, B.; van Ellis, L.; Pauline, M.; Inge, J.; Falkenburg, J.H.F.; Mirjam, H.M.H. A Good Manufacturing Practice procedure to engineer donor virus-specific T cells into potent anti-leukemic effector cells. Haematologica 2014, 99, 759–768. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- van Loenen, M.M.; Hagedoorn, R.S.; Kester, M.G.; Hoogeboom, M.; Willemze, R.; Falkenburg, J.H.; Heemskerk, M.H. Kinetic preservation of dual specificity of coprogrammed minor histocompatibility antigen-reactive virus-specific T cells. Cancer Res. 2009, 69, 2034–2041. [Google Scholar] [CrossRef] [Green Version]
- Hart, D.P.; Xue, S.A.; Thomas, S.; Cesco-Gaspere, M.; Tranter, A.; Willcox, B.; Lee, S.P.; Steven, N.; Morris, E.C.; Stauss, H.J. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther. 2008, 15, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, J.A.; Bajwa, G.; Carreres, B.; Hoyer, E.; Gruber, I.; Martínez-Paniagua, M.A.; Yu, Y.-R.; Nouraee, N.; Sadeghi, F.; Wu, M.; et al. Single-cell transcriptomics identifies multiple pathways underlying antitumor function of TCR- and CD8αβ-engineered human CD4+ T cells. Sci. Adv. 2020, 6, eaaz7809. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Feng, X.; Abhyankar, H.; Romero, E.; Wu, M.F.; Heslop, H.E.; Barth, P.; Dotti, G.; Savoldo, B. Survivin-specific T cell receptor targets tumor but not T cells. J. Clin. Investig. 2015, 125, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, G.; Lanz, I.; Cardenas, M.; Brenner, M.K.; Arber, C. Transgenic CD8αβ co-receptor rescues endogenous TCR function in TCR-transgenic virus-specific T cells. J. Immunother. Cancer 2020, 8, e001487. [Google Scholar] [CrossRef]
- Laugel, B.; van den Berg, H.A.; Gostick, E.; Cole, D.K.; Wooldridge, L.; Boulter, J.; Milicic, A.; Price, D.A.; Sewell, A.K. Different T Cell Receptor Affinity Thresholds and CD8 Coreceptor Dependence Govern Cytotoxic T Lymphocyte Activation and Tetramer Binding Properties. J. Biol. Chem. 2007, 282, 23799–23810. [Google Scholar] [CrossRef] [Green Version]
- Laugel, B.; Cole, D.K.; Clement, M.; Wooldridge, L.; Price, D.A.; Sewell, A.K. The multiple roles of the CD8 coreceptor in T cell biology: Opportunities for the selective modulation of self-reactive cytotoxic T cells. J. Leukoc. Biol. 2011, 90, 1089–1099. [Google Scholar] [CrossRef]
- Helsen, C.W.; Hammill, J.A.; Lau, V.W.C.; Mwawasi, K.A.; Afsahi, A.; Bezverbnaya, K.; Newhook, L.; Hayes, D.L.; Aarts, C.; Bojovic, B.; et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat. Commun. 2018, 9, 3049. [Google Scholar] [CrossRef] [Green Version]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.R.; Flaswinkel, H.; Reth, M.; Scott, D.W. Immunoreceptor tyrosine-based activation motif is required to signal pathways of receptor-mediated growth arrest and apoptosis in murine B lymphoma cells. J. Immunol. 1995, 155, 652–661. [Google Scholar]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Understanding the immunodeficiency in chronic lymphocytic leukemia: Potential clinical implications. Hematol. Oncol. Clin. N. Am. 2013, 27, 207–235. [Google Scholar] [CrossRef]
- te Raa, G.D.; Tonino, S.H.; Remmerswaal, E.B.; van Houte, A.J.; Koene, H.R.; van Oers, M.H.; Kater, A.P. Chronic lymphocytic leukemia specific T-cell subset alterations are clone-size dependent and not present in monoclonal B lymphocytosis. Leuk. Lymphoma 2012, 53, 2321–2325. [Google Scholar] [CrossRef]
- Tonino, S.H.; van de Berg, P.J.; Yong, S.L.; ten Berge, I.J.; Kersten, M.J.; van Lier, R.A.; van Oers, M.H.; Kater, A.P. Expansion of effector T cells associated with decreased PD-1 expression in patients with indolent B cell lymphomas and chronic lymphocytic leukemia. Leuk. Lymphoma 2012, 53, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, A.G.; Johnson, A.J.; Lee, A.M.; Gorgün, G.; Le Dieu, R.; Blum, W.; Byrd, J.C.; Gribben, J.G. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J. Clin. Invest. 2008, 118, 2427–2437. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Görgün, G.; Holderried, T.A.; Zahrieh, D.; Neuberg, D.; Gribben, J.G. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J. Clin. Investig. 2005, 115, 1797–1805. [Google Scholar] [CrossRef]
- Gorgun, G.; Ramsay, A.G.; Holderried, T.A.; Zahrieh, D.; Le Dieu, R.; Liu, F.; Quackenbush, J.; Croce, C.M.; Gribben, J.G. E(mu)-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 6250–6255. [Google Scholar] [CrossRef] [Green Version]
- van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Garfall, A.L.; Dancy, E.K.; Cohen, A.D.; Hwang, W.T.; Fraietta, J.A.; Davis, M.M.; Levine, B.L.; Siegel, D.L.; Stadtmauer, E.A.; Vogl, D.T.; et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv. 2019, 3, 2812–2815. [Google Scholar] [CrossRef] [Green Version]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stüber, T.; Monjezi, R.; Wallstabe, L.; Kühnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wöckel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Ankri, C.; Shamalov, K.; Horovitz-Fried, M.; Mauer, S.; Cohen, C.J. Human T Cells Engineered To Express a Programmed Death 1/28 Costimulatory Retargeting Molecule Display Enhanced Antitumor Activity. J. Immunol. 2013, 1203085. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Samelson, L.E. Signal transduction mediated by the T cell antigen receptor: The role of adapter proteins. Annu. Rev. Immunol. 2002, 20, 371–394. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Daniyan, A.F.; Lopez, A.V.; Purdon, T.J.; Brentjens, R.J. Cytokine IL-36γ improves CAR T-cell functionality and induces endogenous antitumor response. Leukemia 2020. [Google Scholar] [CrossRef] [PubMed]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer. Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Davies, J.S.; Serna, C.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A.; Hinrichs, C.S. Enhanced efficacy and limited systemic cytokine exposure with membrane-anchored interleukin-12 T-cell therapy in murine tumor models. J. Immunother. Cancer 2020, 8, e000210. [Google Scholar] [CrossRef] [Green Version]
- Lapteva, N.; Gilbert, M.; Diaconu, I.; Rollins, L.A.; Al-Sabbagh, M.; Naik, S.; Krance, R.A.; Tripic, T.; Hiregange, M.; Raghavan, D.; et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor–Expressing T Cells. Clin. Cancer. Res. 2019, 25, 7340. [Google Scholar] [CrossRef] [Green Version]
- von Kalle, C.; Deichmann, A.; Schmidt, M. Vector Integration and Tumorigenesis. Hum. Gene Ther. 2014, 25, 475–481. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L. Activation of T cells: Releasing the brakes by proteolytic elimination of Cbl-b. Sci. Signal. 2009, 2, pe38. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Qin, H.; Yates, B.; Su, L.; Shalabi, H.; Raffeld, M.; Ahlman, M.A.; Stetler-Stevenson, M.; Yuan, C.; Guo, S.; et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 2019, 3, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (iPSC)-Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59. [Google Scholar] [CrossRef]
- Zhang, Y.; Wallace, D.L.; de Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265. [Google Scholar] [CrossRef]
- Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef] [Green Version]
- Oei, V.Y.S.; Siernicka, M.; Graczyk-Jarzynka, A.; Hoel, H.J.; Yang, W.; Palacios, D.; Almåsbak, H.; Bajor, M.; Clement, D.; Brandt, L.; et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol. Res. 2018, 6, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Kumagai, A.; Iriguchi, S.; Yasui, Y.; Miyasaka, T.; Nakagoshi, K.; Nakane, K.; Saito, K.; Takahashi, M.; Sasaki, A.; et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti–glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020, 111, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Badeti, S.; Tseng, H.-c.; Ma, M.T.; Liu, T.; Jiang, J.-G.; Liu, C.; Liu, D. Superior Expansion and Cytotoxicity of Human Primary NK and CAR-NK Cells from Various Sources via Enriched Metabolic Pathways. Mol. Ther. Methods Clin. Dev. 2020, 18, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Qasim, W. Allogeneic CAR T cell therapies for leukemia. Am. J. Hematol. 2019, 94, S50–S54. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, R.J.; Prockop, S.; Hasan, A.N.; Koehne, G.; Doubrovina, E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant. 2016, 51, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer. Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.-Z.; Zhu, Y.-Y.; Ruan, M.; Chen, L.; Zhang, Q.-Y. Local Irradiation Sensitized Tumors to Adoptive T Cell Therapy via Enhancing the Cross-Priming, Homing, and Cytotoxicity of Antigen-Specific CD8 T Cells. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bezu, L.; Gomes-da-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial Strategies for the Induction of Immunogenic Cell Death. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Guedan, S.; Alemany, R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 2018, 9, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination with Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular, phenotypic, and functional characteristics of T-cell dysfunctional states. (A) T-cell terminal differentiation, governed by the transcriptional repressor BLIMP1, is phenotypically associated with the re-expression of CD45RA, along with KLRG1, while downregulating IL-7R, leading to an impaired proliferation. (B) T-cell exhaustion is regulated by a gradual decrease in TCF1 and concordant increase in T-bet and TOX expression, associated with the upregulation of many inhibitory receptors. In addition to a diminished proliferative capacity, exhausted T cells become hyporesponsive, mainly shown by an impaired cytokine secretion capacity upon stimulation. (C) Senescent cells may engage various cell cycle regulators, such as p16 or p21 via p53 expression, to inhibit cell cycle progression. Phenotypically, these cells show upregulation of CD57 and/or KLRG1 at the cell surface, and downregulation of co-stimulatory molecules CD27 and CD28. It is associated with impaired mitochondrial functions, an increase in senescence-associated β-galactosidase (SA-β-Gal) activity, and proliferation arrest. (D) Activation-induced cell death requires the activation of a death receptor. Upon TCR stimulation, various transcription factors are induced, such as NF-κB, NFAT, and c-MYC, which can lead to the expression of FASL. FASL may trigger the death receptor FAS on adjacent cells, activating caspase-8 and leading to direct effector caspase activation and/or mitochondrial release of cytochrome c, resulting in apoptosis. This figure was created with BioRender.com.
Figure 1.
Molecular, phenotypic, and functional characteristics of T-cell dysfunctional states. (A) T-cell terminal differentiation, governed by the transcriptional repressor BLIMP1, is phenotypically associated with the re-expression of CD45RA, along with KLRG1, while downregulating IL-7R, leading to an impaired proliferation. (B) T-cell exhaustion is regulated by a gradual decrease in TCF1 and concordant increase in T-bet and TOX expression, associated with the upregulation of many inhibitory receptors. In addition to a diminished proliferative capacity, exhausted T cells become hyporesponsive, mainly shown by an impaired cytokine secretion capacity upon stimulation. (C) Senescent cells may engage various cell cycle regulators, such as p16 or p21 via p53 expression, to inhibit cell cycle progression. Phenotypically, these cells show upregulation of CD57 and/or KLRG1 at the cell surface, and downregulation of co-stimulatory molecules CD27 and CD28. It is associated with impaired mitochondrial functions, an increase in senescence-associated β-galactosidase (SA-β-Gal) activity, and proliferation arrest. (D) Activation-induced cell death requires the activation of a death receptor. Upon TCR stimulation, various transcription factors are induced, such as NF-κB, NFAT, and c-MYC, which can lead to the expression of FASL. FASL may trigger the death receptor FAS on adjacent cells, activating caspase-8 and leading to direct effector caspase activation and/or mitochondrial release of cytochrome c, resulting in apoptosis. This figure was created with BioRender.com.

Figure 2.
Interventions aiming at limiting T-cell dysfunction for adoptive immunotherapy. (A) Conventional αβ T cells are generally stimulated with specific peptides that may be presented by professional antigen-presenting cells such as dendritic cells (DCs) and supported by IL-2 supplementation and/or other cytokines. However, repetitive or chronic antigen stimulation promotes the gradual accumulation of dysfunctional cells over time. Many interventions are under investigation to maintain the cells in a less differentiated state. A shorter culture time allows for the maintenance of a higher percentage of healthy cells, but this may limit the number of cells manufactured. The addition of various factors to promote a memory phenotype differentiation is also evaluated, including the use of IL-21, TGFβ, histone deacetylase (HDAC) inhibitors, or AKT inhibitors. The rejuvenation of dysfunctional T cells by induced pluripotent stem cell technology can also generate less differentiated cells. (B) Unconventional γδ T cells are stimulated with amino-bisphosphonates and phosphoantigens, combined with IL-2. The pre-selection of T cells with a favorable phenotype, excluding cells expressing inhibitory receptors such as PD-1, has been investigated to limit the development of dysfunctional features. (C) Natural killer (NK) cells are mainly activated with IL-2 and feeder cells. Supplementation with various cytokines or agonists of co-stimulatory molecules (e.g., 4-1BB) can shape their differentiation status and modulate their receptor expression pattern. The engineering of feeder cells as well as the NK cells themselves to express these factors is also underway. (D) Tumor-infiltrating lymphocytes are stimulated with anti-CD3 with or without anti-CD28, with IL-2 supplementation. As for conventional T cells, the addition of various cytokines can promote a memory phenotype differentiation. The combination of a 4-1BB agonist and PD-1 blockade can also prevent the development of dysfunction. Inhibition of p38MAPK signaling has further shown to limit development of senescence features. This figure was created with BioRender.com.
Figure 2.
Interventions aiming at limiting T-cell dysfunction for adoptive immunotherapy. (A) Conventional αβ T cells are generally stimulated with specific peptides that may be presented by professional antigen-presenting cells such as dendritic cells (DCs) and supported by IL-2 supplementation and/or other cytokines. However, repetitive or chronic antigen stimulation promotes the gradual accumulation of dysfunctional cells over time. Many interventions are under investigation to maintain the cells in a less differentiated state. A shorter culture time allows for the maintenance of a higher percentage of healthy cells, but this may limit the number of cells manufactured. The addition of various factors to promote a memory phenotype differentiation is also evaluated, including the use of IL-21, TGFβ, histone deacetylase (HDAC) inhibitors, or AKT inhibitors. The rejuvenation of dysfunctional T cells by induced pluripotent stem cell technology can also generate less differentiated cells. (B) Unconventional γδ T cells are stimulated with amino-bisphosphonates and phosphoantigens, combined with IL-2. The pre-selection of T cells with a favorable phenotype, excluding cells expressing inhibitory receptors such as PD-1, has been investigated to limit the development of dysfunctional features. (C) Natural killer (NK) cells are mainly activated with IL-2 and feeder cells. Supplementation with various cytokines or agonists of co-stimulatory molecules (e.g., 4-1BB) can shape their differentiation status and modulate their receptor expression pattern. The engineering of feeder cells as well as the NK cells themselves to express these factors is also underway. (D) Tumor-infiltrating lymphocytes are stimulated with anti-CD3 with or without anti-CD28, with IL-2 supplementation. As for conventional T cells, the addition of various cytokines can promote a memory phenotype differentiation. The combination of a 4-1BB agonist and PD-1 blockade can also prevent the development of dysfunction. Inhibition of p38MAPK signaling has further shown to limit development of senescence features. This figure was created with BioRender.com.

Figure 3.
Interventions aiming at limiting T-cell dysfunction in engineered cells for adoptive immunotherapy. Engineered cells comprise transgenic T-cell receptor (TCR) and chimeric/synthetic antigen receptor T cells. (A) Genetic ablation of negative co-signaling molecules (e.g., PD-1) may be used to circumvent the effects of T-cell dysfunction. (B) Virus-specific T cells (VSTs) have been evaluated as a platform for artificial receptor expression in order to leverage their robust memory differentiation. (C) Another strategy to increase modified T-cell fitness is the use of a T-cell antigen coupler (TAC) to avoid tonic signaling and leverage endogenous TCR signaling. (D) Advanced engineering of CAR molecules involving the inactivation of two out of three ITAM motifs or (E) the blockade of suppressive cytokine signaling, here shown by the overexpression of a dominant negative TGFβ receptor, can also promote better T-cell functions. (F) An increase in c-Jun activity has been shown to further limit tonic signaling and T-cell dysfunction. (G) Disruption of the endogenous TCRα/β chains by CRISPR/Cas9 or specific integration of the CAR vector into the targeted DNA locus (e.g., TRAC) have shown additional efficacy. This was also true when CAR constructs inadvertently disrupted the TET2 and CBL genes. Other strategies include the enhancement of T-cell function through the production of stimulatory cytokines (e.g., IL-36γ, IL-12, and IL-18) or co-stimulatory molecules (e.g., CD28 and 4-1BB). This figure was created with BioRender.com.
Figure 3.
Interventions aiming at limiting T-cell dysfunction in engineered cells for adoptive immunotherapy. Engineered cells comprise transgenic T-cell receptor (TCR) and chimeric/synthetic antigen receptor T cells. (A) Genetic ablation of negative co-signaling molecules (e.g., PD-1) may be used to circumvent the effects of T-cell dysfunction. (B) Virus-specific T cells (VSTs) have been evaluated as a platform for artificial receptor expression in order to leverage their robust memory differentiation. (C) Another strategy to increase modified T-cell fitness is the use of a T-cell antigen coupler (TAC) to avoid tonic signaling and leverage endogenous TCR signaling. (D) Advanced engineering of CAR molecules involving the inactivation of two out of three ITAM motifs or (E) the blockade of suppressive cytokine signaling, here shown by the overexpression of a dominant negative TGFβ receptor, can also promote better T-cell functions. (F) An increase in c-Jun activity has been shown to further limit tonic signaling and T-cell dysfunction. (G) Disruption of the endogenous TCRα/β chains by CRISPR/Cas9 or specific integration of the CAR vector into the targeted DNA locus (e.g., TRAC) have shown additional efficacy. This was also true when CAR constructs inadvertently disrupted the TET2 and CBL genes. Other strategies include the enhancement of T-cell function through the production of stimulatory cytokines (e.g., IL-36γ, IL-12, and IL-18) or co-stimulatory molecules (e.g., CD28 and 4-1BB). This figure was created with BioRender.com.

Figure 4.
Synthetic representation of concerns and interventions aimed at limiting cell dysfunction before, during, and after manufacturing process. This figure was created with BioRender.com.
Figure 4.
Synthetic representation of concerns and interventions aimed at limiting cell dysfunction before, during, and after manufacturing process. This figure was created with BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Janelle, V.; Delisle, J.-S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers 2021, 13, 598. https://doi.org/10.3390/cancers13040598
AMA Style
Janelle V, Delisle J-S. T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies. Cancers. 2021; 13(4):598. https://doi.org/10.3390/cancers13040598
Chicago/Turabian StyleJanelle, Valérie, and Jean-Sébastien Delisle. 2021. "T-Cell Dysfunction as a Limitation of Adoptive Immunotherapy: Current Concepts and Mitigation Strategies" Cancers 13, no. 4: 598. https://doi.org/10.3390/cancers13040598
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.