
Role of Epstein–Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma

 ,
,  , ,
, , 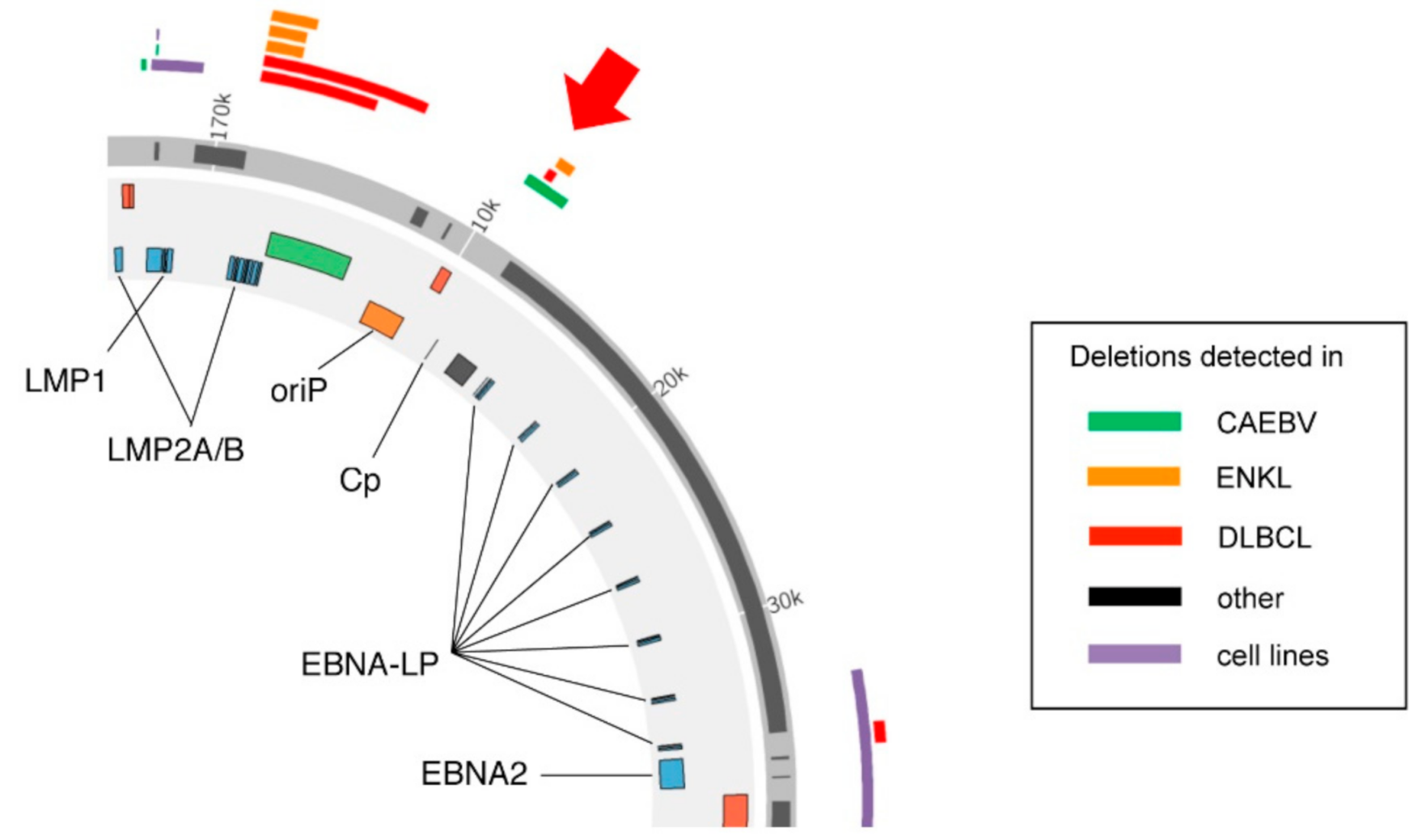{kind=link}
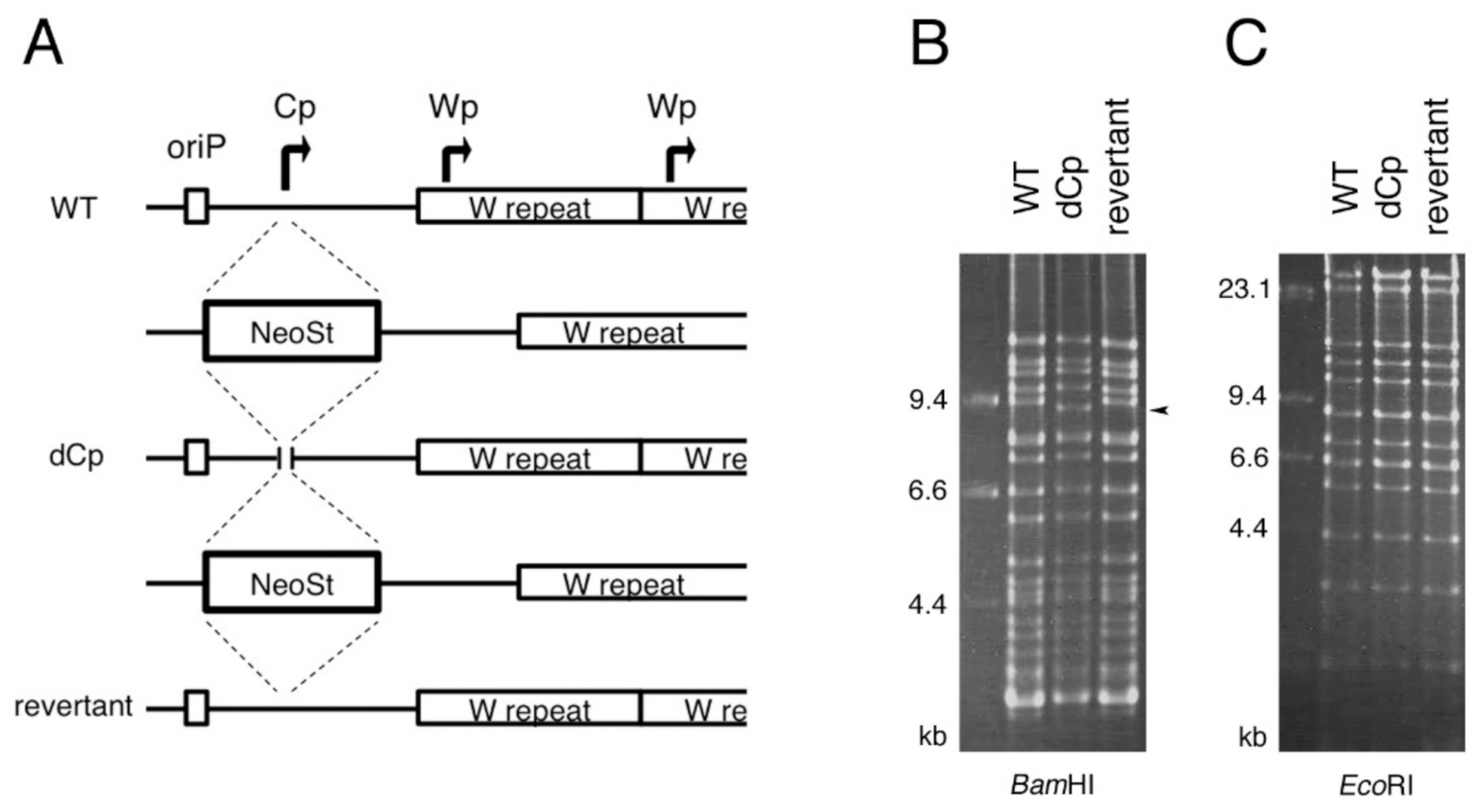{kind=link}
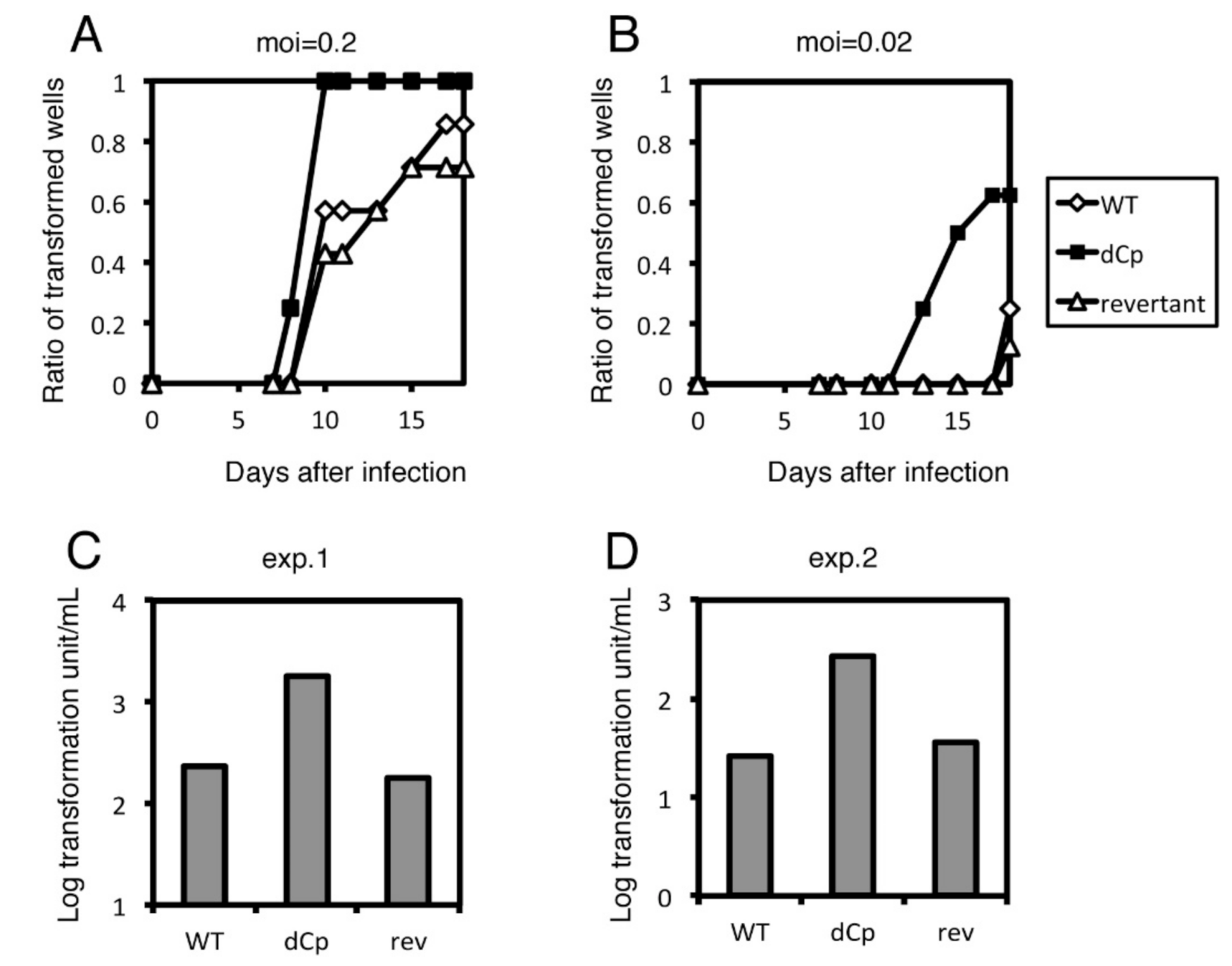{kind=link}
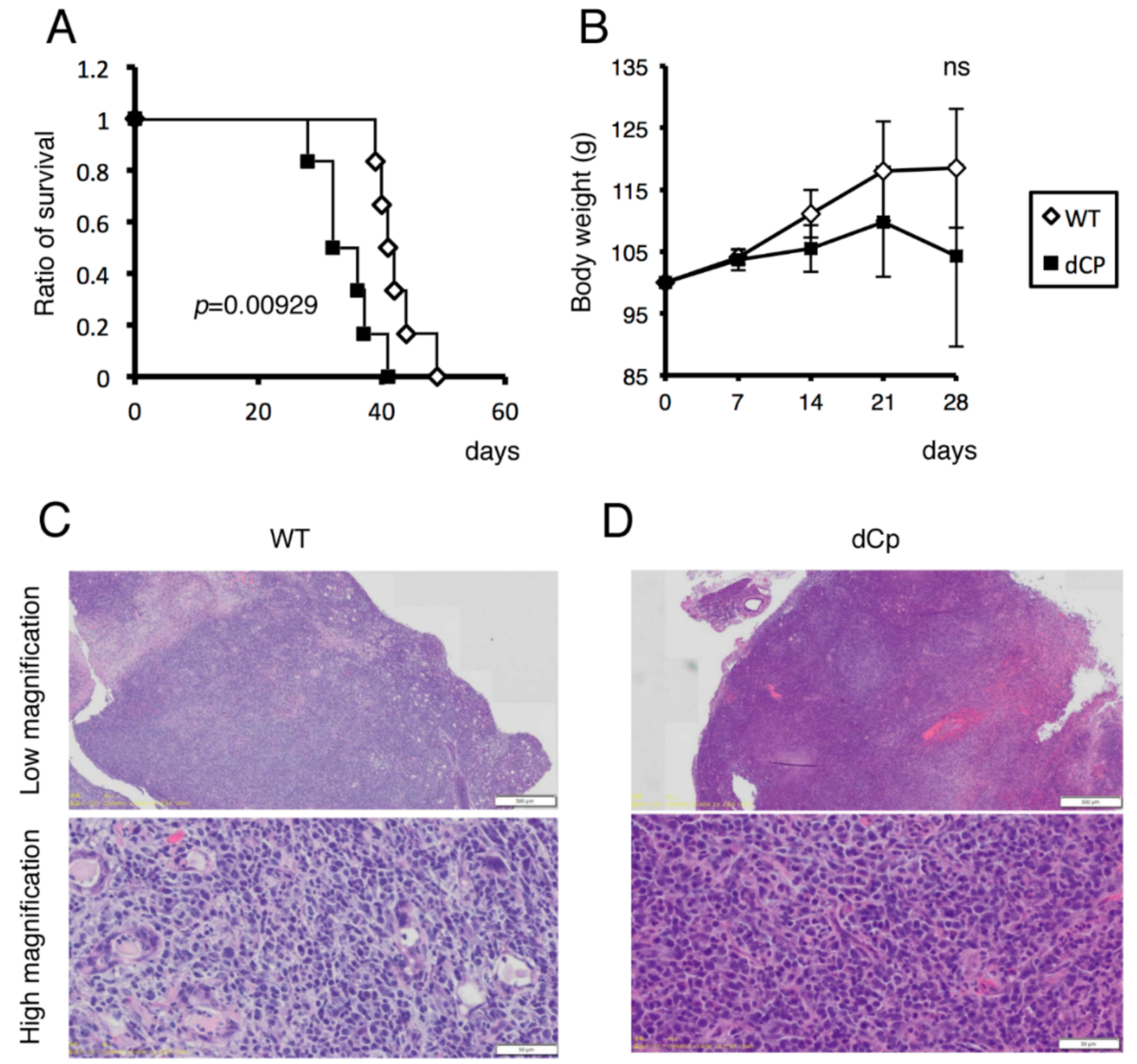{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
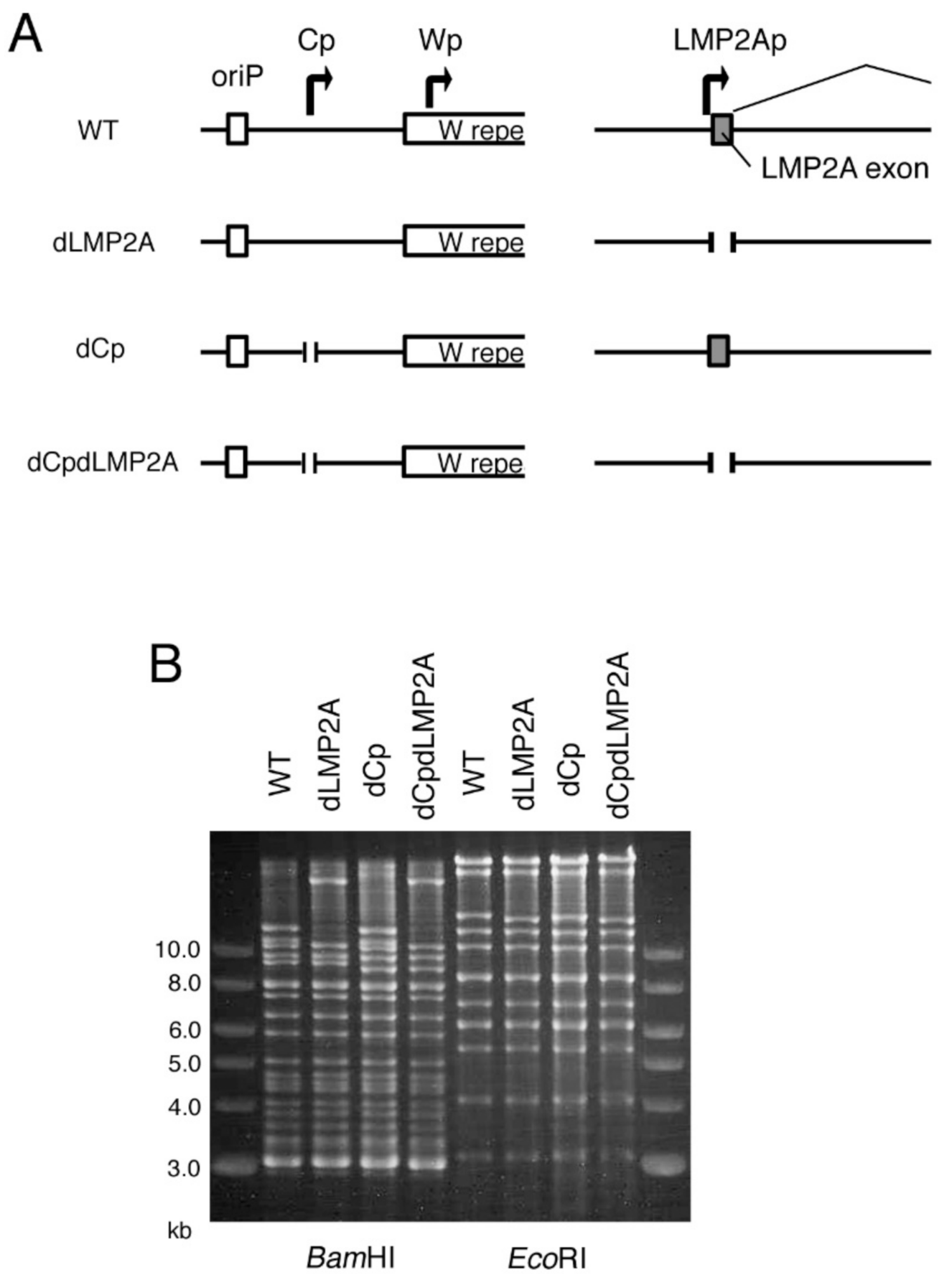
2.1. Construction of Cp-Deficient EBV-BAC Strain
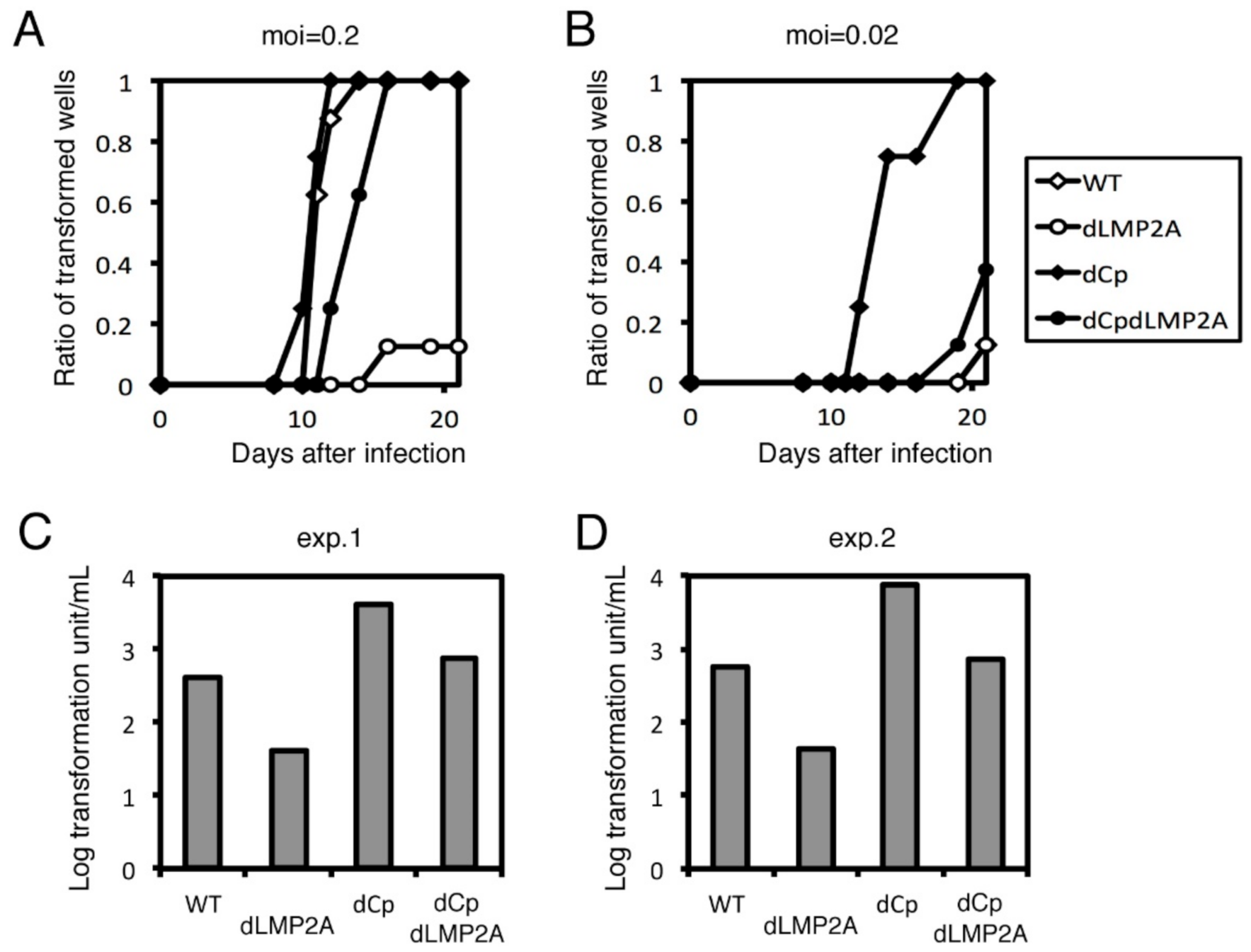
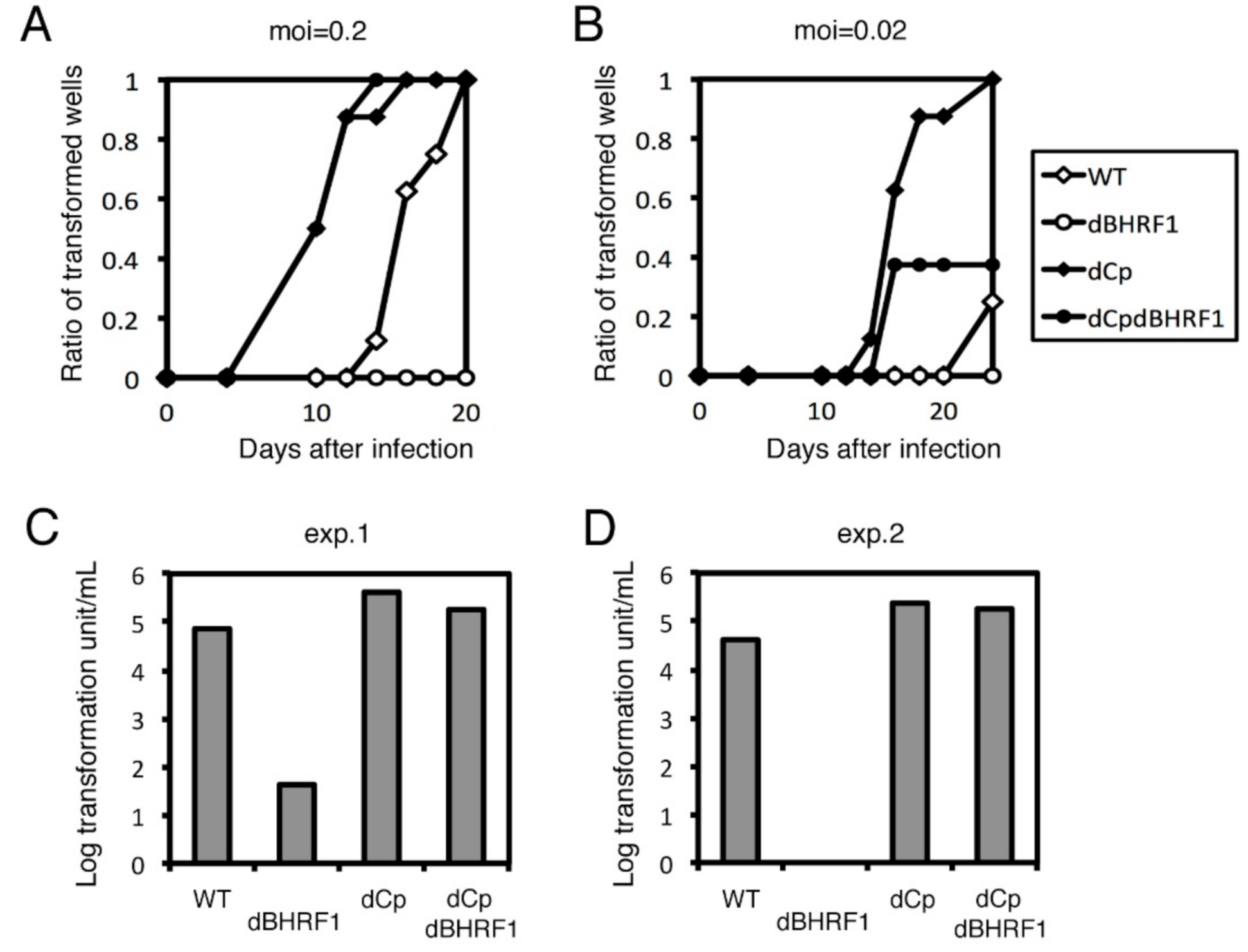
2.2. Cp Deletion Increases B Cell Transformation Efficiency
2.3. dCp Virus Increased Pathogenicity in an EBV-Associated LPD Mouse Model
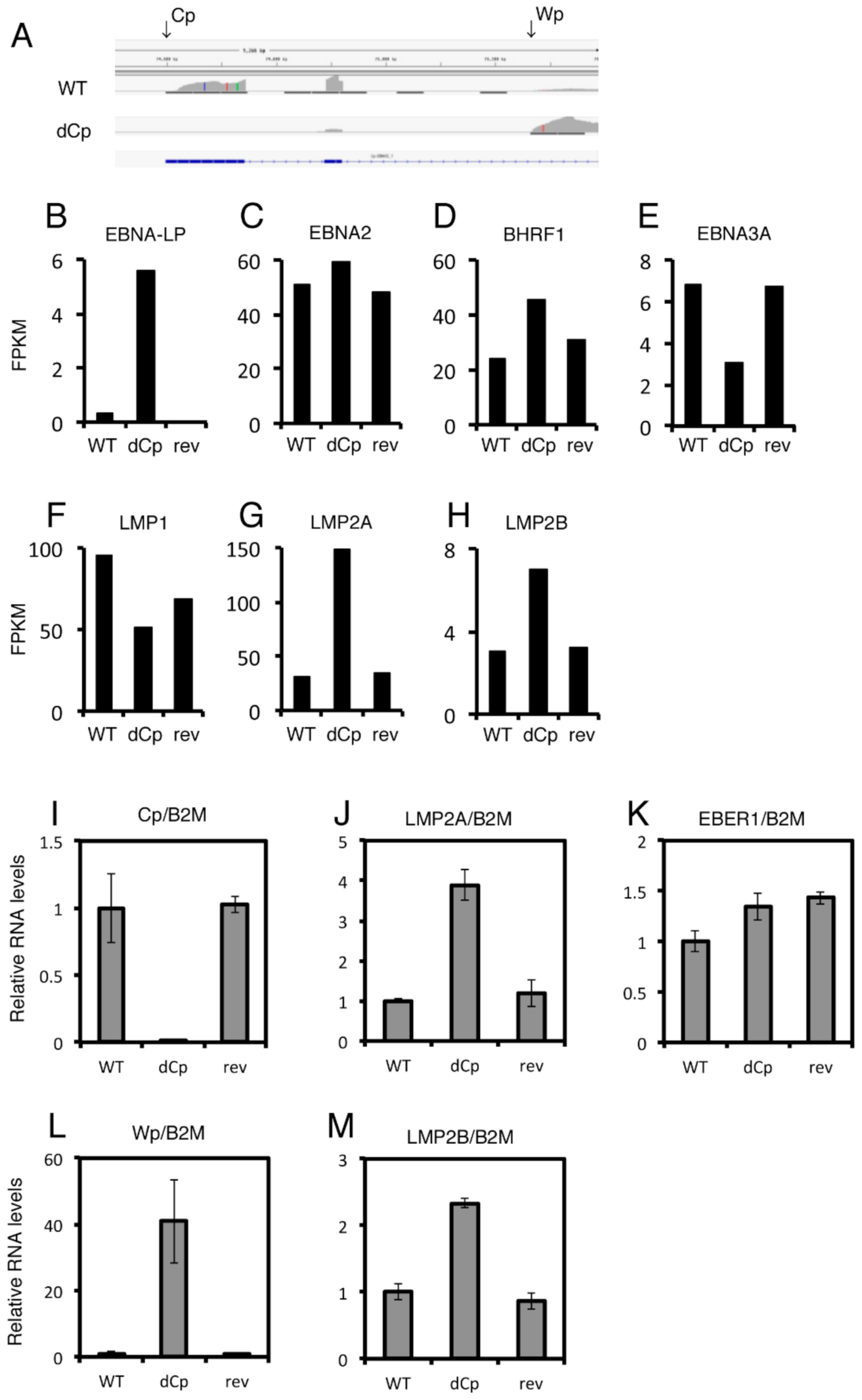
2.4. LCLs with the dCp EBV Show Increased Expression of LMP2A and LMP2B
2.5. LMP2A Does Not Account for the Enhanced Immortalization Efficiency in the dCp
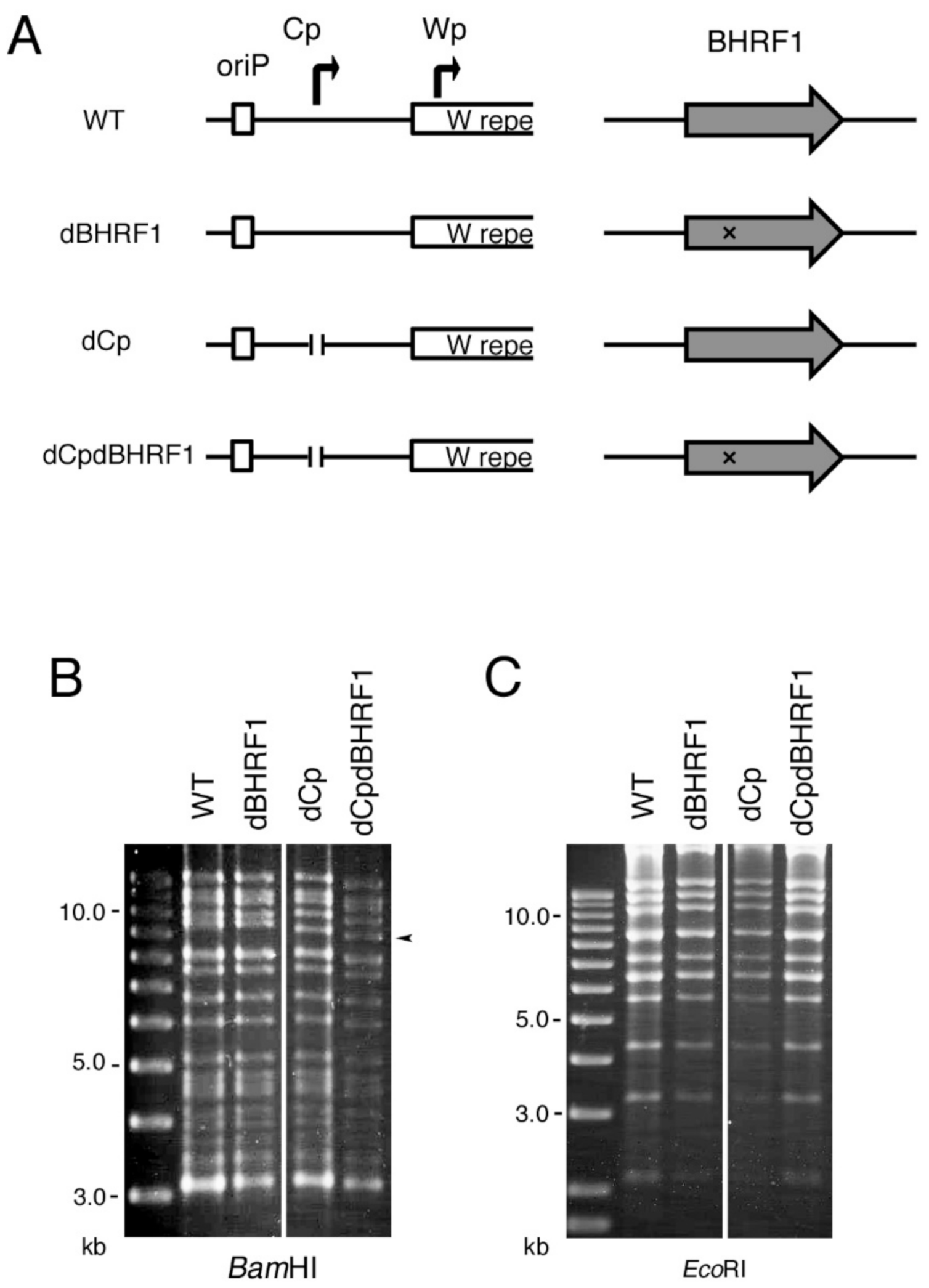
2.6. Viral BCL2 Homolog BHRF1 Does Not Account for the Enhanced Transformation Efficiency Seen in the dCp
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Construction of the dCp, dLMP2A and dBHRF1 EBV-BAC Genome and Transfection into HEK293 Cells
4.3. Lytic Induction and Progeny Virus Titration
4.4. B Cell Growth Transformation Assay
4.5. The EBV-Associated Lymphoproliferative Disorder Mouse Xenograft Model
4.6. RNA Sequencing (RNAseq) and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT–PCR)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev. Med. Virol. 2014, 24, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Schmidt, S.C.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2019, 33, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef]
- Peng, R.J.; Han, B.W.; Cai, Q.Q.; Zuo, X.Y.; Xia, T.; Chen, J.R.; Feng, L.N.; Lim, J.Q.; Chen, S.W.; Zeng, M.S.; et al. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019, 33, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Woisetschlaeger, M.; Jin, X.W.; Yandava, C.N.; Furmanski, L.A.; Strominger, J.L.; Speck, S.H. Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection. Proc. Natl. Acad. Sci. USA 1991, 88, 3942–3946. [Google Scholar] [CrossRef] [Green Version]
- Woisetschlaeger, M.; Yandava, C.N.; Furmanski, L.A.; Strominger, J.L.; Speck, S.H. Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc. Natl. Acad. Sci. USA 1990, 87, 1725–1729. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Tsai, M.H.; Shumilov, A.; Poirey, R.; Bannert, H.; Middeldorp, J.M.; Feederle, R.; Delecluse, H.J. The Epstein-Barr Virus BART miRNA Cluster of the M81 Strain Modulates Multiple Functions in Primary B Cells. PLoS Pathog. 2015, 11, e1005344. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S. Characterization of Epstein-Barr virus recombinants with deletions of the BamHI C promoter. Virology 1996, 217, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.J.; Nagra, J.; Rowe, M.; Bell, A.I.; Rickinson, A.B. The Epstein-Barr virus BamHI C promoter is not essential for B cell immortalization in vitro, but it greatly enhances B cell growth transformation. J. Virol. 2015, 89, 2483–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba Abdullah, M.M.; Palermo, R.D.; Palser, A.L.; Grayson, N.E.; Kellam, P.; Correia, S.; Szymula, A.; White, R.E. Heterogeneity of the Epstein-Barr Virus (EBV) Major Internal Repeat Reveals Evolutionary Mechanisms of EBV and a Functional Defect in the Prototype EBV Strain B95-8. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Longnecker, R.; Miller, C.L.; Tomkinson, B.; Miao, X.Q.; Kieff, E. Deletion of DNA encoding the first five transmembrane domains of Epstein-Barr virus latent membrane proteins 2A and 2B. J. Virol. 1993, 67, 5068–5074. [Google Scholar] [CrossRef] [Green Version]
- Brielmeier, M.; Mautner, J.; Laux, G.; Hammerschmidt, W. The latent membrane protein 2 gene of Epstein-Barr virus is important for efficient B cell immortalization. J. Gen. Virol. 1996, 77 Pt 11, 2807–2818. [Google Scholar] [CrossRef]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, A.M.; Letai, A. BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ. 2008, 15, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef] [Green Version]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [Green Version]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, A.; Maruo, S.; Ito, T.; Ito, M.; Katsumura, K.R.; Takada, K. Epstein-Barr virus-encoded Bcl-2 homologue functions as a survival factor in Wp-restricted Burkitt lymphoma cell line P3HR-1. J. Virol. 2010, 84, 2893–2901. [Google Scholar] [CrossRef] [Green Version]
- Tempera, I.; Lieberman, P.M. Epigenetic regulation of EBV persistence and oncogenesis. Semin. Cancer Biol. 2014, 26, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, D.; Minamitani, T.; Samanta, M. Epstein-Barr virus latent membrane protein 2A contributes to anoikis resistance through ERK activation. J. Virol. 2013, 87, 8227–8234. [Google Scholar] [CrossRef] [Green Version]
- Delecluse, H.J.; Hilsendegen, T.; Pich, D.; Zeidler, R.; Hammerschmidt, W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8245–8250. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Isomura, H.; Yamashita, Y.; Toyama, S.; Sato, Y.; Nakayama, S.; Kudoh, A.; Iwahori, S.; Kanda, T.; Tsurumi, T. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 2009, 389, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Konishi, N.; Narita, Y.; Hijioka, F.; Masud, H.M.A.A.; Sato, Y.; Kimura, H.; Murata, T. BGLF2 Increases Infectivity of Epstein-Barr Virus by Activating AP-1 upon. mSphere 2018, 3, e00138-18. [Google Scholar] [CrossRef] [Green Version]
- Al Masud, H.M.A.; Watanabe, T.; Yoshida, M.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. Epstein-Barr Virus BKRF4 Gene Product Is Required for Efficient Progeny Production. J. Virol. 2017, 91, e00975-17. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3, e00478-18. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabuchi, S.; Hijioka, F.; Watanabe, T.; Yanagi, Y.; Okuno, Y.; Masud, H.M.A.A.; Sato, Y.; Murata, T.; Kimura, H. Role of Epstein–Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 561. https://doi.org/10.3390/cancers13030561
Mabuchi S, Hijioka F, Watanabe T, Yanagi Y, Okuno Y, Masud HMAA, Sato Y, Murata T, Kimura H. Role of Epstein–Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma. Cancers. 2021; 13(3):561. https://doi.org/10.3390/cancers13030561
Chicago/Turabian StyleMabuchi, Seiyo, Fumiya Hijioka, Takahiro Watanabe, Yusuke Yanagi, Yusuke Okuno, H. M. Abdullah Al Masud, Yoshitaka Sato, Takayuki Murata, and Hiroshi Kimura. 2021. "Role of Epstein–Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma" Cancers 13, no. 3: 561. https://doi.org/10.3390/cancers13030561